

Abstract
The application of structural genomics methods and approaches to proteins from organisms causing infectious diseases is making available the three dimensional structures of many proteins that are potential drug targets and laying the groundwork for structure aided drug discovery efforts. There are a number of structural genomics projects with a focus on pathogens that have been initiated worldwide. The Center for Structural Genomics of Infectious Diseases (CSGID) was recently established to apply state-of-the-art high throughput structural biology technologies to the characterization of proteins from the National Institute for Allergy and Infectious Diseases (NIAID) category A–C pathogens and organisms causing emerging, or re-emerging infectious diseases. The target selection process emphasizes potential biomedical benefits. Selected proteins include known drug targets and their homologs, essential enzymes, virulence factors and vaccine candidates. The Center also provides a structure determination service for the infectious disease scientific community. The ultimate goal is to generate a library of structures that are available to the scientific community and can serve as a starting point for further research and structure aided drug discovery for infectious diseases. To achieve this goal, the CSGID will determine protein crystal structures of 400 proteins and protein-ligand complexes using proven, rapid, highly integrated, and cost-effective methods for such determination, primarily by X-ray crystallography. High throughput crystallographic structure determination is greatly aided by frequent, convenient access to high-performance beamlines at third-generation synchrotron X-ray sources.
Keywords: CSGID, structural genomics, infectious diseases, drug discovery, structural biology, protein structure
ANTI-MICROBIAL DRUG DISCOVERY AND STRUCTURE AIDED DRUG DISCOVERY
It is widely recognized that anti-microbial drug discovery has not been very successful in recent years [1]. Coupled with the rise in drug resistance in many pathogens [2, 3], this has resulted in a need for new targets and new approaches to anti-microbial drug discovery. High throughput screening for broad spectrum antibiotics in particular has not been highly successful [1]. There are a number of factors that have limited recent success, but it is clear that discovery of broad spectrum antibacterials is lagging behind society's needs. One alternate direction that is being explored is to focus on new drug targets and approaches that, while unlikely to yield broad-spectrum therapeutics, may be useful for treating specific pathogens [4]. In this context, when one is targeting a specific protein from a specific organism, structure aided drug discovery [5, 6] becomes an important approach.
The development of structure aided approaches to drug discovery has provided a number of advantages, especially for optimization of lead compounds to improve affinity for a structurally characterized target [5]. On the other hand, if the aim is broad-spectrum antimicrobials, the associated disadvantage is that lead compounds are optimized with respect to a specific target protein, not necessarily for broad spectrum effects. A second limitation is the fundamental requirement for a validated target that can be studied using structural biology methods, this is especially a problem for some membrane associated targets.
The main advantage of having structural information on the target protein and its complexes with a variety of compounds is that it can provide important and useful hints for modifications to starting compounds that are likely to improve their affinity for the target protein [5]. Structure aided approaches are important for improving affinity, but their contribution to the related problem of improving specificity is less direct. One can improve the number and strength of the interactions between a small molecule lead and the target protein to improve the affinity. A by-product of this is that the optimized lead compound is unlikely to simultaneously improve its affinity for many other proteins, so that improving affinity for the target will also generally improve its specificity. As structure aided optimization is generally practiced directly optimizing the specificity of a lead compound is not straightforward. This is because only one protein is looked at when it would be beneficial to compare the effects of possible chemical alterations to a lead compound on all proteins that might interact with it. Similarly, optimizing affinity for one target is unlikely to improve the affinity for a range of related targets, as desired for a broad-spectrum antibiotic. In attempts to convert a lead compound into a high affinity, broad spectrum antibiotic, structure aided approaches may, in fact, be counter-productive unless one is extremely careful to favor interactions with highly conserved residues and minimize interactions with residues that are less widely conserved. Screening natural products that help an organism defend itself against competitors is likely to be more effective for identifying broad spectrum leads because evolution has already selected compounds that are useful. However, this also means that natural resistance mechanisms are likely to already exist.
Where structure aided approaches provide great potential is in the discovery and optimization of selective therapeutics that target specific pathogens or disease processes. In this situation one can potentially use multiple structures of homologous proteins from pathogens and nonpathogenic commensal organisms from the microbiome to optimize the specificity of lead compounds to target specific organisms while reducing the effect on others and avoiding general damage to the microbiome. It is here that a structural genomics effort can make a major contribution. Although this project has just begun, it is anticipated that by applying high throughput structure determination methods to a series of proteins (potential drug targets) from a number of organisms, the array of structural information that results can be integrated and will be utilized to optimize lead compounds with the desired specificity. An ecological metaphor for this approach is selectively removing weeds from a forest rather than burning the forest to get rid of the weeds. This should have beneficial effects. One example of a problem associated with the use of broad-spectrum antibiotic treatment is the increase in the likelihood of subsequent infections, as for example, occurs with Clostridium difficile [7].
STRUCTURAL GENOMICS PROJECTS FOCUSED ON INFECTIOUS DISEASES
Structural genomics as it is traditionally practiced, if one can say what is traditional in a field that is less than 10 years old, utilizes the fact that proteins with amino acid sequences that are significantly similar to each other will have very similar structures. Thus, if one wants to obtain structural information for a family of proteins, any one of them can supply it. Applying high throughput structural methods in parallel to a number of members of a protein sequence family allows one to filter out and stop work on any that fail, focusing work on the ones that successfully move from one phase of the work to the next. This approach expends a larger amount of effort, and cost, in the early stages, but increases the likelihood that the structure of some member of the family will be determined. Clearly, this approach is quite different from that applied during most structure aided drug discovery projects, where one wants the structure of a specific validated target protein, not just a protein related to it.
Efficiently applying structural genomics methods to infectious diseases requires a slight modification to the most common approach. High throughput requires the ability to work in parallel on many target proteins. Consequently, instead of working on proteins from a very wide variety of organisms to find the most suitable representative proteins, a relatively large number of proteins which potentially represent drug targets are selected from priority pathogens. Although these may not all be previously validated drug targets, the approach yields many structures of candidate proteins that are amenable to structure-aided methods, can then be further studied and tested to validate them as potential drug targets. An additional important benefit is that even if the protein targets do not fulfill criteria for industrial drug development, their structures will expand our knowledge of the selected pathogens. Still another benefit that is not widely appreciated is the large number of protein expression vectors and purified proteins that are produced and available to the scientific community.
Depending on one's outlook, an advantage, or possible problem, of applying structural genomics approaches and methods to the study of potential drug targets is that you are likely to end up with a narrow range of organisms that are susceptible to the lead compounds. Small differences in the interactions between a small molecule and the protein can result in large differences in affinity. Narrow spectrum antimicrobials require that the physician know what organism is causing an infection, and this requires tests that may delay treatment. A consequence is that the development of such compounds is not economically reasonable unless the disease is wide spread or a rapid diagnostic test is available. However, it seems that such narrow spectrum antimicrobials provide important advantages if the patient is considered as an ecosystem and one wishes to minimize damage to that system.
