

Abstract
Intimal calcification is a feature of advanced atherosclerotic disease that predicts a two- to eightfold increase in the risk of coronary events. Type I collagen promotes vascular smooth muscle cell-mediated calcification, although the mechanism by which this occurs is unknown. The discoidin domain receptor 1 (DDR1) is a collagen receptor that is emerging as a critical mediator of atherosclerosis. To determine whether DDR1 is involved in intimal calcification, we fed male Ddr1−/−;Ldlr−/− and Ddr1+/+;Ldlr−/− mice an atherogenic diet for 6, 12, or 24 weeks. DDR1 deficiency significantly reduced the calcium content of the aortic arch, and microcomputed tomography demonstrated a significant decrease in hydroxyapatite deposition after 24 weeks of atherogenic diet. Reduced calcification was correlated with decreases in macrophage accumulation and tumor necrosis factor α staining, suggesting that the reduction in calcification was in part due to decreased inflammation. The chondrogenic markers type II collagen, type X collagen, and Sox-9 were expressed within the mineralized foci. An in vitro assay performed with vascular smooth muscle cells revealed that DDR1 was required for cell-mediated calcification of the matrix, and Ddr1+/+ smooth muscle cells expressed more alkaline phosphatase activity, whereas Ddr1−/− smooth muscle cells expressed elevated levels of mRNA for nucleotide pyrophosphatase phosphodiesterase 1, an inhibitor of tissue mineralization. Taken together, our results demonstrate that DDR1 mediates an important mechanism for atherosclerotic calcification.
Arterial calcification within the intimal layer is a feature of advanced atherosclerosis that affects plaque mechanics and stability.1 In the context of atherosclerosis, calcification may be mediated by differentiated intimal cells (e.g., vascular smooth muscle cells (VSMCs)) or progenitor cells derived from either the circulation or the vessel wall. Several studies have provided evidence that vascular calcification recapitulates many aspects of bone and cartilage formation and osteoblast- and chondrocyte-like cells have been identified within atherosclerotic lesions.2,3,4,5 There is evidence that VSMC transdifferentiation can occur in vivo,6 and clonal or heterogeneous populations of VSMCs or adventitial myofibroblasts can be stimulated to express osteoblast and chondrocyte proteins in vitro.7,8,9,10
Ldlr−/− mice fed an atherogenic diet develop complex atherosclerotic lesions. These lesions are lipid-laden and rich in VSMCs, macrophages, and extracellular matrix11 and may be complicated by ectopic calcification, depending on the stage of lesion development.9,12,13 Several reports have described the development of medial calcification in these mice as early as 5 weeks after the onset of atherogenic diet-feeding. Medial calcification is attributed to activation of an osteogenic transcriptional program involving tumor necrosis factor (TNF)α, Msx-2, and Wnt signaling.9,14 Intimal calcification, on the other hand, develops only with prolonged diet treatment,12,13 and the molecular mechanism underlying intimal calcification in Ldlr−/− mice is not completely defined.
During atherogenesis, collagens constitute up to 60% of plaque protein, two-thirds of which are represented by the interstitial type I collagen.15 Synthesis of type I collagen occurs predominantly by VSMCs, contributes to plaque expansion, and regulates VSMC migration and proliferation.15,16,17 Type I collagen is also highly expressed by calcifying vascular cells in vitro, and addition of exogenous type I collagen to these cultures enhances their mineralization18; however, little is known about the mechanisms by which collagen affects mineralization. Moreover, the ability of cells to integrate mineralization-promoting cues from the matrix depends on the expression of specific cell surface receptors, but few studies have addressed the roles of collagen matrix receptors in promoting calcification.
The discoidin domain receptor-1 (DDR1) is a receptor tyrosine kinase that is activated by several subtypes of triple helical collagens.19 Through DDR1 activation, type I collagen regulates the proliferation, migration, and differentiation of many cell types,20 and recently, DDR1 has been implicated in the pathogenesis of atherosclerosis. DDR1 is expressed in atheromatous lesions of nonhuman primates,21 in low-density lipoprotein receptor-deficient (Ldlr−/−) mice,22 and in the rat carotid artery after balloon catheter injury.23 DDR1 serves as a positive regulator of neointimal formation and plays a pivotal role in regulating vascular inflammation and matrix accumulation mediating the responses of macrophages and VSMCs.22,23 Considering its central role in mediating plaque expansion through collagen signaling, we reasoned that DDR1 might promote intimal calcification in atherogenesis.
To determine the role of DDR1 in atherosclerotic calcification, mice deficient in DDR1 (Ddr1−/−;Ldlr−/−) and control mice (Ddr1+/+;Ldlr−/−) were fed a high-fat/high-cholesterol diet for 6, 12, or 24 weeks and analyzed for intimal calcification. Our findings support a role for DDR1 in promoting mineralization, because decreased incidence and extent of atherosclerotic mineralization were found in Ddr1−/−;Ldlr−/− mice. The decrease in calcification was related to decreased plaque inflammation and also to altered expression of the calcification regulators alkaline phosphatase and nucleotide pyrophosphatase phosphodiesterase (NPP1) in Ddr1−/− VSMCs. These findings identify DDR1 in VSMCs as a likely candidate for the local regulation of atherosclerotic calcification.
Materials and Methods
Chemicals and Reagents
All chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise stated.
Animals
Animal experiments were performed in accordance with the guidelines of the Canadian Council on Animal Care, with the approval of the University of Toronto Faculty of Medicine Animal Care Committee. Male Ddr1−/− mice on a Sv/129 background were bred with female Ldlr−/− mice on a C57BL/6 background. Double heterozygotes were crossed to generate Ddr1−/−;Ldlr−/− mice and littermate Ddr1+/+;Ldlr−/− controls. Genomic DNA was isolated from tail clippings, and genotyping was performed by PCR analysis as described previously.22 At 3 weeks of age, mice were weaned to a standard chow diet. Between 8 and 12 weeks of age, male mice were placed on an atherogenic diet composed of 40% kCal fat and 1.25% cholesterol (Research Diets D12108). Mice had access to water ad libitum. All animals were sacrificed by lethal injection of 67 mg/kg xylaxine and 333 mg/kg ketamine, administered i.p. 6, 12, or 24 weeks after the onset of the atherogenic diet. All animals were perfused with 0.9% NaCl at 100 mmHg to clean the vasculature, and for histological analysis, aortas were perfusion fixed using 4% paraformaldehyde (pH 7.4) and subsequently cleared of adipose and connective tissue. Aortic arches used for calcium extraction were harvested after perfusion with 0.9% NaCl, rinsed in PBS, blotted dry, snap frozen in liquid nitrogen, and transferred to −80°C.
Histochemical Staining
Mineralization was visualized by staining 5.0-μm-thick sections with Alizarin Red S (60 minutes with 0.5% Alizarin Red S (pH 9.0) and subsequently rinsed briefly in 0.1 M borate buffer) or alternatively with Von Kossa (1 hour in 5% silver nitrate, followed by 3-minute incubation in 5% sodium thiosulfate). Von Kossa-stained sections were counterstained with methyl green for 5 minutes.
