

Abstract
Fatty acid synthase (FASN) is an emerging tumor-associated marker and a promising antitumor therapeutic target. In this study, we analyzed the expression of FASN in normal and molar placentas, as well as gestational trophoblastic neoplasia, and assessed the effects of a new FASN inhibitor, C93, on cellular proliferation and apoptosis in choriocarcinoma cells. Using a FASN-specific monoclonal antibody, we found that FASN immunoreactivity was detected in the cytotrophoblast and intermediate (extravillous) trophoblast of normal and molar placentas, as well as in placental site nodules. All choriocarcinomas (n = 33), 90% of epithelioid trophoblastic tumors (n = 20), and 60% of placental site trophoblastic tumors (n = 10) exhibited FASN positivity. FASN expression was further confirmed in vitro by Western blot and real-time PCR. Treatment of JEG3 and JAR cells with C93 induced significant apoptosis through the caspase-3/caspase-9/poly(ADP)ribose polymerase pathway. Cell cycle progression was not affected by the inhibitor. In summary, the data indicate that FASN is expressed in the majority of gestational trophoblastic neoplasias, and is essential for choriocarcinoma cells to survive and escape from apoptosis. FASN inhibitors such as C93 warrant further investigation as targeted therapeutic agents for metastatic and chemoresistant gestational trophoblastic neoplasia.
Gestational trophoblastic neoplasms (GTNs) represent a relatively uncommon type of gynecological tumor that behaves differently from other cancers. GTNs include choriocarcinoma, placental site trophoblastic tumor (PSTT), and epithelioid trophoblastic tumor (ETT).1 Clinically, GTNs are one of the few human tumors that are often cured by chemotherapy and/or local tumor resection. More specifically, nonmetastatic, low-risk GTNs treated with methotrexate or actinomycin D are almost always successfully treated, but cure rates in high-risk metastatic disease decrease to 80% to 90%, despite combination chemotherapy, surgery, and radiation.2,3 Also, approximately 5% of low-risk and 25% of high-risk patients respond poorly to initial treatment and require salvage chemotherapeutic regimens that include platinum or paclitaxel. Unfortunately, high-risk patients who fail or relapse after first-line EMA-CO (etoposide, methotrexate, dactinomycin, vincristine, and cyclophosphamide) therapy demonstrate an overall survival of 60% to 80%.2,4,5 Therefore, newer therapeutic regimens are needed to reduce the toxicity associated with current multi-agent chemotherapies and to salvage the occasional nonoperable patient with recurrent or chemoresistant disease.6 Until the fundamental biology of GTNs becomes more clearly understood, development of more novel therapies remains empirical.
Clinical promise has been shown by target-based therapies designed to inactivate molecular pathways that are essential for tumor cell growth and survival. Unlike standard chemotherapy, which indiscriminately affects proliferating cells, whether normal or neoplastic, inhibitors that target specific pathways in cancer have the potential to selectively eliminate tumor cells, thereby achieving maximal therapeutic effect with minimal adverse side effects. Recent examples of successful anticancer agents include gefitinib, a small kinase inhibitor that targets epidermal growth factor receptors, and trastuzumab (Herceptin), a humanized antibody that targets HER2/neu receptors. Given the success of molecular targeting in previous clinical trials, targeted therapy in the treatment of metastatic GTNs might be applied by tailoring management based on the expression profile of tumor’s specific markers.
Fatty acid synthase (FASN) is an intracellular enzyme that promotes the NADPH-dependent condensation of malonyl-CoA and acetyl-CoA to palmitate in endogenous lipogenesis.7,8 In normal cells, FASN levels are generally low due to the presence of abundant dietary lipids, but in neoplastic cells, FASN expression is up-regulated despite the presence of dietary lipids. Upregulation of FASN is observed in several types of human cancer including carcinomas of the breast, colon, ovary, and prostate.9,10,11,12,13,14 In this study, we assessed the biological role of FASN in GTN and in normal and molar placentas. We found that FASN expression in cytotrophoblast and intermediate (extravillous) trophoblastic cells is unique in that both normal and neoplastic trophoblastic cells express FASN. This is in contrast to most other tissue types, which preferentially express FASN in tumor cells, but not in their normal or benign counterparts. Moreover, inactivation of FASN led to massive apoptosis in choriocarcinoma cell lines, suggesting that FASN expression is required for the survival of choriocarcinoma cells. These results suggest that FASN inhibitor may be potentially useful as a new therapeutic reagent for advanced stage choriocarcinoma.