A number of large-scale projects have been undertaken over the past ten years that have had as their focus organisms causing infectious diseases. Among them are two the National Institute for Allergy and Infectious Diseases (NIAID) has recently established to work on proteins from their priority pathogens. These include the Seattle Structural Genomics Center for Infectious Diseases, SSGCID (http://ssgcid.org/, see article by P. Myler), and the Center for Structural Genomics of Infectious Diseases, CSGID (http://www.csgid.org/), discussed here. The organisms that are the focus of work in the CSGID are given in Table 1.
Table 1.
Initial Organism Focus of CSGID
| Super-kingdom | Class (or Phylum) | Genus (or Family) |
|---|---|---|
| Bacteria | Bacilli | Bacillus, Listeria, Staphyloccus, Streptococcus |
| Clostridia | Clostridium | |
| Epsilon-proteobacteria | Campylobacter, Helicobacter | |
| Gamma-proteobacteria | Coxiella, Escherichia, Francisella, Salmonella, Shigella, Vibrio, Yersinia | |
| Viruses | dsDNA viruses | Orthopoxvirus, Rhadinovirus, Roseolovirus, Erythrovirus |
| ssRNA + strand viruses | (Calicivirdiae), Alphavirus, Coronavirus, Enterovirus, Flavivirus, Hepacivirus, Hepatovirus, Hepevirus |
Although not all of the structural genomics projects that work on proteins from pathogens will be mentioned, a few with the central aim of examining proteins that are potential drug targets should be noted. Among them are an international consortium aimed at tuberculosis [8]. This effort has probably been the most successful, in terms of the number of protein structures and Protein Data Bank (PDB, http://www.rcsb.org/ [9]) deposits. There are now 607 PDB deposits of Mycobacterium tuberculosis proteins. The international consortium, TBSGC (http://www.doe-mbi.ucla.edu/TB/), is listed as author of 194 of those deposits, though this underestimates the impact this consortium has had on the field.
Projects that focused primarily on pathogenic protozoa, include two related projects, the Structural Genomics of Pathogenic Protozoa, SGPP (http://www.sgpp.org/) and its continuation, the Medical Structural Genomics of Pathogenic Protozoa, MSGPP (http://www.msgpp.org/) [10] have a total of 69 PDB deposits. Another, larger, structural genomics project with an effort on pathogenic protozoa is the Structural Genomics Consortium (SGC, http://www.sgc.utoronto.ca/). Although the primary SGC focus has been on human proteins, it has deposited 102 structures from pathogenic protozoa in the PDB [11].
A number of efforts have been aimed at viral pathogens. One example of such an effort, is a European initiative, Vizier (http://www.vizier-europe.org/), that is focused primarily on the single stranded RNA viruses [12] and has deposited a dozen structures in the PDB. Another is the SARS consortium which has determined the structures of many of the functional domains of proteins from the SARS coronavirus (http://visp.scripps.edu/SARS/).
HIGH-THROUGHPUT STRUCTURE DETERMINATION
Essentially all structural genomics projects have been initiated within the past ten years. This relatively young field was made possible by two scientific developments. The first was the availability of genome sequences, the other was the improvement in structural biology methods and the associated technology for rapid protein production, purification and structure determination [13, 14]. Many improvements in crystallographic structure determination have been coupled to the development of modern beamlines at third generation synchrotron sources. Consequently, frequent, convenient access to such beamlines is an advantage to a structural genomics effort.
The CSGID uses a variety of methods for obtaining phase information for X-ray crystallographic structure determination. The most common is single, or multiple, wave-length anomalous diffraction (SAD/MAD) using selenomethionine substituted proteins. Although this requires access to synchrotron radiation, the advantage of this method is that it provides experimental phase information that is not dependent on similarity to a model structure. A smaller number, about 40%, of the CSGID structures are determined using molecular replacement methods, primarily where the structure of a close homolog exists, and for protein-ligand complexes.
The CSGID has adopted the methods developed by other structural genomics efforts, especially the Protein Structure Initiative [13]. Further improvements in methods are being developed, particularly in the rapid structural characterization of protein-ligand complexes and in improving the overall success rate.
TARGET SELECTION
The selection of proteins for a focused structural genomics project is critical to its success. For the application of structural genomics to infectious disease drug discovery there are a number of criteria that can be used to select protein targets. One is whether the protein is already known to be the target of an existing drug. Such a protein is already validated as a drug target. The downside is that it represents a system that is already well characterized and has been explored. Proteins that are homologous to known drug targets are an obvious extension. Though they may not have been validated in a particular organism, it is a reasonable supposition that they would be of interest. Proteins that are coded for by essential genes are another category of proteins to select. They have been shown to be essential to the organism (or a close relative) under some set of conditions. These proteins are even more attractive if the function is absent, or nonessential, in humans. A potential problem is that the conditions under which the gene is essential may have little to do with the conditions for establishment or maintenance of an infection. One would really prefer to examine the proteins whose functions are necessary to maintain an infection (the infection will generally already be established before treatment is explored). Proteins that are needed for the uptake or metabolism of limiting nutrients in vivo are still another, possibly overlapping, set of protein targets.
As it must be, the initial step in the production pipeline is the selection of targets for structure determination. Target selection priorities for the NIAID Structural Genomics Centers can be summarized by the following categories:
Known drug target - The gene product is the target of a drug that is currently on the market or in development. Minimally, inhibitors of the target will have been shown to block cell growth in the genus/species listed.
Potential drug target - An ortholog of a known drug target, an essential gene or a homolog of an essential gene.
Targets associated with virulence and/or pathogenesis - A gene product that is associated with pathogen adhesion, colonization, invasion, immune response inhibition or toxicity of the host cells.
Vaccine candidate - A gene product identified as a vaccine candidate.
Targets involved in drug resistance- The gene product that is confirmed or suggested to be involved (directly or indirectly) in drug-target binding, drug degradation or drug removal, all of which result in the evolution over time of drug resistance.
Targets showing phylogenetic evidence for essentiality, virulence or pathogenesis- targets for which orthologs are present in a pathogenic species/strain and absent in nonpathogenic species/strains.
Targets associated with innate immunity- Human proteins associated with non-adaptive immune mechanisms that recognize, and respond to, microorganisms, microbial products and antigens.
Marker of infection- A biomarker, antigen or other protein used to detect pathogen infection.
Targets of other special biological interest that fall outside the categories described above.
There are obviously many proteins from any specific pathogen that fall into one, or even several, of these categories. This, together with a focus on a number of pathogens, produce a sufficient number of target proteins to apply highly parallel, high throughput structural genomics methods.