Immunostaining
Chondrocyte-like cells were identified by positive staining for Sox-9, type II collagen, and type X collagen. Briefly, antigens were retrieved from 5.0-μm-thick sections by incubation with 0.1% trypsin (Zymed Laboratories, San Francisco, CA) for 10 minutes at 37°C. Endogenous peroxidase activity was quenched by 0.3% hydrogen peroxide. Endogenous biotin was blocked by treatment with avidin and biotin blocking reagents (Vector Laboratories, Burlingame, CA) for 30 minutes each, washing thoroughly between treatments with PBS. Nonspecific binding was blocked by 4% normal goat or horse serum and 0.25% BSA. Sections were incubated overnight at 4°C with rabbit polyclonal antibodies raised against type II collagen (4 μg/ml, Ab761; Chemicon International, Temecula, CA) or Sox-9 (3 μg/ml; H-90, Santa Cruz Biotechnology, Santa Cruz, CA) or a mouse monoclonal antibody raised against type X collagen (4 μg/ml, C7974; Sigma-Aldrich). As a control, adjacent sections were incubated with the same concentration of rabbit or mouse IgG. The following day, sections were washed thoroughly with PBS and incubated at room temperature for 30 minutes with biotinylated goat-anti-rabbit (Jackson Laboratories) diluted 1/500 (for collagen type II) or 1/1000 (for Sox-9) or biotinylated horse-anti-mouse (Jackson Immuno Research Laboratories) diluted 1/500 (for collagen type X) in PBS containing 2% normal goat serum and 0.25% BSA. Sections were washed thoroughly with PBS and incubated at room temperature for 30 minutes with avidin-biotin–horseradish peroxidase complex (Vector Laboratories) before incubation with the chromogenic substrate 3,3′-diaminobenzidine (DakoCytomation, Carpinteria, CA). Sections were washed thoroughly with PBS and counterstained with hematoxylin QS (Vector Laboratories) or methyl green.
Serial sections were stained for TNFα. Briefly, antigen retrieval was performed by boiling in 10 mmol/L sodium citrate (pH 6.0) for 10 minutes using a pressure cooker. Endogenous peroxidase activity was quenched with 0.3% hydrogen peroxide. Endogenous biotin was blocked by treatment with avidin and biotin blocking reagents for 20 minutes each, washing between treatments with PBS. Nonspecific binding was blocked by 4% normal goat serum and 0.25% BSA. Sections were incubated with a rabbit polyclonal antibody directed against mouse TNFα (100 μg/ml, ab6671; Abcam). Subsequently, sections were washed thoroughly with PBS and incubated at room temperature for 30 minutes with biotinylated goat anti-rabbit antibody diluted 1/1000. Negative control sections were incubated without the primary antibody.
Incidence of Vascular Calcification
Animals were scored as either positive or negative for Alizarin Red S staining. Consistency between animals was maintained by using sections that contained the lesser curvature of the aortic arch and as landmarks, the branches of the common carotid and the left subclavian arteries. Two to three sections were stained and analyzed from each animal. The proportion of positive animals in each group was calculated.
Quantification of Von Kossa Staining
The extent of intimal calcification was quantified by image analysis of Von Kossa-stained sections. Sections were visualized using light microscopy (E600; Nikon, Melville, NY) and images captured using a Digital CCD Camera (C4742-95-12NRB; Hamamatsu, Middlesex, NJ). Using simple PCI Imaging software (Compix), the area of the atherosclerotic lesion along the lesser curvature of the aortic arch, the length of the arch, and the area of the plaque that was positive for Von Kossa staining were measured. Consistency between animals was maintained by using sections that contained the lesser curvature of the aortic arch and, as landmarks, the branches of the common carotid and the left subclavian arteries. Two to three sections were stained and analyzed from each animal. Data are presented as the percentage of lesion area positive for Von Kossa stain (calcified lesion area percentage).
Measurement of the Ratio of Von Kossa-Stained Area to Mac-2-Stained Area
Serial sections of the aortic arch from Ddr1+/+;Ldlr−/− and Ddr1−/−;Ldlr−/− mice fed the high fat diet for 24 weeks were stained with Von Kossa stain, as described above, or with an antibody against Mac-2 to identify macrophages as described previously.22 To determine whether intimal calcification was correlated with inflammation, the ratio of Von Kossa-stained area to Mac-2-stained area within the plaque was calculated.
Quantification of Aortic Calcium Content
Aortic arches were dissected from the proximal aorta to just past the left subclavian artery, cleaned in situ, excised, immersed in PBS, blotted dry, and snap frozen in liquid nitrogen. Frozen vessels were lyophilized for 48 hours, weighed, and incubated in 0.6 N HCl (50 μl/mg dry weight) for 24 hours. Calcium concentration was measured using the o-cresolphthalein complexone colorimetric assay (Clinotech Diagnostics). Standard solutions ranging from 0 (blank) to 7.5 mg/ml calcium were used and samples were diluted 1/10 or 1/20 with 0.6 N HCl and measured in triplicate. Averages of each triplicate were calculated and normalized to tissue dry weight.
Micro Computerized Tomography Scanning
Paraffin blocks containing aortic arches from Ddr1+/+;Ldlr−/− and Ddr1−/−;Ldlr−/− mice fed the high-fat diet for 24 weeks were placed in a 36.9-mm polymethylmethacrylate holder in a micro computerized tomography scanner (μCT 40; Scanco Medical, Sweden) and scanned using high resolution at 55 KVp and 145 μA. Each block scan was composed of 828 slices, each 18 μm thick, and aortic arch tissue within the block spanned between 172 and 265 slices. Slices containing calcified regions were digitally reconstructed into three-dimensional images, and the density of hydroxyapatite and volume of calcified tissue were determined by the software provided (μCT 40, Scanco Medical; Evaluation Protocol number 5 – Full Morphometry). Threshold values were set between 220 and 1000.