Materials and Methods
Case Selection and Immunohistochemistry
Formalin-fixed, paraffin-embedded tissues from 63 GTNs (33 choriocarcinomas, 20 epithelioid trophoblastic tumors, and 10 placental site trophoblastic tumors), ten placental site nodules, eight early placentas, four term placentas, and eight complete hydatidiform moles were retrieved from the surgical pathology files of the Gynecologic Pathology Division at the Johns Hopkins Hospital. The lesions were arranged on tissue microarrays, which contained three representative 1.5-mm cores constructed at the Johns Hopkins Tissue Microarray Facility. Specimens were anonymized and tissues were collected in compliance with institutional review board regulations. Paraffin sections were incubated overnight with a mouse monoclonal anti-FASN antibody (clone 6E7, FASgen Inc., Baltimore, MD) at a dilution of 1:50. Two observers independently scored the FASN immunoreactivity based on 10 different randomly selected high-power fields (×40 magnification). Scoring (H-score) was based on the percentage of positively stained cells and the intensity of nuclear staining, which ranged from 0 to 3+.15 The H-score was calculated using the following equation: H-score = ∑Pi (i +1), where i is the intensity of the stained tumor cells (0 to 3+), and Pi is the percentage of stained tumor cells for each intensity varying from 0% to 100%. This semiquantitative analysis has been shown to have high intra-observer and inter-observer reproducibility.16 In addition to H-score, we also used the percentage of stained cells as a criterion to measure FASN expression in tissue sections.
Cell Culture and Western Blot Analysis
Choriocarcinoma cell lines, JEG3 and JAR, were obtained from American Type Culture Collection (Rockville, MD). Both cell lines were incubated at 95% humidity, 5% CO2, and 37°C in RPMI media with 5% heat-inactivated fetal bovine serum (HyClone, Logan, UT) and 2% penicillin and streptomycin (Gibco, Rockville, MD). Western blot analysis was performed on the protein lysate of JAR and JEG3 cells and was compared with benign serous epithelium, which is known to minimally express FASN. Similar amounts of total protein from each lysate were separated on 10% Tris-glycine-SDS polyacrylamide gels (Novex, San Diego, CA) and then electroblotted to Millipore Immobilon-P polyvinylidene difluoride membranes. The membranes were probed with an anti-FASN mouse monoclonal antibody (1:100) followed by a peroxidase conjugated goat anti-mouse immunoglobulin (1:6000). To better delineate the regulation of apoptosis in JAR and JEG3 cells treated with FASN inhibitors, we used caspase and bcl-2 family sampler kits (Cell Signaling, Danvers, MA) including the antibodies reacting to caspase-3, caspase-7, caspase-9, poly(ADP)ribose polymerase (PARP), BIK, BOK, Bad, and PUMA to determine the levels of the apoptosis-related proteins. Western blots were developed by chemiluminescence (Pierce, Rockford, IL), using glyceraldehyde-3-phosphate dehydrogenase as a loading control (1:6000).
Real-Time PCR to Quantify FASN mRNA Expression
Total RNA was isolated using the TRIzol method (Invitrogen), and cDNA was synthesized using 2 to 5 μg total RNA template. Primer sequences to amplify the FASN cDNA were designed using the Primer 3 program and were: 5′-CATCCAGATAGGCCTCATAGAC-3′ (forward) and 5′-CTCCATGAAGTAGGAGTGGAAG-3′ (reverse). PCR conditions were as follows: one cycle at 95°C for 1 minute, 40 cycles at 95°C for 20 s, 60°C for 20 s, 70°C for 15 seconds, and 80°C for 10 seconds. The expression of FASN was normalized to that of human amyloid β precursor protein with threshold cycle numbers calculated from duplicate measurements. Mean fold expression differences were further normalized to that of benign ovarian surface epithelium, OSE10.