Exactly what information should be used in selecting targets and the relative weight that should be assigned to different types of information is still being defined. There may be advantages to developing drugs that would end the infectious process without major damage to the existing microbiome [15]. One could imagine that a careful “systems biology” approach [16], that considered the complete host-pathogen system, could be an important consideration in target selection. Preventing pathogens from evading innate immunity could also provide important therapeutic advantages [17, 18]. For these reasons, proteins that are essential for virulence, or are associated with virulence are attractive target proteins.
Target selection is a highly multidisciplinary process and greatly benefits from soliciting as wide a range of community involvement as possible. For this reason, the CSGID actively solicits requests for structure determination from the scientific community. The bro ader scientific community can provide expertise on specific organisms that is not present within the center. Specifically, CSGID offers a protein gene to structure service to the infectious disease scientific community. All materials and informatio n generated from these services will become publicly available through the Biodefense & Emerging Infections (BEI) Research Resources Repository (http://www.beiresources.org/), and through structure and data deposition with the Protein Data Bank. Individuals or collaborative groups of investigators interested in proposing a target for structure determination can do so by submitting their requests online on the CSGID website (http://csgid.org/csgid under “Community Requests”). Proposed targets will be reviewed by the CSGID target selection team, and they will then be submitted to NIAID for approval. Approved applications will be given the highest priority for entry into the high-throughput structure determination pipeline, and the applicants will receive periodic e-mails updating their progress. The distribution of currently approved targets among the focus organisms is shown in Fig. (1A). These target proteins include both those selected by CSGID investigators and those that have come through community requests. At present the CSGID has received community requests, obtained approval for, and started work on >150 requested target proteins. The number of community requests is increasing and there are a few hundred protein requests being processed. Frequently community requests are for proteins that the CSGID has already identified and selected after an analysis of existing data.
Fig. (1).

Current CSGID Organism Distribution: Targets and Structures. (A) Targets: Bacillus anthracis (544), Salmonella typhimurium (437), Yersinia pestis (406), Francisella tularensis (264), Vibrio cholerae (262), Staphylococcus aureus (261), Coxiella burnetii (166), Campylobacter jejuni (156), Listeria monocytogenes (93), Monkeypox virus (50), Cowpox virus (38), Shigella flexneri (27), Dengue virus (20), Escherichia coli (19), Myxoma virus (16), Clostridium difficile (7), others (18). (B) Structures: Salmonella typhimurium (19), Vibrio cholerae (14), Bacillus anthracis (11), Staphylococcus aureus (12), Yersinia pestis (11), Shigella flexneri (2), Dengue virus (1), Listeria monocytogenes (1), Francisella tularensis (1).
There are a very small number of cases where the structure of a protein was known and a complex of the identical protein with a ligand has been, or will be, determined by the CSGID. There are also some that have no homologous structures with >25% sequence identity. However, most of the targeted proteins are between these extremes. For the majority there are homologous structures in the PDB with sequence identity between 25 and 90%. This distinguishes the NIAID structural genomics of infectious disease centers from other efforts, such as the PSI.
CURRENT CSGID PROGRESS AND EXPECTATIONS
The CSGID has only recently fully established its production pipeline for structure determination and begun depositing new structures in the PDB. The current numbers of approved target proteins, clones, expressed and soluble proteins, purified, crystallized and structures in PDB are shown in Table 2. After target selection, the CSGID structure determination pipeline includes high throughput cloning, expression and solubility testing, followed by protein production, purification and crystallization trials. For those target proteins that successfully pass these steps, crystallization conditions may need to be optimized and when crystals suitable for structure determination are obtained, diffraction data is collected at the Advanced Photon Source synchrotron (APS, http://www.aps.anl.gov/) at the beamlines constructed and managed by the Structural Biology Center (SBC, http://www.sbc.anl.gov/) or the Life Sciences Collaborative Access Team (LS-CAT, http://lscat.org/). Immediately after structure determination and refinement, all structures are deposited in the Protein Databank (PDB, [9]) and made available.
Table 2.
CSGID Progress on Target Proteins (as May 2009)
| Targets selected | Targets cloned successfully | Proteins expressed | Soluble proteins | Proteins purified | Proteins crystallized | Target structures in PDB | Total structures in PDB |
|---|---|---|---|---|---|---|---|
| 2784 | 2603 | 1252 | 1025 | 492 | 112 | 67 | 72 |
The best way to gain information on the progress being made at the CSGID is through its web site (http://www.csgid.org/). Database reports generated in the web site provide current detailed information on the targets that have been selected, their progress through the structure determination process and summary information about the proteins whose structures have been determined. The information can be filtered and sorted in different ways to allow access to whatever information is desired.
Although the project has only recently begun, there are already 72 structures of 67 different target proteins deposited in the PDB. Among them are 19 from Salmonella typhimurium, 14 from Vibrio cholerae, 12 from Staphylococcus aureus and 11 each Bacillus anthracis and Yersinia pestis. There are also small number (one or two) from Shigella flexneri, Francisella tularensis, Listeria monocytogenes and Dengue virus (Fig. 1B). Some of these organisms, such as Salmonella typhimurium and Staphylococcus aureus are already highly represented by structures in the PDB, while others, such as Yersinia pestis and Francisella tularensis have very limited numbers of structures. Among these protein structures (Fig. 2), the functional categories include proteins involved in cellular processes such as toxin production, resistance and detoxification (11), biosynthesis of cofactors (8), central energy metabolism (6), fatty acid biosynthesis (6), amino acid metabolism (5), purine/pyrimidine biosynthesis (5) and cell envelope synthesis (5).
Fig. (2).

Structures of 67 unique targets solved by CSGID as May 2009. The PDB codes for these structures are: 3FZY [27], 3G1Z [38], 3FF1 [39], 3G25 [40], 3FTT [24], 3FWX [41], 3E9A [42], 3FWW [43], 3G48 [44], 3FPK [45], 3G0A [46], 3GOM [47], 3G0S [48], 3EEV [20], 3EFV [49], 3F0I [50], 3ERP [51], 3F09 [52], 3HL3 [95], 3EME [53], 3EFB [54], 3F6M [55], 3F4N [56], 3FOB [57], 3DQQ [58], 34EF [19], 3EDN [59], 3EFE [60], 3E7N [61], 3DR6 [21], 3H07 [62], 3EGJ [63], 3EC6 [64], 3EG7 [26], 3EGP [65], 3DZC [66], 3GRI [67], 3DR3 [68], 3ECT [22], 3ED6 [69], 3EER [70], 3GSD [71], 3GBX [72], 3GEU [73], 3GHZ [74], 3GJZ [75], 3GC2 [76], 3HJJ [25], 3GA7 [77], 3H1S [78], 3H0P [79], 3GO9 [80], 3H2Y [81], 3H5Q [82], 3H88 [83], 3H83 [84], 3GSE [85], 3H02 [86], 3CWC [87], 3HFR [88], 3GVD [23], 3HHO [89], 3HJB [90], 3GOS [91], 3HID [92], 3GIU [93], and 3HJV [96].