Serum Calcium and Inorganic Phosphorus Measurement
Blood samples were collected into heparinized capillary tubes from animals fed an atherogenic diet for 6, 12, and 24 weeks. Blood was obtained either by capillary tube aspiration of the saphenous vein or by ventricular puncture at the time of sacrifice. After centrifugation (10,000 × g for 10 minutes at 4°C), serum was removed from plasma and stored at −80°C. Serum calcium was measured using the o-cresophthalein complexone-based colorimetric assay (Clinotech Diagnostics), according to the manufacturer’s protocol. Four microliters of undiluted serum was used for the analysis. To correct for hemolysis or lipemia, a serum blank (1 μl of serum diluted in 59 μl of distilled water) was also used for each sample. The absorbances at 570 nmol/L (Abs570) of the samples, serum blanks, blank and standard, were read, the average of each triplicate was calculated, and the concentration of total calcium in the serum was calculated using the following equation:
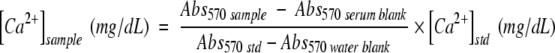 |
Serum concentration of inorganic phosphorus (Pi) was measured using a commercially available molybdenate-based colorimetric assay (Clinotech Diagnostics) according to the manufacturer’s protocol. Four microliters of diluted serum (diluted 1/1 with saline) was used for the analysis. To correct for hemolysis or lipemia, a serum blank (1 μl serum diluted in 49 μl of 0.9% saline) was also used for each sample. The absorbance at 675 nmol/L (Abs675) of samples, serum blanks, blank and standard (std) were read, the average of each triplicate was calculated, and the concentration of Pi in the serum was calculated using the following equation:
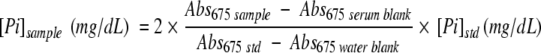 |
In Vitro Calcification Assay
Mouse VSMCs were isolated from the carotid arteries of Ddr1+/+ and Ddr−/− mice on C57B/L6 background. VSMCs were propagated in growth medium composed of Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 2% penicillin/streptomycin at 37°C with 5% CO2. For calcification experiments, subcultured VSMCs between passages 6 and 10 were seeded in growth medium at a density of 20,000 cells/well in 6-well dishes. Forty-eight hours later, growth medium was replaced with high-phosphate medium (Dulbecco’s Modified Eagle’s Medium, supplemented with 25 mmol/L glucose, 3% fetal bovine serum, 1% penicillin/streptomycin, and 3 mmol/L Pi, final concentrations) to induce matrix calcification.24 Calcium was extracted from the matrix using 0.6 N HCl 4, 7 and 12 days after the addition of high-phosphate medium. Calcium content was measured using the o-cresolphthalein complexone colorimetric assay (Clinotech Diagnostics) as described under Quantification of Aortic Calcium Content (above) with the following exception: samples were prepared by diluting 1/1 with 0.6 N HCl and were normalized to cellular protein as described previously.24
In a parallel experiment, VSMCs were seeded at a density of 168,000 cells in a 10-cm tissue culture dish, then total cell RNA was isolated 13 days after the onset of calcification, and NPP1 mRNA expression was measured by quantitative real time PCR as described below.
Alkaline Phosphatase Activity
Alkaline phosphatase activity was detected by directly incubating Ddr1+/+ and Ddr−/− VSMCs for 45 minutes with the working solution used for detecting alkaline phosphatase-conjugated antibodies (Red Alkaline Phosphatase Substrate Kit I, Vector Laboratories). Cells were counterstained with Hoescht nuclear dye for 5 minutes, rinsed briefly in PBS, and visualized by fluorescence microscopy (Nikon). Images were captured using a Digital CCD Camera (C4742-95-12NRB; Hamamatsu).
Reverse Transcription and Quantitative Real-Time PCR
Total RNA from cultures of VSMCs was isolated by TRIzol extraction (Invitrogen, Carlsbad, CA), treated with DNase I to remove contaminating genomic DNA (Fermentas), and reverse transcribed using the Superscript First-Strand Synthesis System for RT-PCR (Invitrogen). Expression of DDR1 was determined using quantitative real-time PCR. The number of copies of DDR1 sequence was determined by comparison with a standard curve using known amounts of a DDR1-containing plasmid. Primers for DDR1 were described previously.22 Expression of Enpp1 was determined using the comparative Ct (2−ΔΔCt) method. The primers for NPP1 were as follows: forward (5′-CTCGGTTGAGACCCACTGATG-3′) and reverse (5′-GCTCCCGGCAAGAAAGATTT-3′). The acidic ribosomal protein gene [primers, forward (5′-AGACCTCCTTCTTCCAGGCTTT-3′) and reverse (5′-CCCACCTTGTCTCCAGTCTTTATC-3′)] was used as an endogenous internal control.
Statistical Analysis
Statistical analyses were performed using Sigma Stat (SyStat Software, Evanston, IL) and data are presented as means ± SEM. For the analyses of vessel calcium content, percent calcified area, serum calcium and Pi, and VSMC calcification in vitro, a two-way analysis of variance (analysis of variance) was used to incorporate all available data into each analysis. Pairwise comparisons between groups were made following the two-way analysis of variance using Tukey’s posthoc test. Student’s t-test was used to compare plaque density, the levels of NPP1 mRNA, and the Von Kossa/Mac-2 ratios between genotypes. For all tests, statistical significance was set at P < 0.05.
Results
DDR1 Deficiency Attenuates the Extent of Intimal Calcification in Mice
To test the hypothesis that DDR1 is involved in intimal calcification, we engineered mice with a combined deficiency in both LDLR and DDR1 (Ddr1−/−;Ldlr−/−) as well as control animals deficient only in LDLR (Ddr1+/+;Ldlr−/−). Mice were fed an atherogenic diet for 6, 12, or 24 weeks, and as reported in a separate study, no significant differences in body weight, plasma triglycerides, or plasma cholesterol were observed between groups.22
Biochemical and imaging approaches were used to measure calcium and hydroxyapatite in the aortic arch. Calcium content per unit vessel weight increased between 12 and 24 weeks (Figure 1, A and B). After 12 weeks on the atherogenic diet, calcium content of the aortic arches from Ddr1−/−;Ldlr−/− mice was reduced by 60% compared with the aortic arches of Ddr1+/+;Ldlr−/− mice, although the difference was not statistically significant (Figure 1A). By 24 weeks on the atherogenic diet, a significant 40% attenuation in vessel calcium content persisted in Ddr1−/−;Ldlr−/− mice compared with Ddr1+/+;Ldlr−/− mice (Figure 1B). Microcomputerized tomography was used to measure the plaque density in the aortic arches from mice fed the high-fat diet for 24 weeks. Plaque density was significantly higher in the Ddr1+/+;Ldlr−/− mice compared with Ddr1−/−;Ldlr−/− mice (Figure 1C). Taken together, this evidence, showing the attenuation of vascular calcification in the Ddr1−/−;Ldlr−/− mice, supports a role for DDR1 in the process of intimal calcification.
Figure 1.

DDR1-deficiency attenuates the accumulation of calcium in the aortic arches of Ldlr−/− mice. The extent of vascular calcification in Ddr1+/+;Ldlr−/− (black bars) and Ddr1−/−;Ldlr−/− mice (grey bars) after 12 (n = 14 and n = 13) (A) and 24 weeks (n = 8 and n = 7) (B) of atherogenic diet treatment was quantified by measuring the amount of calcium retained within the aortic arch. C: Microcomputerized tomography was used to measure the plaque density in the aortic arches from mice fed the high-fat diet for 24 weeks (n = 5 and 8). Data are mean ± SEM. *P < 0.05.