Cell Viability and Apoptosis Detection
Cancer cells were seeded in 96-well plates at a density of 2.5 × 103 per well and then incubated with 100 μl of the FASN inhibitor, C93 (FASgen Inc. Baltimore, MD), at a series of concentrations from 2.5 μg/ml to 50 μg/ml. Forty-eight hours after incubation, cell proliferation and viability were measured by the CellTiter-Blue assay (Promega, Madison, WI). Cells were incubated with 40 μl of the CellTiter reagent for 4 hours at 37°C. The fluorescent signal was then measured using a microplate reader with excitation/emission at 560/590 nm. Cell numbers were plotted against concentration of FASN inhibitor, and IC50 values (ie, the concentration at which cell growth was inhibited by 50% of the control level) was estimated. For apoptosis detection, 5 × 105 cells were grown in 6-well plates and treated with C93 at the calculated IC50 concentration or equivalent dimethyl sulfoxide (DMSO) control. Apoptotic cells were subsequently quantified using the Annexin V-FITC Detection Kit (Biovision, Mountain View, CA). Annexin binding was determined by flow cytometry and propidium iodide staining by phycoerythrin emission signaling.
Flow Cytometry
In preparation for flow cytometry 5 × 105 choriocarcinoma cells were seeded in 6-well plates and incubated for at least 24 hours until 50% to 60% cell confluence was reached. Cells were then treated with C93 at the calculated IC50 or with the equivalent DMSO control for 0 to 48 hours. After incubation, the cells were trypsinized, washed, and resuspended in a solution containing 0.6% NP-40, 3.7% formaldehyde, and 11 mg/ml Hoechst 33258 in PBS before being quantified using a BD LSR cytometer (Becton Dickinson, Franklin Lakes, NJ). Cell cycle analysis and the subG1 (apoptotic) fraction were determined using CellQuest software (Becton Dickinson).
Results
Expression of FASN in Gestational Trophoblastic Neoplasms and Tumor-Like Lesions
Immunohistochemistry was performed to assess the protein expression levels of FASN in GTNs (including choriocarcinoma, ETT, and PSTT) as well as in placental site nodules, and in normal and molar placentas. We found that FASN immunoreactivity was exhibited by 100% of choriocarcinomas (n = 33), 90% of epithelioid trophoblastic tumors (n = 20), 60% of placental site trophoblastic tumors (n = 10), and 90% of placental site nodules (n = 10). H-scores for the different types of GTNs, placental site nodules, and trophoblasts from normal and molar placentas are shown and summarized in Figure 1. The median H-scores in choriocarcinomas ranged from 10 to 130. The highest mean H-score (130) among trophoblastic tumors was observed in a choriocarcinoma. Representative immunostained GTNs are shown in Figure 2. In choriocarcinoma, FASN staining was observed in both cytotrophoblast and intermediate (extravillous) trophoblast, whereas FASN immunoreactivity was not detected in syncytiotrophoblast (Figure 2). Representative immunostained normal placenta and placental site nodule are shown in Figure 3. Similar to choriocarcinoma, FASN immunoreactivity in normal early placentas was detected in both cytotrophoblast and intermediate (extravillous) trophoblast, but was not detected in syncytiotrophoblast (Figure 3A). Term placentas exhibited only focal FASN positivity, which was exclusively located in cytotrophoblastic cells (Figure 3B). Intermediate trophoblastic cells in normal placentas and placental site nodules were also positive for FASN stain (Figure 3, C and D). Similarly, FASN immunoreactivity was detected in both cytotrophoblast and intermediate trophoblast in eight complete hydatidiform moles (Figure 3E). The H-score or the percentage of FASN positive cells in choriocarcinoma was significantly higher than that in PSTTs (P = 0.035, t-test), but was not significantly different from H-scores of ETT, complete hydatidiform mole, or trophoblastic cells in normal placentas (P > 0.05). There was no statistically significant difference between choriocarcinoma and PSTT, between ETT and PSTT, or between ETT and placental site IT. There was also no statistically significant difference in H-score or percentage of FASN positive cells between PSTTs and placental site trophoblastic cells in normal placentas, or between ETTs and placental site nodules (P > 0.05).
Figure 1.