As examples of the CSGID structures, among the proteins involved in drug resistance are two acetyltransferases, an aminoglycoside N-acetyltransferase from Bacillus anthracis (PDB code 3E4F [19]), and a chloramphenicol acetyltransferase from Vibrio cholerae (PDB code 3EEV [20]) (Fig. (2)). The underlying protein structures that are used to form the active sites are quite different in spite of the similarity in the reactions that they catalyze. A number of acetyltransferases involved in other cellular functions have also been determined by CSGID (PDB codes 3DR6 [21], 3ECT [22], 3GVD [23], 3FTT [24], 3HJJ [25] and 3EG7 [26]). Comparison of the structures and properties of the multiple families of acetyltransferases with those involved in drug resistance will aid understanding of the evolution of enzymes now involved in drug resistance and possibly suggest ways to inhibit their action.
A case of a pathway where CSGID structures are beginning to provide more complete information is the menaquinone pathway (Fig. 3). There are already three structures of enzymes in this pathway that have been determined by the CSGID. At present two of these are from Salmonella typhimurium (menB and menC) and one from Yersinia pestis (menF), but as work proceeds more structures from more organisms will be added.
Fig. (3).

Structures from the menaquinone biosynthetic pathway determined by CSGID as of May 2009. Adapted from [94].
The first structure determined for a community request was a cysteine protease domain (CPD) of a Vibrio cholerae toxin (Fig. 4, PDB code 3FZY [27]). The MARTX toxin is a multifunctional protein that undergoes autoproteolytic processing catalyzed by its cysteine protease domain. The full-length toxin is over 450 kDa and has extensive repeat regions at the N- and C-termini that are predicted to transfer the effector domains across the eukaryotic host cell membrane. At least four functional domains are translocated. They include an actin crosslinking domain and a Rhoinactivating domain that inhibits small GTPases, as well as the autoprocessing CPD. Because the cytopathic domains are within the protein, autoprocessing must occur after translocation in order to release them to the cytosol. The CPD is activated by binding Inositol hexakisphosphate (InsP6) upon translocation into the eukaryotic cell. The structure of the pro-form of a catalytically inactive mutant (C3568S) CPD was determined and reveals the structural basis for the InsP6 activation and the mechanism of proteolytic processing. The 1.95Å crystal structure shows that the CPD has the leucine of its N-terminal cleavage site bound in a hydrophobic specificity pocket and the scissile peptide bond is located between the serine that was substituted for the essential cysteine and the conserved histidine residue. The InsP6 activating ligand is bound by a number of positively charged lysine and arginine residues.
Fig. (4).
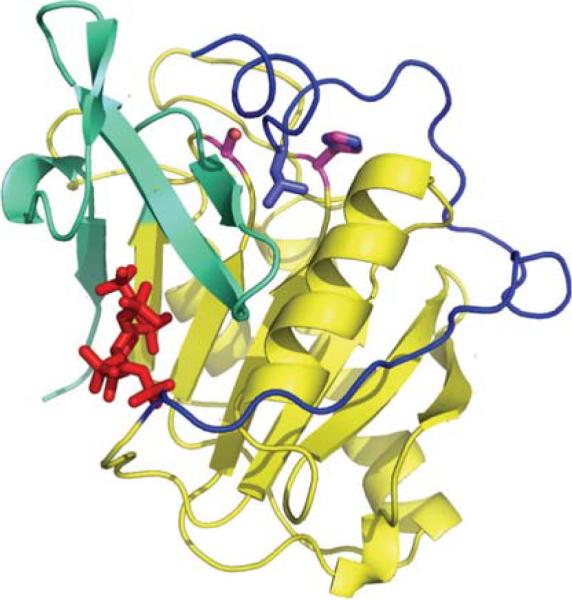
Structure of the cysteine protease domain (CPD) of the Vibrio cholerae MARTX. The CPD is activated after translocation into the host cell by the binding of inositol hexaphosphate (InsP6, red in the picture). CPD is a caspase-like protein that has the leucine (purple) of its N-terminal cleavage site bound in a hydrophobic specificity pocket and the scissile peptide bond located between the serine that was substituted for the essential cysteine (C3568S, catalytically inactive mutant) and the conserved histidine residue (both shown with magenta carbon atoms). The InsP6 activating ligand is bound by a number of positively charged lysine and arginine residues from the N-terminal region (blue), the main body of the CDP (yellow) and the C-terminal region (green). Related toxins with Cysteine Protease Domains, but other cytopathic domains, occur in a number of other gram-negative bacteria.
Structural data is not the only product of the CSGID. The E. coli over-expression strains for CSGID targets are being made available through the Biodefense & Emerging Infections (BEI) Research Resources Repository (http://www.beiresources.org/). When protein targets are requested by the community and genomic DNA suitable for preparing expression clones can be obtained, clones for these proteins will be made available through the BEI Repository. Thus even if the target fails to proceed through the structure determination pipeline, materials will be available for those interested in further studies of the target protein.
One problem area that the CSGID is trying to address is the low overall success rate between selected targets and protein structures. This is particularly important for a subset of organisms that are being found to have unusually low success rates for steps such as obtaining soluble protein. The fraction of cloned proteins that produce sufficient soluble protein varies significantly. For those organisms that have had over 200 proteins cloned, the CSGID has obtained soluble protein for 50–70% for Salmonella typhimurium, Vibrio cholerae, and Staphylococcus aureus proteins but only 11% for Francisella tularensis and 36% for Yersinia pestis. Initial trials indicate that success for some organisms is likely to be even lower. This tendency has already been seen by other structural genomics projects. However, the usual structural genomics approach of finding a homologous protein that is more amenable isn't useful when the eventual aim is structure aided drug discovery. Modifications to the current experimental protocols will have to be tested to determine if the organism specific success rates can be increased. To improve the success rate for crystallization, surface mutagenesis [28, 29], chemical [30] or proteolytic modification of the protein [31], and modification of the expression construct [32] have all been found useful for recovering a fraction of the recalcitrant proteins. Another approach, that is being tested, is the identification of small molecule ligands that stabilize a targeted protein [33, 34] and can then be used in co-crystallization experiments. The rationale is that crystallization is favored by conditions that stabilize a protein. Thus, if the presence of an additive stabilizes the protein, it is likely to have a positive effect on crystallization. The CSGID uses changes in the thermal denaturation temperature to detect stabilizing ligands or solvent conditions [34]. This has the advantage that it works for most proteins and does not require setting up a different assay for each of hundreds of target proteins. The Structural Genomics of Pathogenic Protozoa, mentioned earlier, has carried out initial tests using a relatively small set of cocrystallants (compounds that may form a specific complex with a protein or become involved in crystal packing interactions) [10]. Although the number of proteins and compounds used in the trials were too small to judge the generality of the approach, the results were promising. Small molecule additives that favor protein crystal formation may be involved in crystal packing, but they may not bind to the protein in solution strongly enough to stabilize the protein structure in the thermal denaturation assay. Small molecules that might favor weak crystal packing interactions can also be used in screening crystallization conditions using mixtures of such additives [35].