To investigate the role of DDR1 in the progression and severity of atherosclerotic intimal calcification, we quantified the percentage of atherosclerotic lesion area that was mineralized. Longitudinal aortic arch sections were stained with Von Kossa to identify mineralized tissues; cell-associated mineralization was apparent in areas with the appearance of cartilaginous tissue, and mineral was also present in amorphous deposits (Figure 2A). Image analysis software was used to measure 1) the area of the intimal lesion along the lesser curvature and 2) the area of Von Kossa-positive tissue within the intimal lesion along the lesser curvature. This information was used to calculate the percentage of lesion area that was mineralized (Figure 2B). Mineralization within the medial layers of the Ldlr−/−-deficient mice has been reported9,12 but was only rarely observed in the present study. Therefore, we focused our analysis on intimal calcification. After 6 weeks on the atherogenic diet, no calcification was observed in either genotype (Figure 2B), although small atherosclerotic lesions were detected. After 12 weeks on the atherogenic diet, evidence of intimal calcification was observed in lesions of Ddr1+/+;Ldlr−/− but not Ddr1−/−;Ldlr−/− mice. After 24 weeks on the diet, both genotypes exhibited intimal calcification; however, the extent of lesion calcification was significantly reduced by 57% in the Ddr1−/−;Ldlr−/− compared with Ddr1+/+;Ldlr−/− mice (Figure 2B). These data show a significant attenuation of mineralization within the atherosclerotic lesions in response to the deletion of the Ddr1 gene. However, when we measured calcified lesion area in mice after 48 weeks on the atherogenic diet, there was no significant difference between the two genotypes (data not shown). Thus, there may be a compensatory mechanism that leads to a catch-up in arterial calcification in the Ddr1−/−;Ldlr−/− mice.
Figure 2.

DDR1-deficiency attenuates the extent of intimal calcification in the aortas of Ldlr−/− mice. A: Representative micrographs of vessels from Ddr1+/+;Ldlr−/− and Ddr1−/−;Ldlr−/− mice fed an atherogenic diet for 24 weeks stained by Von Kossa and methyl green counterstain. Scale bars represent 500 μm. High magnification micrographs are inset to illustrate tissue morphology; scale bars in inset micrographs represent 50 μm. B: The percentage of intimal lesion area that was stained by Von Kossa in Ddr1+/+;Ldlr−/− (black bars) and Ddr1−/−;Ldlr−/− mice (grey bars) after 6 (n = 8 for both), 12 (n = 7 and n = 4), and 24 (n = 5 for both) weeks of atherogenic diet. Data are mean ± SEM; *P < 0.05.
DDR1 Deficiency Reduces the Incidence of Intimal Calcification in Ldlr−/− Mice
Longitudinal sections of the aortic arch were stained with Alizarin Red S (Figure 3A) and used to determine the frequency of calcification in Ddr1+/+;Ldlr−/− and Ddr1−/−;Ldlr−/− mice. Sections demonstrating positive Alizarin Red S staining within the intima of the lesser curvature lesion were scored as positive and sections without Alizarin Red S staining were scored as negative; the proportion of animals that exhibited intimal calcification was calculated (Figure 3B). Both Ddr1+/+;Ldlr−/− and Ddr1−/−;Ldlr−/− animals demonstrated a time-dependent increase in the occurrence of intimal calcification when fed an atherogenic diet. After 6 weeks of diet treatment, there was no evidence of intimal calcification in either genotype, but after 12 weeks of diet treatment, 33% of the Ddr1+/+;Ldlr−/− mice developed calcification in the lesions, and by 24 weeks, 75% of the mice showed calcified lesions. By contrast, none of the Ddr1−/−;Ldlr−/− mice had developed intimal calcification at 12 weeks, and by 24 weeks, only 60% of the mice showed calcification in the lesions, demonstrating a delayed development of calcification in the atherosclerotic lesion when Ddr1 is deleted.
Figure 3.

DDR1 deficiency reduces the incidence of intimal calcification in Ldlr−/− mice. A: Representative micrographs of vessels from Ddr1+/+;Ldlr−/− and Ddr1−/−;Ldlr−/− mice fed an atherogenic diet for 24 weeks stained with Alizarin Red S. Scale bars represent 500 μm. B: After 6 (n = 8 for both), 12 (Ddr1+/+;Ldlr−/−, n = 7 and Ddr1−/−;Ldlr−/−, n = 5) and 24 (n = 6 for both) weeks of atherogenic diet, the proportion of Ddr1+/+;Ldlr−/− (•) and Ddr1−/−;Ldlr−/− (▿) animals positive for intimal calcification was plotted.
Attenuation of Intimal Calcification in DDR1-Deficient Mice Is Not Due to Reduced Serum Calcium or Pi
Systemic factors such as increased serum calcium and/or inorganic Pi levels have been linked to the calcification of atherosclerotic lesions.25 To determine whether the attenuation in intimal calcification of Ddr1−/−;Ldlr−/− mice was the result of reduced serum calcium or Pi in these mice, total serum calcium and Pi were measured in animals fed the atherogenic diet for 6, 12, or 24 weeks. Ddr1−/−;Ldlr−/− mice exhibited slightly higher serum calcium concentrations than Ddr1+/+;Ldlr−/− mice after 6 and 12 weeks of the atherogenic diet; however, this difference was not significant at the 24-week time point (Figure 4A). Serum Pi concentrations were similar between Ddr1+/+;Ldlr−/− and Ddr1−/−;Ldlr−/− mice after 6 weeks of the atherogenic diet, however, by 12 weeks, DDR1-deficient mice exhibited hyperphosphatemia that persisted to the 24-week time point (Figure 4B). An elevation in serum calcium or Pi would be expected to increase intimal calcification, which stands in contrast to our findings that DDR1-deficient mice developed less calcification in the atherosclerotic lesions, and therefore, the alterations in serum calcium and Pi levels did not contribute to the decrease in the intimal calcification observed in the Ddr1−/−;Ldlr−/− mice. The levels of serum Pi that we measured are consistent with those reported for Ldlr−/− mice fed a high-fat diet.26
Figure 4.

DDR1 deficiency increases serum calcium and phosphorus. Serum calcium (A) and inorganic (B) Pi concentrations in Ddr1+/+;Ldlr−/− (black bars) and Ddr1−/−;Ldlr−/− mice (grey bars) after 6 (n = 7 and n = 10), 12 (n = 11 for both), and 24 (n = 7 and n = 11) weeks of atherogenic diet treatment. Data are mean ± SEM. *P < 0.05.
Reduced Intimal Calcification in DDR1-Deficient Mice Is Correlated with Decreased Inflammation
Our previous studies demonstrated a decrease in intimal macrophage accumulation in Ddr1−/−;Ldlr−/− compared with Ddr1+/+;Ldlr−/− mice, evidenced by a decrease in Mac-2-stained area in Ddr1−/−;Ldlr−/− mice.22 To investigate a potential link between inflammation and calcification in these mice, serial sections of the aortic arch stained with Von Kossa and Mac-2 were used to calculate the ratio of Von Kossa-stained intimal area to Mac-2-stained intimal area. There was no significant difference in the Von Kossa/Mac-2 ratio between the genotypes (Figure 5A), indicating that calcification was reduced in proportion to the extent of inflammation.
Figure 5.