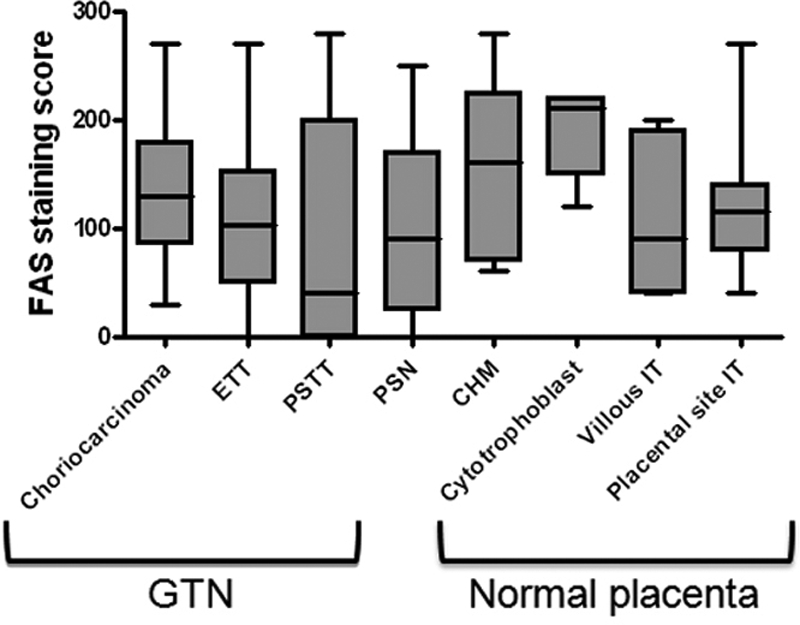
The H-scores of FASN immunoreactivity in gestational trophoblastic neoplasms (GTN), including choriocarcinoma, placental site trophoblastic tumor (PSTT), and epithelioid trophoblastic tumor (ETT), hydatidiform moles (CHM), placental site nodules (PSN), and normal trophoblastic cells in placentas. IT: intermediate (extravillous) trophoblast.
Figure 2.

Distribution of FASN immunoreactivity in choriocarcinoma, epithelioid trophoblastic tumor (ETT), and placental site trophoblastic tumor (PSTT). Upper panels: H&E stains; lower panels: corresponding FASN immunostains.
Figure 3.

FASN immunoreactivity in normal placenta, placental site nodule and molar placentas. A: In normal early placenta, FASN immunoreactivity is detectable in both cytotrophoblast (CT) and intermediate trophoblast (IT), whereas FASN is not detected in syncytiotrophoblast (ST). B: Similarly, only cytotrophoblast expresses FASN in a term placenta. Intermediate trophoblast is almost absent in a term placenta. C: Implantation site intermediate trophoblastic cells are immunoreactive to FASN. D: The intermediate trophoblastic cells in a placental site nodule are positive for FASN staining. E: FASN expression in a complete hydatidiform mole (CHM). A portion of a hydropic villus is shown. FASN immunoreactivity is present in both cytotrophoblast (CT) and intermediate trophoblast (IT) on the villous surface but is not detected in syncytiotrophoblast (ST).
C93 Suppresses Cancer Cell Growth
To determine whether FASN expression was essential for cell growth and survival in GTN, we treated two well-established choriocarcinoma cell lines, JAR and JEG3, with a new generation of FASN inhibitor, C93. Both cell lines expressed FASN proteins based on Western blot analysis. As shown in Figure 4A, a single protein band with a molecular mass corresponding to FASN protein was detected in JAR and JEG3 cells, but not in the negative control, OSE-10, a cell line established from benign ovarian surface epithelium. This result indicated the specificity of the anti-FASN monoclonal antibody used in this study. Quantitative real-time PCR confirmed this finding, revealing 3.5- to fourfold greater mean expression in JAR and JEG cells compared with normal ovarian surface epithelium (data not shown). These data demonstrated that both choriocarcinoma cell lines expressed FASN proteins. CellTiter Blue assay further demonstrated that C93 inhibited proliferation in a dose- dependent manner. The IC50 values were 8.7 and 9.5 μg/ml for JAR and JEG3, respectively (Figure 4, B and C).
Figure 4.

Expression of FASN and growth inhibition by C93 in choriocarcinoma cell lines. A: Western blot analysis shows a single protein band with a molecular mass (∼230 kDa) corresponding to FASN protein, in both JAR and JEG3 cells, but not in the negative control, OSE-10, established from ovarian surface epithelium. B and C: The IC50 curves demonstrate a C93 dose-dependent growth inhibition in JAR and JEG3 cells.