Many of the ligands tested in protein stabilization or co-crystallization screens are chosen based on the known activities or functions of each protein. Independent of whether ligands that are identified improve crystallization success, they are likely to provide useful information for drug discovery efforts. Co-crystal structures will be determined with ligands that are identified from annotated functions of the proteins or from thermal denaturation shift assays. For a subset of the selected protein targets, crystallographic or NMR screens of fragment-based libraries of compounds will be carried out [36, 37].
As structures of target proteins are determined more rapidly and the cost and effort expended on each target protein are reduced, a problem that arises is how to ensure that other investigators make use of the results. Just as with target selection, interactions with the scientific community and continuing community involvement are essential to the success of the initiative. As in most other structural genomics efforts, the emphasis on high throughput and efficiency has the consequence that publication of the results becomes rate limiting and much of the work is available in public databases long before it appears in scientific journals. In this situation, stimulating follow-up work requires that the CSGID make the broader scientific community aware of its results and that it seeks to involve other investigators who may make use of the information that the center has generated.
The broad aims of a focused structural genomics project, like the CSGID, can best be fulfilled through a multidisciplinary effort. The wide range of expertise required, from microbiology and pathogenesis, through crystallographic and NMR structure determination, to computational aspects of bioinformatics or drug discovery, means that a team approach is critical. The full potential of the project can only be achieved if the broader scientific community is involved in the target selection process and in the use of the experimental results. These two stages especially benefit from a detailed understanding of the biology of each pathogen and its unique interactions with the host.
ACKNOWLEDGEMENTS
The author would like to acknowledge the efforts of all of the members of the Center for Structural Genomics of Infectious Diseases, which have enabled the Center's activities and Dr. Elisabetta Sabini for assistance with the manuscript. The Center for Structural Genomics of Infectious Diseases is funded by the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract Number HHSN272200700058C.
REFERENCES
- [1].Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Nat. Rev. Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- [2].Taubes G. Science. 2008;321:356–361. doi: 10.1126/science.321.5887.356. [DOI] [PubMed] [Google Scholar]
- [3].Alekshun MN, Levy SB. Cell. 2007;128:1037–1050. doi: 10.1016/j.cell.2007.03.004. [DOI] [PubMed] [Google Scholar]
- [4].Payne DJ. Science. 2008;321:1644–1645. doi: 10.1126/science.1164586. [DOI] [PubMed] [Google Scholar]
- [5].Barker JJ. Drug Discov. Today. 2006;11:391–404. doi: 10.1016/j.drudis.2006.03.001. [DOI] [PubMed] [Google Scholar]
- [6].Congreve M, Murray CW, Blundell TL. Drug Discov. Today. 2005;10:895–907. doi: 10.1016/S1359-6446(05)03484-7. [DOI] [PubMed] [Google Scholar]
- [7].Johnson S. J. Infect. 2009;58:403–410. doi: 10.1016/j.jinf.2009.03.010. [DOI] [PubMed] [Google Scholar]
- [8].Baker EN. J. Struct. Funct. Genomics. 2007;8:57–65. doi: 10.1007/s10969-007-9020-9. [DOI] [PubMed] [Google Scholar]
- [9].Berman H, Henrick K, Nakamura H. Nat. Struct. Biol. 2003;10:980. doi: 10.1038/nsb1203-980. [DOI] [PubMed] [Google Scholar]
- [10].Fan E, Baker D, Fields S, Gelb MH, Buckner FS, Van Voorhis WC, Phizicky E, Dumont M, Mehlin C, Grayhack E, Sullivan M, Verlinde C, Detitta G, Meldrum DR, Merritt EA, Earnest T, Soltis M, Zucker F, Myler PJ, Schoenfeld L, Kim D, Worthey L, Lacount D, Vignali M, Li J, Mondal S, Massey A, Carroll B, Gulde S, Luft J, Desoto L, Holl M, Caruthers J, Bosch J, Robien M, Arakaki T, Holmes M, Le Trong I, Hol WG. Methods Mol. Biol. 2008;426:497–513. doi: 10.1007/978-1-60327-058-8_33. [DOI] [PubMed] [Google Scholar]
- [11].Weigelt J, McBroom-Cerajewski LD, Schapira M, Zhao Y, Arrowmsmith CH. Curr. Opin. Chem. Biol. 2008;12:32–39. doi: 10.1016/j.cbpa.2008.01.045. [DOI] [PubMed] [Google Scholar]