Inflammation and TNFα staining are decreased in the plaques of DDR1-deficient mice. A, The ratio of Von Kossa stained area/Mac-2-stained area in Ddr1+/+;Ldlr−/− (black bar) and Ddr1−/−;Ldlr−/− mice (gray bar) after 24 weeks of atherogenic diet (n = 6). Data are mean ± SEM. *P < 0.05. Β–D: Representative micrographs of sections from the lesser curvature of the aortic arch from Ddr1+/+;Ldlr−/− and Ddr1−/−;Ldlr−/− mice fed an atherogenic diet for 12 weeks, and immunostained with an antibody against TNFα. B: Control section from a Ddr1+/+;Ldlr−/− mouse stained without primary antibody. C: Section from a Ddr1+/+;Ldlr−/− mouse. D, Section from a Ddr1−/−;Ldlr−/− mouse.
Previous studies have demonstrated that TNFα produced by macrophages can stimulate SMC calcification in vitro and in vivo.14,27 We immunostained sections from Ddr1+/+;Ldlr−/− and Ddr1−/−;Ldlr−/− mice fed the atherogenic diet for 12 weeks, and observed a marked reduction in TNFα staining in the Ddr1−/−;Ldlr−/− mice (Figure 5, B–D).
Calcified Regions of Atherosclerotic Plaques Contain Cells Elaborating a Chondrogenic Phenotype
Chondrocyte-like cells have been demonstrated in the intimal lesions of ApoE-deficient mice,4 LDLR-deficient mice,13 and humans.2 To determine whether intimal cells were elaborating a chondrogenic phenotype, we stained serial longitudinal tissue sections of the aortic arch with antibodies against the chondrocyte-specific proteins Sox-9, type II collagen, and type X collagen. As demonstrated in Figure 6A, many of the cells within the calcified foci of 24-week Ddr1+/+;Ldlr−/− mice were positive for Sox-9 (arrows). Moreover, several of these cells were surrounded by dense rings of type II collagen (Figure 6B, arrows), and there was also evidence of type X collagen production (Figure 6C, arrows). Cells elaborating this chondroid phenotype were also evident within the aortic arches of Ddr1−/−;Ldlr−/− mice where calcification occurred, although these sites were much smaller and less numerous in the DDR1-deficient mice (Figure 6, D–F and G–I). This is consistent with the notion that inactivation of Ddr1 attenuates intimal calcification but suggests that it does not determine the underlying mechanism by which the phenotypic transition is achieved.
Figure 6.

Sox-9, type II collagen and type X collagen are expressed by intimal cells in Ddr1+/+;Ldlr−/− mice. Representative micrographs demonstrating staining of sections of the lesser curvature of the aortic arch from Ddr1+/+;Ldlr−/− mice (A–C) and from Ddr1−/−;Ldlr−/− mice (D–F) after 24 weeks of atherogenic diet, along with control sections from Ddr1+/+;Ldlr−/− mice stained with IgG alone (G–I). Sox-9 staining (A and D), type II collagen staining (B and E), and type X collagen staining (C and F). Arrows indicate cells stained with the antibodies. Scale bars represent 50 μm. Sections were counterstained with methyl green or hematoxylin (B, E, H).
DDR1 Is SMC-Autonomous Regulator of Calcification
VSMCs retain considerable phenotypic plasticity as they may transition from a contractile to a synthetic or to an osteo/chondrogenic phenotype. To determine whether DDR1 deficiency altered calcification by a cell-autonomous mechanism, we investigated the ability of isolated VSMCs to calcify matrix in vitro. VSMCs isolated from wild-type or DDR1-deficient mice were cultured under high phosphate conditions for 4, 7, and 12 days. Expression of DDR1 mRNA in Ddr1+/+ VSMCs, but not Ddr1−/− VSMCs, was confirmed using quantitative real-time PCR (Figure 7A). There was marked and progressive calcification of the matrix by Ddr1+/+ VSMCs between 4 and 12 days of culture; by contrast, Ddr1−/− VSMCs did not calcify the matrix (Figure 7B). Pyrophosphate levels, which are regulated by the antagonistic activities of alkaline phosphatase and NPP1, represent an important mechanism by which vascular calcification may be controlled.28 Notably, when Ddr1+/+ and Ddr1−/− VSMCs were cultured in high phosphate for 12 days, alkaline phosphatase activity was readily detectible in Ddr1+/+ but not in Ddr1−/− VSMCs (Figure 7C). Conversely, the mRNA expression of NPP1 was significantly elevated in Ddr1−/− compared with Ddr1+/+ VSMCs after 13 days in culture (Figure 7D).
Figure 7.

DDR1-deficiency attenuates VSMC-mediated calcification and alkaline phosphatase activity in vitro and increases NPP1 mRNA expression. A: DDR1 mRNA expression of Ddr1+/+ and Ddr1−/− VSMCs was measured by quantitative real-time PCR. B: Ddr1+/+ (black bars) and Ddr1−/− (grey bars) VSMCs were induced to calcify under high-phosphate culture conditions (3.0 mmol/L Pi). After 4, 7, and 12 days of high-phosphate treatment, calcium was extracted and measured using o-cresolphthalein complexone. Calcium content was normalized to total cellular protein. Data shown are means ± SEM; n = 3. *P < 0.05. C: Ddr1+/+ and Ddr1−/− VSMCs were cultured in the presence of 3.0 mmol/L Pi for 12 days, after which they were stained for alkaline phosphatase (red) and counterstained with Hoescht nuclear dye (blue). Scale bar represents 500 μm. D: Ddr1+/+ and Ddr1−/− VSMCs were cultured in the presence of 3.0 mmol/L Pi for 13 days, after which mRNA levels for NPP1 were measured using quantitative real-time PCR. Data shown are means ± SEM; n = 7. *P < 0.05.
Discussion
In the present study, we have demonstrated robust calcification within intimal atherosclerotic lesions of Ldlr−/− mice fed a high-fat/high-cholesterol diet. This phenotype develops with increasing severity over the course of diet treatment and is correlated with the development of inflammation in the vessel wall. Probing the mechanisms involved, we identified DDR1 as an important mediator of calcification. Deletion of Ddr1 significantly attenuated the development of mineralization in vivo, as well as the ability of VSMCs to calcify in vitro, further confirming that DDR1 is a local regulator of VSMC calcification.
Atherosclerotic lesions are sites of formidable matrix accumulation and remodeling, and the composition of the extracellular matrix provides important biochemical and mechanical cues that locally regulate vascular cell behavior.29 Collagens have been linked to vascular cell-mediated calcification such that type I collagen has been shown to promote mineralization, whereas type IV collagen inhibits this process18; however, the receptors involved and the mechanisms by which collagens are able to mediate these effects remain unclear. In this study, deletion of the collagen receptor DDR1 attenuated atherosclerotic calcification in vivo and VSMC-mediated mineralization in vitro, providing the first clues as to the mechanism by which type I collagen stimulates calcification. Consistent with this notion, we observed that Ddr1−/− VSMCs failed to exhibit alkaline phosphatase activity in vitro and expressed high levels of NPP1, an endogenous inhibitor of hydroxyapatite deposition. Thus, our studies indicate that DDR1 on the surface of VSMCs is important for the transduction of mineralization-promoting cues from the local extracellular matrix.