The Effects of C93 on Cellular Survival and Apoptosis in Choriocarcinoma Cells in Vitro
To further assess the mechanism of C93-induced growth arrest in choriocarcinoma cell lines, we applied flow cytometry, annexin-V staining, and Western blot analysis for caspase-3. C93 failed to affect the cell cycle in JAR and JEG3 48 hours after treatment. In contrast, a considerable percentage of subG1 (apoptotic) cells appeared in both cell lines starting at 12 hours and became more pronounced at 36 hours. After 12-hour and 48-hour incubations with C93, the subG1 cells accounted for 10.5% and 34% in JEG3, and 4.6% and 44% in JAR, respectively. In contrast, the subG1 fraction was only 1.9% and 3.7% for DMSO controls (P < 0.05, Figure 5A). As compared with mock (DMSO)-treated cells, C93 treatment resulted in an increase in the percentage of Annexin V stained cells in JAR and JEG3 cells in a time-dependant fashion. The percentage of early apoptotic cells was 78.3% and 78.2% for JAR and JEG3 after 48 hours, respectively, but was only 9.9% and 5.9% for DMSO controls (P < 0.01, Figure 5B). Next, after C93 treatment we analyzed several proteins critical in orchestrating apoptotic pathways in choriocarcinoma cells. Cleavage of caspase-3, caspase-9, and PARP was detected in choriocarcinoma cells 4 hours and 12 hours after C93 treatment (Figure 5C). In contrast, the protein levels for other apoptosis-related proteins including caspase-7, BIK, BOK, Bad, and PUMA did not change following C93 treatment in either of the cell lines (data not shown).
Figure 5.

The effects of C93 on cell cycle progression and induction of apoptosis in JAR and JEG3 cell lines. A: DNA flow cytometry analysis shows that the cell cycle distribution of choriocarcinoma cells is not altered after C93 treatment. In contrast, the late apoptotic cells, as defined by the subG1 fraction, significantly increase 48 hours after C93 treatment. B: The percentage of early apoptotic cells as detected by positive Annexin V staining increase in a time-dependent manner. DMSO was used as the negative control. C: Western blot analysis demonstrates the cleavage of caspase-3, caspase-9, and PARP (asterisks) is detected as early as 4 hours after C93 treatment in both cell lines. The upper bands in both caspase-9 and PARP represent the uncleaved forms of the proteins. Glyceraldehyde-3-phosphate dehydrogenase was used as the loading control.
Discussion
Although choriocarcinoma is highly responsive to combined chemotherapy, a small but significant proportion of patients develop recurrent disease after primary chemotherapy.5,17 Malignant PSTT and ETT, on the other hand, generally appear to be refractory to conventional chemotherapy.18,19,20,21,22,23,24,25,26 Consequently, patients with PSTT and ETT may develop chemoresistant tumors and eventually succumb to their disease.18,24 Thus, identification and characterization of molecular therapeutic targets are fundamental in designing new therapeutic interventions for GTN patients in the future. In this study, we showed that most GTNs expressed FASN, which was essential for cellular survival in choriocarcinoma cells. Our findings suggest that FASN inhibitors may be useful for treating GTN patients with advanced stage diseases that are resistant to conventional chemotherapy.
As compared with other tumor types that express FASN, such as breast, lung, and ovarian carcinoma, the expression pattern of FASN in human trophoblast is highly interesting and unique as both benign and neoplastic trophoblastic cells express FASN. One of the possible explanations for FASN expression in normal trophoblast is that trophoblastic cells may use de novo lipid synthesis due to FASN up-regulation in maintaining placental functions essential for fetal development. This strategy appears to be an evolutionarily favored compensatory mechanism, as the lipid supply from food intake may become limited during pregnancy. As an adjunct to lipid transportation across the maternal-fetal interface, FASN equips trophoblast cells with an alternative mechanism to ensure a continuous supply of fatty acids that are required for fetal organ development. In the tumorigenesis of GTN, tumor cells may take advantage of FASN expression for cell survival and growth. Nonetheless, the presence of FASN in normal trophoblast should not abate enthusiasm for developing FASN targeted therapy in GTN patients who are not pregnant because there is no normal trophoblastic tissue in those patients that would be affected by anti-FASN treatment.