- [12].Coutard B, Gorbalenya AE, Snijder EJ, Leontovich AM, Poupon A, De Lamballerie X, Charrel R, Gould EA, Gunther S, Norder H, Klempa B, Bourhy H, Rohayem J, L'Hermite E, Nordlund P, Stuart DI, Owens RJ, Grimes JM, Tucker PA, Bolognesi M, Mattevi A, Coll M, Jones TA, Aqvist J, Unge T, Hilgenfeld R, Bricogne G, Neyts J, La Colla P, Puerstinger G, Gonzalez JP, Leroy E, Cambillau C, Romette JL, Canard B. Antiviral Res. 2008;78:37–46. doi: 10.1016/j.antiviral.2007.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Graslund S, Nordlund P, Weigelt J, Hallberg BM, Bray J, Gileadi O, Knapp S, Oppermann U, Arrowsmith C, Hui R, Ming J, Dhe-Paganon S, Park HW, Savchenko A, Yee A, Edwards A, Vincentelli R, Cambillau C, Kim R, Kim SH, Rao Z, Shi Y, Terwilliger TC, Kim CY, Hung LW, Waldo GS, Peleg Y, Albeck S, Unger T, Dym O, Prilusky J, Sussman JL, Stevens RC, Lesley SA, Wilson IA, Joachimiak A, Collart F, Dementieva I, Donnelly MI, Eschenfeldt WH, Kim Y, Stols L, Wu R, Zhou M, Burley SK, Emtage JS, Sauder JM, Thompson D, Bain K, Luz J, Gheyi T, Zhang F, Atwell S, Almo SC, Bonanno JB, Fiser A, Swaminathan S, Studier FW, Chance MR, Sali A, Acton TB, Xiao R, Zhao L, Ma LC, Hunt JF, Tong L, Cunningham K, Inouye M, Anderson S, Janjua H, Shastry R, Ho CK, Wang D, Wang H, Jiang M, Montelione GT, Stuart DI, Owens RJ, Daenke S, Schutz A, Heinemann U, Yokoyama S, Bussow K, Gunsalus KC. Nat. Methods. 2008;5:135–146. [Google Scholar]
- [14].Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. Acta. Crystallogr D. Biol. Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- [15].Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Science. 2005;307:1915–1920. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- [16].Gerdes S, Edwards R, Kubal M, Fonstein M, Stevens R, Osterman A. Curr. Opin. Biotechnol. 2006;17:448–456. doi: 10.1016/j.copbio.2006.08.006. [DOI] [PubMed] [Google Scholar]
- [17].Clatworthy AE, Pierson E, Hung DT. Nat. Chem. Biol. 2007;3:541–548. doi: 10.1038/nchembio.2007.24. [DOI] [PubMed] [Google Scholar]
- [18].Schwegmann A, Brombacher F. Sci. Signal. 2008;1:re8. doi: 10.1126/scisignal.129re8. [DOI] [PubMed] [Google Scholar]
- [19].Klimecka MM, Chruszcz M, Skarina T, Onopryienko O, Cymborowski M, Savchenko A, Edwards A, Anderson W, Minor W, CSGID Protein Data Bank (PDB) 2008 DOI 10.2210/pdb3e4f/pdb. [Google Scholar]
- [20].Kim Y, Maltseva N, Kwon K, Anderson WF, Joachimiak A, CSGID Protein Data Bank (PDB) 2008 DOI 10.2210/pdb3eev/pdb. [Google Scholar]
- [21].Singer AU, Skarina T, Onopriyenko O, Edwards AM, Anderson WF, Savchenko A, CSGID Protein Data Bank (PDB) 2008 DOI 10.2210/pdb3dr6/pdb. [Google Scholar]
- [22].Kim Y, Maltseva N, Kwon K, Papazisi L, Hasseman J, Peterson S, Anderson WF, Joachimiak A, CSGID Protein Data Bank, (PDB) 2008 DOI 10.2210/pdb3ect/pdb. [Google Scholar]
- [23].Kim Y, Zhou M, Peterson S, Anderson WF, Joachimiak A. Protein Data Bank (PDB) 2009 DOI 10.2210/pdb3gvd/pdb. [Google Scholar]
- [24].Knapik AA, Shumilin IA, Cui H, Xu X, Chruszcz M, Zimmerman MD, Cymborowski M, Anderson WF, Savchenko A, Minor W. Protein Data Bank (PDB) 2009 DOI 10.2210/pdb3ftt/pdb. [Google Scholar]
- [25].Kim Y, Maltseva N, Papazisi L, Anderson W, Joachimiak A. Protein Data Bank (PDB) 2009 DOI:10.2210/pdb3hjj/pdb. [Google Scholar]
- [26].Osipiuk J, Volkart L, Moy S, Anderson WF, Joachimiak A, CSGID Protein Data Bank (PDB) 2008 DOI 10.2210/pdb3eg7/pdb. [Google Scholar]
- [27].Prochazkova K, Shuvalova L, Minasov G, Anderson WF, Satchell KJF. Protein Data Bank (PDB) 2009 DOI 10.2210/pdb3fzy/pdb. [Google Scholar]
- [28].Cooper DR, Boczek T, Grelewska K, Pinkowska M, Sikorska M, Zawadzki M, Derewenda Z. Acta. Crystallogr. D. Biol. Crystallogr. 2007;63:636–645. doi: 10.1107/S0907444907010931. [DOI] [PubMed] [Google Scholar]
- [29].Derewenda ZS, Vekilov PG. Acta Crystallogr. D. Biol. Crystallogr. 2006;62:116–124. doi: 10.1107/S0907444905035237. [DOI] [PubMed] [Google Scholar]
- [30].Kim Y, Quartey P, Li H, Volkart L, Hatzos C, Chang C, Nocek B, Cuff M, Osipiuk J, Tan K, Fan Y, Bigelow L, Maltseva N, Wu R, Borovilos M, Duggan E, Zhou M, Binkowski TA, Zhang RG, Joachimiak A. Nat. Methods. 2008;5:853–854. doi: 10.1038/nmeth1008-853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dong A, Xu X, Edwards AM, Chang C, Chruszcz M, Cuff M, Cymborowski M, Di Leo R, Egorova O, Evdokimova E, Filippova E, Gu J, Guthrie J, Ignatchenko A, Joachimiak A, Klostermann N, Kim Y, Korniyenko Y, Minor W, Que Q, Savchenko A, Skarina T, Tan K, Yakunin A, Yee A, Yim V, Zhang R, Zheng H, Akutsu M, Arrowsmith C, Avvakumov GV, Bochkarev A, Dahlgren LG, Dhe-Paganon S, Dimov S, Dombrovski L, Finerty P, Jr., Flodin S, Flores A, Graslund S, Hammerstrom M, Herman MD, Hong BS, Hui R, Johansson I, Liu Y, Nilsson M, Nedyalkova L, Nordlund P, Nyman T, Min J, Ouyang H, Park HW, Qi C, Rabeh W, Shen L, Shen Y, Sukumard D, Tempel W, Tong Y, Tresagues L, Vedadi M, Walker JR, Weigelt J, Welin M, Wu H, Xiao T, Zeng H, Zhu H. Nat. Methods. 2007;4:1019–1021. doi: 10.1038/nmeth1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Graslund S, Sagemark J, Berglund H, Dahlgren LG, Flores A, Hammarstrom M, Johansson I, Kotenyova T, Nilsson M, Nordlund P, Weigelt J. Protein Expr. Purif. 2008;58:210–221. doi: 10.1016/j.pep.2007.11.008. [DOI] [PubMed] [Google Scholar]