Limited information exists regarding the role of other collagen receptors in the regulation of atherosclerotic calcification, although Schapira et al30 have reported increased cartilaginous metaplasia in ApoE-null mice deficient in the α1 integrin subunit. These opposing effects of DDR1 and α1β1 in atherosclerotic calcification imply a complex mechanism by which collagens contribute to this pathological process. There is evidence that the two receptors bind preferentially to different collagen subtypes, with DDR1 more responsive to type I collagen and α1β1 binding to type IV collagen.19,31 In light of earlier findings by Watson et al,18 it is tempting to speculate that during atherogenesis, type I collagen promotes mineralization through binding to DDR1, whereas type IV collagen limits this process through ligation of the α1β1 integrin.
There are differences in the matrix composition of plaques between Ddr1+/+;Ldlr−/− and Ddr1−/−;Ldlr−/− mice; however, these cannot account for the differences in calcification that we have observed between the genotypes. Our previous studies have demonstrated robust type I collagen accumulation in the lesions of Ddr1−/−;Ldlr−/− mice22; thus, even in the context of increased type I collagen accumulation, we observed attenuated vascular calcification in these mice. Furthermore, VSMCs deficient in DDR1 express much more type I collagen and much less type IV collagen (data not shown) than Ddr1+/+ VSMCs, conditions that should promote, rather than inhibit, calcification.
Previous studies have demonstrated a strong link between inflammation and calcification of vessels. In particular, macrophages produce TNFα, which influences VSMC phenotypic transition and VSMC-mediated calcification.14,27 In the present study, we observed a marked decrease in TNFα staining in atherosclerotic lesions of Ddr1−/−;Ldlr−/− mice compared with Ddr1+/+;Ldlr−/− mice, and in a previous study, we demonstrated decreased macrophage accumulation and MCP-1 expression in lesions from Ddr1−/−;Ldlr−/− mice.22 Notably, the decreased mineralization in Ddr1−/−;Ldlr−/− mice was proportional to the decrease in inflammation in these lesions, because the ratios of Von Kossa-stained area/Mac-2-stained area were similar between Ddr1+/+;Ldlr−/− and Ddr1−/−;Ldlr−/− mice. Thus, the current study is consistent with a growing body of literature suggesting evidence of a proinflammatory role for DDR1 in other pathologies.32,33
Another novel finding of our work is that in Ldlr1−/− mice, the development of intimal calcification shares molecular features with the process of endochondral mineralization. Specifically, expression of Sox-9, a transcription factor involved in chondrocytic differentiation,34 was evident in rounded cells located within or near mineralized foci, as was the expression of type II collagen, an extracellular matrix protein involved in chondrogenesis.35 Type X collagen, a protein expressed uniquely by hypertrophic chondrocytes,36 was also detected in mineralizing lesions. In human atherosclerotic disease, chondrocyte-like cells and chondrocyte-specific proteins are seen in intimal lesions,2,5,7 and some of these proteins have also been documented in calcified atherosclerotic lesions of mice.4,13,25 Our research extends the list of chondrocyte-specific proteins identified in intimal calcification. Evidence of a chondrogenic phenotype was also observed in intimal lesions of Ddr1−/−;Ldlr−/− mice, indicating that while deletion of Ddr1 decreased the extent of this pathological process, it did not determine the fundamental mechanisms by which the phenotypic transition was accomplished.
DDR1-deficient VSMCs did not calcify the extracellular matrix, suggesting that there are cell-autonomous defects in the calcification process. Notably, our findings indicate that deletion of DDR1 may disrupt the local regulation of pyrophosphate, an important molecule that inhibits hydroxyapatite deposition. Ddr1−/− cells did not express alkaline phosphatase activity, an enzyme that reduces pyrophosphate levels. Furthermore, the DDR1-deficient cells expressed high levels of mRNA for NPP1, which encodes an enzyme that generates pyrophosphate.37 NPP1 also antagonizes the effects of alkaline phosphatase by competing for ATP substrate.38 Enpp1-deficient mice manifest with robust vascular calcification secondary to a change in VSMC phenotype characterized by the expression of chondrocytic markers Sox-9, type II, and type X collagen.28 It is noteworthy that in our studies, expression of these chondrocytic marker proteins was more extensive in the Ddr1+/+;Ldlr−/− mice compared with Ddr1−/−;Ldlr−/− mice.
In contrast to intimal calcification, medial calcification in Ldlr−/− mice fed a Western diet elaborates through an osteogenic transcriptional program, dependent on TNF-α, Msx-2 signaling, and paracrine Wnt signaling from the adventitial layers.9 Recently, Al-Aly et al14 demonstrated that pharmacological antagonism of TNFα by Infliximab treatment attenuated the expression of Msx-2 and abrogated the development of medial calcification in these mice. In our study, medial calcification was rare and was always adjacent to regions of intimal calcification, making it difficult to determine whether medial calcification in our study resulted from encroachment of intimal calcification into the medial layers or whether it originated there independently. Differences between our study and these previous reports may be attributed to different diet composition; for example, the diet used in our study (Research Diets D12108) contains 1.25% cholesterol, whereas the diet used in the previous report (Harlan Teklad 88137) contains only 0.15% cholesterol. Importantly, the diet used in previous studies contained twice as much vitamin D3 as the diet used in the current report (2200 versus 1000 IU/kg diet, respectively), and ingested vitamin D leads to elevations in serum calcium, which can initiate medial calcification.39 It is also possible that the differences we observed in our model were influenced by the mixed genetic background of the mice.
Although mineralization of the intima in Ldlr−/− mice can proceed in the context of normal serum Pi,13 intimal calcification in these mice has been attributed in some studies to elevated serum Pi.25,26 We observed normal levels of serum calcium and Pi in Ddr1+/+;Ldlr−/− mice; however, increased serum Pi was observed in DDR1-deficient mice after 12 and 24 weeks of atherogenic diet feeding. The reason for this increase is not clear. Measurements of blood urea nitrogen and serum creatinine in both Ddr1+/+;Ldlr−/− and Ddr1−/−;Ldlr−/− mice were within the normal range (data not shown), suggesting that elevated serum Pi in the Ddr1−/−;Ldlr−/− mice was not likely due to impaired renal function. Moreover, we observed similar levels of circulating Pi in 6-month-old Ddr1−/−;Ldlr−/− mice fed a standard rodent chow diet (data not shown). Thus, the elevation in serum Pi in these mice does not appear to be related to the high-cholesterol diet. Importantly, elevated serum Pi in Ddr1−/−;Ldlr−/− mice did not correlate with intimal calcification. These findings further support the notion that DDR1 acts locally, rather than systemically, to promote mineralization within the vessel wall.