FASN inhibitors, such as C93, may exert antitumor effects through multiple mechanisms. For example, the depletion of end product fatty acids accompanied by accumulation of toxic intracellular malonyl-CoA, or altered production of phospholipids resulting in diminished membrane synthesis,10,27 are thought to play major roles. FASN inhibitors may also suppress tumor growth by metabolism-independent mechanisms. FASN inhibition has been shown to selectively activate AMP-activated protein kinase in ovarian cancer cells causing cytotoxicity, while sparing most normal human tissues from these pleiotropic effects of AMP-activated protein kinase activation.28 A positive feedback regulation between AKT activation and FASN expression has been reported in ovarian carcinoma cells.29,30,31 The high frequency of FASN expression in GTNs and the essential role of FASN in maintaining cell growth are consistent with previous studies focusing on other types of human cancer, and indicate that FASN is a promising therapeutic target for neoplastic diseases including GTNs. Indeed, the anti-tumor effects of the first-generation FASN inhibitors (eg, cerulenin and Orlistat) have been well established.8,32,33,34 However, the original FASN inhibitors lacked chemical stability and caused undesirable weight loss in a mouse model, limiting their clinical application in cancer patients.8 A second generation of more selective FASN inhibitors that eliminate the concurrent stimulation of CPT-1, which is the rate-controlling catalyst in mitochondrial fatty acid oxidation, has been developed. These include C93, which has been reported to potently inhibit the enzyme activity of FASN.28,35
The most remarkable effect of exposure of choriocarcinoma cells to C93 is apoptosis, which is likely a point of convergence of different antitumor mechanisms consequent to FASN inhibition. Significantly lower caspase levels were observed in choriocarcinomas than in normal placentas.36 This is important because it has been shown that caspases initiate DNA fragmentation and cytoskeletal reorganization in cytotrophoblast and syncytiotrophoblast. Because a variety of apoptosis-associated proteins initiate and execute cytotoxic stress-induced apoptosis, we further delineated the molecules that were involved in C93-induced apoptosis. Among several candidate proteins that participate in apoptosis, we found that cleavage of caspase-9, caspase-3, and PARP occurred in response to FASN inhibition by C93. Thus, FASN inhibitors appear to trigger this apoptotic pathway in GTN cells, and it can be speculated that suppression of the caspase-9/casepase-3/PARP cascade by FASN may be responsible, at least in part, for the pathogenesis of GTNs. The finding that C93 rapidly induced apoptosis in choriocarcinoma cell lines, but failed to induce cell cycle arrest is different from observations made in other cancers including renal cell carcinoma,37 glioma,38 and ovarian cancer,27 suggesting that the antitumor effects of FASN inhibitors are cell-type specific.
In summary, this study provides new evidence that FASN is a novel trophoblast-associated enzyme in lipid metabolism. FASN is expressed in the majority of GTNs and is essential for choriocarcinoma cells to survive and escape from apoptosis. These findings suggest that FASN represents a potential therapeutic target in GTNs. Second generation FASN inhibitors, therefore, deserve special consideration in future clinical studies as potential therapeutics for patients with refractory and recurrent GTN.
Footnotes
Address reprint requests to Ie-Ming Shih, M.D., Ph.D. Johns Hopkins University School of Medicine, 1550 Orleans Street, Room 305 Baltimore, MD 21231. E-mail: ishih@jhmi.edu.
Supported by NIH grant CA129080 (to I.M.S.).