- [33].Pantoliano MW, Petrella EC, Kwasnoski JD, Lobanov VS, Myslik J, Graf E, Carver T, Asel E, Springer BA, Lane P, Salemme FR. J. Biomol. Screen. 2001;6:429–440. doi: 10.1177/108705710100600609. [DOI] [PubMed] [Google Scholar]
- [34].Vedadi M, Niesen FH, Allali-Hassani A, Fedorov OY, Finerty PJ, Jr., Wasney GA, Yeung R, Arrowsmith C, Ball LJ, Berglund H, Hui R, Marsden BD, Nordlund P, Sundstrom M, Weigelt J, Edwards AM. Proc. Natl. Acad. Sci. USA. 2006;103:15835–15840. doi: 10.1073/pnas.0605224103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].McPherson A, Cudney B. J. Struct. Biol. 2006;156:387–406. doi: 10.1016/j.jsb.2006.09.006. [DOI] [PubMed] [Google Scholar]
- [36].Nienaber VL, Richardson PL, Klighofer V, Bouska JJ, Giranda VL, Greer J. Nat. Biotechnol. 2000;18:1105–1108. doi: 10.1038/80319. [DOI] [PubMed] [Google Scholar]
- [37].Carr RA, Congreve M, Murray CW, Rees DC. Drug Discov. Today. 2005;10:987–992. doi: 10.1016/S1359-6446(05)03511-7. [DOI] [PubMed] [Google Scholar]
- [38].Singer AU, Evdokimova E, Ali S, Edwards AM, Navarre W, Savchenko A. Protein Data Bank, (PDB) 2009 DOI 10.2210/pdb3g1z/pdb. [Google Scholar]
- [39].Anderson SM, Brunzelle JS, Onopriyenko O, Peterson S, Anderson WF, Savchenko A, CSGID Protein Data Bank, (PDB) 2008 DOI 10.2210/pdb3ff1/pdb. [Google Scholar]
- [40].Minasov G, Skarina T, Onopriyenko O, Savchenko A, Anderson WF. Protein Data Bank, (PDB) 2009 10.2210/pdb3g25/pdb. [Google Scholar]
- [41].Zhang R, Zhou M, Stam J, Anderson W, Joachimiak A. Protein Data Bank, (PDB) 2009 DOI 10.2210/pdb3fwx/pdb. [Google Scholar]
- [42].Nocek B, Mulligan R, Kwon K, Joachimiak A, Anderson WF, CSGID Protein Data Bank, (PDB) 2008 DOI 10.2210/pdb3e9a/pdb. [Google Scholar]
- [43].Zhang R, Gu M, Anderson W, Joachimiak A. Protein Data Bank, (PDB) 2009 DOI 10.2210/pdb3fww/pdb. [Google Scholar]
- [44].Nocek B, Zhou M, Stam J, Anderson W, Joachimiak A. Protein Data Bank, (PDB) 2009 DOI 10.2210/pdb3g48/pdb. [Google Scholar]
- [45].Kim Y, Gu M, Stam J, Anderson WF, Joachimiak A. Protein Data Bank, (PDB) 2009 DOI 10.2210/pdb3fpk/pdb. [Google Scholar]
- [46].Anderson SM, Skarina T, Onopriyenko O, Wawrzak Z, Papazisi L, Savchenko A, Anderson WF. Protein Data Bank, (PDB) 2009 DOI 10.2210/pdb3goa/pdb. [Google Scholar]
- [47].Nocek B, Maltseva N, Stam J, Anderson W, Joachimiak A. Protein Data Bank, (PDB) 2009 DOI 10.2210/pdb3g0m/pdb. [Google Scholar]
- [48].Osipiuk J, Maltseva N, Stam J, Anderson WF, Joachimiak A. Protein Data Bank, (PDB) 2009 DOI 10.2210/pdb3ff1/pdb. [Google Scholar]
- [49].Brunzelle JS, Evdokimova E, Kudritska M, Anderson WF, Savchenko A. Protein Data Bank, (PDB) 2009 DOI 10.2210/pdb3efv/pdb. [Google Scholar]
- [50].Osipiuk J, Gu M, Stam J, Anderson WF, Joachimiak A, CSGID Protein Data Bank, (PDB) 2008 DOI 10.2210/pdb3f0i/pdb. [Google Scholar]
- [51].Singer AU, Minasov G, Evdokimova E, Brunzelle JS, Kudritsdka M, Edwards AM, Anderson WF, Savchenko A, CSGID Protein Data Bank, (PDB) 2008 DOI 10.2210/pdb3erp/pdb. [Google Scholar]
- [52].Halavaty AS, Minasov G, Shuvalova L, Dubrovska I, Papazisi L, Anderson WF, CSGID Protein Data Bank, (PDB) 2008 DOI 10.2210/pdb3f09/pdb. [Google Scholar]
- [53].Kim Y, Edwards A, Anderson WF, Joachimiak A, CSGID Protein Data Bank, (PDB) 2008 DOI 10.2210/pdb3eme/pdb. [Google Scholar]
- [54].Kim Y, Evdokimova E, Kudritska M, Savchenko A, Edwards A, Joachimiak A, CSGID Protein Data Bank (PDB) 2008 DOI 10.2210/pdb3efb/pdb. [Google Scholar]
- [55].Kim Y, Maltseva N, Stam J, Anderson WF, Joachimiak A, CSGID Protein Data Bank (PDB) 2008 DOI 10.2210/pdb3f6m/pdb. [Google Scholar]
- [56].Kim Y, Maltseva N, Stam J, Anderson WF, Joachimiak A, CSGID Protein Data Bank (PDB) 2008 DOI 10.2210/pdb3f4n/pdb. [Google Scholar]
- [57].Osipiuk J, Gu M, Stam J, Anderson WF, Joachimiak A, CSGID Protein Data Bank (PDB) 2008 DOI: 10.2210/pdb3fob/pdb. [Google Scholar]
- [58].Zhang R, Gu M, Zhou M, Anderson W, Joachimiak A, CSGID Protein Data Bank (PDB) 2008 DOI 10.2210/pdb3dqq/pdb. [Google Scholar]
- [59].Anderson SM, Brunzelle JS, Onopriyenko O, Savchenko A, Anderson WF, CSGID Protein Data Bank (PDB) 2008 DOI 10.2210/pdb3edn/pdb. [Google Scholar]
- [60].Zhang R, Xu X, Cui H, Savchenko A, Edwards A, Anderson W, Joachimiak A, CSGID Protein Data Bank (PDB) 2008 DOI 10.2210/pdb3efe/pdb. [Google Scholar]
- [61].Nocek B, Maltseva N, Gu M, Joachimiak A, Anderson W, CSGID Protein Data Bank (PDB) 2008 DOI 10.2210/pdb3e7n/pdb. [Google Scholar]
- [62].Nocek B, Gu M, Kwon K, Joachimiak A, Anderson WF. Protein Data Bank (PDB) 2009 DOI 10.2210/pdb3h07/pdb. [Google Scholar]
- [63].Osipiuk J, Maltseva N, Stam J, Anderson WF, Joachimiak A, CSGID Protein Data Bank (PDB) 2008 DOI 10.2210/pdb3egj/pdb. [Google Scholar]
- [64].Kim Y, Xu X, Cui H, Savchenko A, Edwards A, Anderson WF, Joachimiak A, CSGID Protein Data Bank (PDB) 2008 DOI 10.2210/pdb3ec6/pdb. [Google Scholar]