In conclusion, our results demonstrate that DDR1 is an important positive regulator of atherosclerotic calcification, a process sharing molecular features with endochondral mineralization. Deletion of DDR1 resulted in a sustained reduction in the extent and incidence of atherosclerotic calcification and attenuated VSMC-mediated calcification of the extracellular matrix. Importantly, the reductions in vascular calcification that we have observed in Ddr1−/−;Ldlr−/− mice represent specific reductions in intimal calcification, a feature of advanced atherosclerosis that is correlated with important causes of clinical events, such as increased risk of plaque dissection and rupture. Defining the precise mechanisms by which DDR1 regulates this process will be the focus of future studies in an attempt to further understand the role of this receptor in disease progression.
Footnotes
Address reprint requests to Michelle P. Bendeck, Ph.D., Professor, Department of Laboratory Medicine and Pathobiology, University of Toronto, Medical Sciences Bldg., Rm. 6213, 1 King’s College Circle, Toronto, Ontario M5S 1A8, Canada. E-mail: michelle.bendeck@utoronto.ca.
Supported by grants from the Canadian Institutes of Health Research (MOP37847) and the Heart and Stroke Foundation of Canada (NA6096; to M.P.B). M.P.B. is a Career Investigator of the Heart and Stroke Foundation of Ontario. P.J.A. was supported by a Canada Graduate Scholarship Doctoral Research Award from the Canadian Institutes of Health Research. S.X. was supported by a John Schultz Summer Fellowship from the Heart and Stroke Foundation of Ontario. C.F. was supported by a Canada Graduate Scholarship Doctoral Research Award from the Canadian Institutes of Health Research and a Doctoral Research Award from the Heart and Stroke Foundation of Canada.
References
- Abedin M, Tintut Y, Demer LL. Vascular calcification: mechanisms and clinical ramifications. Arterioscler Thromb Vasc Biol. 2004;24:1161–1170. doi: 10.1161/01.ATV.0000133194.94939.42. [DOI] [PubMed] [Google Scholar]
- Bobryshev YV. Transdifferentiation of smooth muscle cells into chondrocytes in atherosclerotic arteries in situ: implications for diffuse intimal calcification. J Pathol. 2005;205:641–650. doi: 10.1002/path.1743. [DOI] [PubMed] [Google Scholar]
- Qiao JH, Fishbein MC, Demer LL, Lusis AJ. Genetic determination of cartilaginous metaplasia in mouse aorta. Arterioscler Thromb Vasc Biol. 1995;15:2265–2272. doi: 10.1161/01.atv.15.12.2265. [DOI] [PubMed] [Google Scholar]
- Rattazzi M, Bennett BJ, Bea F, Kirk EA, Ricks JL, Speer M, Schwartz SM, Giachelli CM, Rosenfeld ME. Calcification of advanced atherosclerotic lesions in the innominate arteries of ApoE-deficient mice: potential role of chondrocyte-like cells. Arterioscler Thromb Vasc Biol. 2005;25:1420–1425. doi: 10.1161/01.ATV.0000166600.58468.1b. [DOI] [PubMed] [Google Scholar]
- Tyson KL, Reynolds JL, McNair R, Zhang Q, Weissberg PL, Shanahan CM. Osteo/chondrocytic transcription factors and their target genes exhibit distinct patterns of expression in human arterial calcification. Arterioscler Thromb Vasc Biol. 2003;23:489–494. doi: 10.1161/01.ATV.0000059406.92165.31. [DOI] [PubMed] [Google Scholar]
- Speer MY, Yang HY, Brabb T, Leaf E, Look A, Lin WL, Frutkin A, Dichek D, Giachelli CM. Smooth muscle cells give rise to osteochondrogenic precursors and chondrocytes in calcifying arteries. Circ Res. 2009;104:733–741. doi: 10.1161/CIRCRESAHA.108.183053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostrom K, Watson KE, Horn S, Wortham C, Herman IM, Demer LL. Bone morphogenetic protein expression in human atherosclerotic lesions. J Clin Invest. 1993;91:1800–1809. doi: 10.1172/JCI116391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyemere VP, Proudfoot D, Weissberg PL, Shanahan CM. Vascular smooth muscle cell phenotypic plasticity and the regulation of vascular calcification. J Intern Med. 2006;260:192–210. doi: 10.1111/j.1365-2796.2006.01692.x. [DOI] [PubMed] [Google Scholar]
- Towler DA, Bidder M, Latifi T, Coleman T, Semenkovich CF. Diet-induced diabetes activates an osteogenic gene regulatory program in the aortas of low density lipoprotein receptor-deficient mice. J Biol Chem. 1998;273:30427–30434. doi: 10.1074/jbc.273.46.30427. [DOI] [PubMed] [Google Scholar]
- Cheng SL, Shao JS, Charlton-Kachigian N, Loewy AP, Towler DA. Msx2 promotes osteogenesis and suppresses adipogenic differentiation of multipotent mesenchymal progenitors. J Biol Chem. 2003;278:45969–45977. doi: 10.1074/jbc.M306972200. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Clinton SK, Iiyama K, Connelly PW, Libby P, Cybulsky MI. Hyperlipidemia and atherosclerotic lesion development in LDL receptor-deficient mice fed defined semipurified diets with and without cholate. Arterioscler Thromb Vasc Biol. 1999;19:1938–1944. doi: 10.1161/01.atv.19.8.1938. [DOI] [PubMed] [Google Scholar]
- Davies MR, Lund RJ, Hruska KA. BMP-7 is an efficacious treatment of vascular calcification in a murine model of atherosclerosis and chronic renal failure. J Am Soc Nephrol. 2003;14:1559–1567. doi: 10.1097/01.asn.0000068404.57780.dd. [DOI] [PubMed] [Google Scholar]
- Morony S, Tintut Y, Zhang Z, Cattley RC, Van G, Dwyer D, Stolina M, Kostenuik PJ, Demer LL. Osteoprotegerin inhibits vascular calcification without affecting atherosclerosis in ldlr−/− mice. Circulation. 2008;117:411–420. doi: 10.1161/CIRCULATIONAHA.107.707380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Aly Z, Shao JS, Lai CF, Huang E, Cai J, Behrmann A, Cheng SL, Towler DA. Aortic Msx2-Wnt calcification cascade is regulated by TNF-α-dependent signals in diabetic Ldlr−/− mice. Arterioscler Thromb Vasc Biol. 2007;27:2589–2596. doi: 10.1161/ATVBAHA.107.153668. [DOI] [PubMed] [Google Scholar]
- Smith EB. The influence of age and atherosclerosis on the chemistry of aortic intima. 2. Collagen and mucopolysaccharides. J Atheroscler Res. 1965;5:241–248. doi: 10.1016/s0368-1319(65)80065-5. [DOI] [PubMed] [Google Scholar]