References
- Shih IM. Gestational trophoblastic neoplasia—pathogenesis and potential therapeutic targets. Lancet Oncol. 2007;8:642–650. doi: 10.1016/S1470-2045(07)70204-8. [DOI] [PubMed] [Google Scholar]
- Lurain JR, Nejad B. Secondary chemotherapy for high-risk gestational trophoblastic neoplasia. Gynecol Oncol. 2005;97:618–623. doi: 10.1016/j.ygyno.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Foulmann K, Guastalla JP, Caminet N, Trillet-Lenoir V, Raudrant D, Golfier F, Schott AM. What is the best protocol of single-agent methotrexate chemotherapy in nonmetastatic or low-risk metastatic gestational trophoblastic tumors? A review of the evidence. Gynecol Oncol. 2006;102:103–110. doi: 10.1016/j.ygyno.2006.02.038. [DOI] [PubMed] [Google Scholar]
- Powles T, Savage PM, Stebbing J, Short D, Young A, Bower M, Pappin C, Schmid P, Seckl MJ. A comparison of patients with relapsed and chemo-refractory gestational trophoblastic neoplasia. Br J Cancer. 2007;96:732–737. doi: 10.1038/sj.bjc.6603608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngan HY, Tam KF, Lam KW, Chan KK. Relapsed gestational trophoblastic neoplasia: a 20-year experience. J Reprod Med. 2006;51:829–834. [PubMed] [Google Scholar]
- Soper JT. Gestational trophoblastic disease. Obstet Gynecol. 2006;108:176–187. doi: 10.1097/01.AOG.0000224697.31138.a1. [DOI] [PubMed] [Google Scholar]
- Kuhajda FP, Pizer ES, Li JN, Mani NS, Frehywot GL, Townsend CA. Synthesis and antitumor activity of an inhibitor of fatty acid synthase. Proc Natl Acad Sci USA. 2000;97:3450–3454. doi: 10.1073/pnas.050582897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizer ES, Thupari J, Han WF, Pinn ML, Chrest FJ, Frehywot GL, Townsend CA, Kuhajda FP. Malonyl-coenzyme-A is a potential mediator of cytotoxicity induced by fatty-acid synthase inhibition in human breast cancer cells and xenografts. Cancer Res. 2000;60:213–218. [PubMed] [Google Scholar]
- Kuhajda FP, Jenner K, Wood FD, Hennigar RA, Jacobs LB, Dick JD, Pasternack GR. Fatty acid synthesis: a potential selective target for antineoplastic therapy. Proc Natl Acad Sci USA. 1994;91:6379–6383. doi: 10.1073/pnas.91.14.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizer ES, Wood FD, Heine HS, Romantsev FE, Pasternack GR, Kuhajda FP. Inhibition of fatty acid synthesis delays disease progression in a xenograft model of ovarian cancer. Cancer Res. 1996;56:1189–1193. [PubMed] [Google Scholar]
- Gansler TS, Hardman W, 3rd, Hunt DA, Schaffel S, Hennigar RA. Increased expression of fatty acid synthase (OA-519) in ovarian neoplasms predicts shorter survival. Hum Pathol. 1997;28:686–692. doi: 10.1016/s0046-8177(97)90177-5. [DOI] [PubMed] [Google Scholar]
- Epstein JI, Carmichael M, Partin AW. OA-519 (fatty acid synthase) as an independent predictor of pathologic state in adenocarcinoma of the prostate. Urology. 1995;45:81–86. doi: 10.1016/s0090-4295(95)96904-7. [DOI] [PubMed] [Google Scholar]
- Alo PL, Visca P, Marci A, Mangoni A, Botti C, Di Tondo U. Expression of fatty acid synthase (FAS) as a predictor of recurrence in stage I breast carcinoma patients. Cancer. 1996;77:474–482. doi: 10.1002/(SICI)1097-0142(19960201)77:3<474::AID-CNCR8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Rashid A, Pizer ES, Moga M, Milgraum LZ, Zahurak M, Pasternack GR, Kuhajda FP, Hamilton SR. Elevated expression of fatty acid synthase and fatty acid synthetic activity in colorectal neoplasia. Am J Pathol. 1997;150:201–208. [PMC free article] [PubMed] [Google Scholar]
- Budwit-Novotny DA, McCarty KS, Cox EB, Soper JT, Mutch DG, Creasman WT, Flowers JL, McCarty KS., Jr Immunohistochemical analyses of estrogen receptor in endometrial adenocarcinoma using a monoclonal antibody. Cancer Res. 1986;46:5419–5425. [PubMed] [Google Scholar]