- [65].Nelson CA, Kim T, Warren JT, Fremont DH, CSGID Protein Data Bank (PDB) 2008 DOI 10.2210/pdb3egp/pdb. [Google Scholar]
- [66].Minasov G, Shuvalova L, Dubrovska I, Winsor J, Papazisi L, Kwon K, Hasseman J, Peterson SN, Anderson WF, CSGID Protein Data Bank (PDB) 2008 DOI 10.2210/pdb3dzc/pdb. [Google Scholar]
- [67].Brunzelle JS, Wawrzak Z, Skarina T, Onopriyenko O, Savchenko A, Anderson WF. Protein Data Bank (PDB) 2009 DOI 10.2210/pdb3gri/pdb. [Google Scholar]
- [68].Singer AU, Skarina T, Onopriyenko O, Edwards AM, Anderson WF, Savchenko A, CSGID Protein Data Bank (PDB) 2008 DOI 10.2210/pdb3dr3/pdb. [Google Scholar]
- [69].Halavaty AS, Shuvalova L, Minasov G, Dubrovska I, Winsor J, Peterson SN, Anderson WF, CSGID Protein Data Bank (PDB) 2008 DOI 10.2210/pdb3ed6/pdb. [Google Scholar]
- [70].Nocek B, Maltseva N, Kwon K, Anderson WF, Joachimiak A, CSGID Protein Data Bank (PDB) 2008 DOI 10.2210/pdb3eer/pdb. [Google Scholar]
- [71].Minasov G, Wawrzak Z, Skarina T, Onopriyenko O, Scott P, Savchenko A, Anderson WF. Protein Data Bank (PDB) 2009 DOI 10.2210/pdb3gsd/pdb. [Google Scholar]
- [72].Osipiuk J, Nocek B, Zhou M, Stam J, Anderson WF, Joachimiak A. Protein Data Bank (PDB) 2009 DOI 10.2210/pdb3gbx/pdb. [Google Scholar]
- [73].Anderson SM, Brunzelle JS, Wawrzak Z, Skarina T, Papazisi L, Anderson WF, Savchenko A. Protein Data Bank (PDB) 2009 DOI 10.2210/pdb3geu/pdb. [Google Scholar]
- [74].Osipiuk J, Gu M, Peterson J, Anderson WF, Joachimiak A. Protein Data Bank (PDB) 2009 DOI 10.2210/pdb3ghz/pdb. [Google Scholar]
- [75].Nocek B, Zhou M, Kwon K, Anderson W, Joachimiak A. Protein Data Bank (PDB) 2009 DOI 10.2210/pdb3gjz/pdb. [Google Scholar]
- [76].Minasov G, Wawrzak Z, Skarina T, Onopriyenko O, Scott P, Savchenko A, Anderson WF. Protein Data Bank (PDB) 2009 DOI 10.2210/pdb3gc2/pdb. [Google Scholar]
- [77].Minasov G, Wawrzak Z, Brunzelle J, Onopriyenko O, Skarina T, Scott P, Savchenko A, Anderson WF. Protein Data Bank (PDB) 2009 DOI 10.2210/pdb3ga7/pdb. [Google Scholar]
- [78].Nocek B, Zhou M, Papazisi L, Anderson WF, Joachimiak A. Protein Data Bank (PDB) 2009 DOI 10.2210/pdb3h1s/pdb. [Google Scholar]
- [79].Minasov G, Wawrzak Z, Skarina T, Onopriyenko O, Scott P, Savchenko A, Anderson WF. Protein Data Bank (PDB) 2009 DOI 10.2210/pdb3h0p/pdb. [Google Scholar]
- [80].Osipiuk J, Maltseva N, Onopriyenko O, Peterson S, Anderson WF, Joachimiak A. Protein Data Bank (PDB) 2009 DOI 10.2210/pdb3go9/pdb. [Google Scholar]
- [81].Brunzelle JS, Anderson SM, Xu X, Savchenko A, Anderson WF. Protein Data Bank (PDB) 2009 DOI: 10.2210/pdb3h2y/pdb. [Google Scholar]
- [82].Shumilin IA, Zimmerman M, Cymborowski M, Skarina T, Onopriyenko O, Anderson WF, Savchenko A, Minor W. Protein Data Bank (PDB) 2009 DOI 10.2210/pdb3h5q/pdb. [Google Scholar]
- [83].Kim Y, Zhou M, Kwon K, Anderson WF, Joachimiak A. Protein Data Bank (PDB) 2009 [Google Scholar]
- [84].Halavaty AS, Shuvalova L, Minasov G, Dubrovska I, Peterson SN, Anderson WF. Protein Data Bank (PDB) 2009 DOI 10.2210/pdb3h83/pdb. [Google Scholar]
- [85].Nocek B, Gu M, Papazisi L, Anderson WF, Joachimiak A. Protein Data Bank (PDB) 2009 DOI 10.2210/pdb3gse/pdb. [Google Scholar]
- [86].Minasov G, Wawrzak Z, Skarina T, Onopriyenko O, Scott P, Savchenko A, Anderson WF. Protein Data Bank (PDB) 2009 DOI 10.2210/pdb3h02/pdb. [Google Scholar]
- [87].Osipiuk J, Zhou M, Holzle D, Anderson W, Joachimiak A, CSGID Protein Data Bank (PDB) 2008 DOI 10.2210/pdb3cwc/pdb. [Google Scholar]
- [88].Majorek KA, Chruszcz M, Zimmerman MD, Klimecka MM, Cymborowski M, Skarina T, Onopriyenko O, Stam J, Otwinowski Z, Anderson WF, Savchenko A, Minor W. Protein Data Bank (PDB) 2009 DOI:10.2210/pdb3hfr/pdb. [Google Scholar]
- [89].Osipiuk J, Gu M, Papazisi L, Anderson WF, Joachimiak A. Protein Data Bank (PDB) 2009 DOI 10.2210/pdb3hho/pdb. [Google Scholar]
- [90].Minasov G, Halavaty A, Shuvalova L, Dubrovska I, Winsor J, Papazisi L, Anderson WF. Protein Data Bank (PDB) 2009 DOI:10.2210/pdb3hjb/pdb. [Google Scholar]
- [91].Zhang R, Maltseva N, Kwon K, Anderson W, Joachimiak A. Protein Data Bank (PDB) 2009 DOI 10.2210/pdb3gos/pdb. [Google Scholar]
- [92].Zhang R, Zhou M, Stam J, Anderson SM, Joachimiak A. Protein Data Bank (PDB) 2009 DOI:10.2210/pdb3hid/pdb. [Google Scholar]
- [93].Minasov G, Wawrzak Z, Skarina T, Onopriyenko O, Scott P, Savchenko A, Anderson WF. Protein Data Bank (PDB) 2009 DOI 10.2210/pdb3giu/pdb. [Google Scholar]
- [94].Hiratsuka T, Furihata K, Ishikawa J, Yamashita H, Itoh N, Seto H, Dairi T. Science. 2008;321:1670–1673. doi: 10.1126/science.1160446. [DOI] [PubMed] [Google Scholar]
- [95].Minasov G, Shuvalova L, Halavaty A, Dubrovska I, Winsor J, Papazisi L, Anderson WF. Protein Data Bank (PDB) 2009 DOI:10.2210/pdb3hl3/pdb. [Google Scholar]
- [96].Halavaty AS, Wawrzak Z, Anderson S, Skarina T, Onopriyenko O, Kwon K, Savchenko A, Anderson WF. Protein Data Bank (PDB) 2009 DOI:10.2210/pdb3hjv/pdb. [Google Scholar]