- Koyama H, Raines EW, Bornfeldt KE, Roberts JM, Ross R. Fibrillar collagen inhibits arterial smooth muscle proliferation through regulation of Cdk2 inhibitors. Cell. 1996;87:1069–1078. doi: 10.1016/s0092-8674(00)81801-2. [DOI] [PubMed] [Google Scholar]
- Rocnik EF, Chan BMC, Pickering JG. Evidence for a role of collagen synthesis in arterial smooth muscle cell migration. J Clin Invest. 1998;101:1889–1898. doi: 10.1172/JCI1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson KE, Parhami F, Shin V, Demer LL. Fibronectin and collagen I matrixes promote calcification of vascular cells in vitro, whereas collagen IV matrix is inhibitory. Arterioscler Thromb Vasc Biol. 1998;18:1964–1971. doi: 10.1161/01.atv.18.12.1964. [DOI] [PubMed] [Google Scholar]
- Vogel W, Gish GD, Alves F, Pawson T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell. 1997;1:13–23. doi: 10.1016/s1097-2765(00)80003-9. [DOI] [PubMed] [Google Scholar]
- Vogel WF, Abdulhussein R, Ford CE. Sensing extracellular matrix: an update on discoidin domain receptor function. Cell Signal. 2006;18:1108–1116. doi: 10.1016/j.cellsig.2006.02.012. [DOI] [PubMed] [Google Scholar]
- Ferri N, Carragher NO, Raines EW. Role of discoidin domain receptors 1 and 2 in human smooth muscle cell-mediated collagen remodeling: potential implications in atherosclerosis and lymphangioleiomyomatosis. Am J Pathol. 2004;164:1575–1585. doi: 10.1016/S0002-9440(10)63716-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco C, Hou G, Ahmad PJ, Fu EY, Koh L, Vogel WF, Bendeck MP. Discoidin domain receptor 1 (Ddr1) deletion decreases atherosclerosis by accelerating matrix accumulation and reducing inflammation in low-density lipoprotein receptor deficient mice. Circ Res. 2008;102:1202–1211. doi: 10.1161/CIRCRESAHA.107.170662. [DOI] [PubMed] [Google Scholar]
- Hou G, Vogel W, Bendeck MP. The discoidin domain receptor tyrosine kinase DDR1 in arterial wound repair. J Clin Invest. 2001;107:727–735. doi: 10.1172/JCI10720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steitz SA, Speer MY, Curinga G, Yang HY, Haynes P, Aebersold R, Schinke T, Karsenty G, Giachelli CM. Smooth muscle cell phenotypic transition associated with calcification: up-regulation of Cbfa1 and down-regulation of smooth muscle lineage markers. Circ Res. 2001;89:1147–1154. doi: 10.1161/hh2401.101070. [DOI] [PubMed] [Google Scholar]
- Mathew S, Tustison KS, Sugatani T, Chaudhary LR, Rifas L, Hruska KA. The mechanism of phosphorus as a cardiovascular risk factor in CKD. J Am Soc Nephrol. 2008;19:1092–1105. doi: 10.1681/ASN.2007070760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies MR, Lund RJ, Mathew S, Hruska KA. Low turnover osteodystrophy and vascular calcification are amenable to skeletal anabolism in an animal model of chronic kidney disease and the metabolic syndrome. J Am Soc Nephrol. 2005;16:917–928. doi: 10.1681/ASN.2004100835. [DOI] [PubMed] [Google Scholar]
- Tintut Y, Patel J, Territo M, Saini T, Parhami F, Demer LL. Monocyte/macrophage regulation of vascular calcification in vitro. Circulation. 2002;105:650–655. doi: 10.1161/hc0502.102969. [DOI] [PubMed] [Google Scholar]
- Johnson K, Polewski M, van ED, Terkeltaub R. Chondrogenesis mediated by PPi depletion promotes spontaneous aortic calcification in NPP1−/− mice. Arterioscler Thromb Vasc Biol. 2005;25:686–691. doi: 10.1161/01.ATV.0000154774.71187.f0. [DOI] [PubMed] [Google Scholar]
- Adiguzel E, Ahmad PJ, Franco C, Bendeck MP. Collagens in the progression and complications of atherosclerosis. Vasc Med. 2009;14:73–89. doi: 10.1177/1358863X08094801. [DOI] [PubMed] [Google Scholar]
- Schapira K, Lutgens E, de FA, Sprague A, Roemen A, Gardner H, Koteliansky V, Daemen M, Heeneman S. Genetic deletion or antibody blockade of α1β1 integrin induces a stable plaque phenotype in ApoE−/− mice. Arterioscler Thromb Vasc Biol. 2005;25:1917–1924. doi: 10.1161/01.ATV.0000174807.90292.2f. [DOI] [PubMed] [Google Scholar]
- Tulla M, Pentikainen OT, Viitasalo T, Kapyla J, Impola U, Nykvist P, Nissinen L, Johnson MS, Heino J. Selective binding of collagen subtypes by integrin α 1I, α 2I, and α 10I domains. J Biol Chem. 2001;276:48206–48212. doi: 10.1074/jbc.M104058200. [DOI] [PubMed] [Google Scholar]
- Avivi-Green C, Singal M, Vogel WF. Discoidin domain receptor 1 deficient mice are resistant to bleomycin-induced lung fibrosis. Am J Respir Crit Care Med. 2006;174:420–427. doi: 10.1164/rccm.200603-333OC. [DOI] [PubMed] [Google Scholar]
- Flamant M, Placier S, Rodenas A, Curat CA, Vogel WF, Chatziantoniou C, Dussaule JC. Discoidin domain receptor 1 null mice are protected against hypertension-induced renal disease. J Am Soc Nephrol. 2006;17:3374–3381. doi: 10.1681/ASN.2006060677. [DOI] [PubMed] [Google Scholar]
- Bi W, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B. Sox9 is required for cartilage formation. Nat Genet. 1999;22:85–89. doi: 10.1038/8792. [DOI] [PubMed] [Google Scholar]
- von der MK, von der MH. Immunological and biochemical studies of collagen type transition during in vitro chrondrogenesis of chick limb mesodermal cells. J Cell Biol. 1977;73:736–747. doi: 10.1083/jcb.73.3.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichenberger E, Aigner T, von der MK, Stoss H, Bertling W. In situ hybridization studies on the expression of type X collagen in fetal human cartilage. Dev Biol. 1991;148:562–572. doi: 10.1016/0012-1606(91)90274-7. [DOI] [PubMed] [Google Scholar]
- Terkeltaub RA. Inorganic pyrophosphate generation and disposition in pathophysiology. Am J Physiol Cell Physiol. 2001;281:C1–C11. doi: 10.1152/ajpcell.2001.281.1.C1. [DOI] [PubMed] [Google Scholar]
- Johnson KA, Hessle L, Vaingankar S, Wennberg C, Mauro S, Narisawa S, Goding JW, Sano K, Millan JL, Terkeltaub R. Osteoblast tissue-nonspecific alkaline phosphatase antagonizes and regulates PC-1. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1365–R1377. doi: 10.1152/ajpregu.2000.279.4.R1365. [DOI] [PubMed] [Google Scholar]
- Bonucci E, Sadun R. Dihydrotachysterol-induced aortic calcification: a histochemical and ultrastructural investigation. Clin Orthop Relat Res. 1975:283–294. [PubMed] [Google Scholar]