- Lessey BA, Castelbaum AJ, Wolf L, Greene W, Paulson M, Meyer WR, Fritz MA. Use of integrins to date the endometrium. Fertil Steril. 2000;73:779–787. doi: 10.1016/s0015-0282(99)00604-4. [DOI] [PubMed] [Google Scholar]
- Newlands ES. The management of recurrent and drug-resistant gestational trophoblastic neoplasia (GTN). Best Pract Res Clin Obstet Gynaecol. 2003;17:905–923. doi: 10.1016/s1521-6934(03)00092-0. [DOI] [PubMed] [Google Scholar]
- Chang YL, Chang TC, Hsueh S, Huang KG, Wang PN, Liu HP, Soong YK. Prognostic factors and treatment for placental site trophoblastic tumor—report of 3 cases and analysis of 88 cases. Gynecol Oncol. 1999;73:216–222. doi: 10.1006/gyno.1999.5344. [DOI] [PubMed] [Google Scholar]
- Denny LA, Dehaeck K, Nevin J, Soeters R, van Wijk AL, Megevand E, Bloch B. Placental site trophoblastic tumor: three case reports and literature review. Gynecol Oncol. 1995;59:300–303. doi: 10.1006/gyno.1995.0026. [DOI] [PubMed] [Google Scholar]
- Twiggs LB, Hartenbach E, Saltzman AK, King LA. Metastatic placental site trophoblastic tumor. Int J Gynaecol Obstet. 1998;60 Suppl 1:S51–55. doi: 10.1016/S0020-7292(98)80005-2. [DOI] [PubMed] [Google Scholar]
- Swisher E, Drescher CW. Metastatic placental site trophoblastic tumor: long-term remission in a patient treated with EMA/CO chemotherapy. Gynecol Oncol. 1998;68:62–65. doi: 10.1006/gyno.1997.4888. [DOI] [PubMed] [Google Scholar]
- Feltmate CM, Genest DR, Goldstein DP, Berkowitz RS. Advances in the understanding of placental site trophoblastic tumor. J Reprod Med. 2002;47:337–341. [PubMed] [Google Scholar]
- Macdonald MC, Palmer JE, Hancock BW, Tidy JA. Diagnostic challenges in extrauterine epithelioid trophoblastic tumours: a report of two cases. Gynecol Oncol. 2008;108:452–454. doi: 10.1016/j.ygyno.2007.11.015. [DOI] [PubMed] [Google Scholar]
- Shih I-M, Kurman RJ. Epithelioid trophoblastic tumor—A neoplasm distinct from choriocarcinoma and placental site trophoblastic tumor simulating carcinoma. Am J Surg Pathol. 1998;22:1393–1403. doi: 10.1097/00000478-199811000-00010. [DOI] [PubMed] [Google Scholar]
- Mazur MT. Metastatic gestational choriocarcinoma. Unusual pathologic variant following therapy. Cancer. 1989;63:1370–1377. doi: 10.1002/1097-0142(19890401)63:7<1370::aid-cncr2820630723>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Silva EG, Tornos C, Lage J, Ordonez NG, Morris M, Kavanagh J. Multiple nodules of intermediate trophoblast following hydatidiform moles. Int J Gynecol Pathol. 1993;12:324–332. doi: 10.1097/00004347-199310000-00007. [DOI] [PubMed] [Google Scholar]
- Pizer ES, Chrest FJ, DiGiuseppe JA, Han WF. Pharmacological inhibitors of mammalian fatty acid synthase suppress DNA replication and induce apoptosis in tumor cell lines. Cancer Res. 1998;58:4611–4615. [PubMed] [Google Scholar]
- Zhou W, Han WF, Landree LE, Thupari JN, Pinn ML, Bililign T, Kim EK, Vadlamudi A, Medghalchi SM, El Meskini R, Ronnett GV, Townsend CA, Kuhajda FP. Fatty acid synthase inhibition activates AMP-activated protein kinase in SKOV3 human ovarian cancer cells. Cancer Res. 2007;67:2964–2971. doi: 10.1158/0008-5472.CAN-06-3439. [DOI] [PubMed] [Google Scholar]
- Wang HQ, Altomare DA, Skele KL, Poulikakos PI, Kuhajda FP, Di Cristofano A, Testa JR. Positive feedback regulation between AKT activation and fatty acid synthase expression in ovarian carcinoma cells. Oncogene. 2005;24:3574–3582. doi: 10.1038/sj.onc.1208463. [DOI] [PubMed] [Google Scholar]
- Grunt TW, Wagner R, Grusch M, Berger W, Singer CF, Marian B, Zielinski CC, Lupu R. Interaction between fatty acid synthase- and ErbB-systems in ovarian cancer cells. Biochem Biophys Res Commun. 2009;385:454–459. doi: 10.1016/j.bbrc.2009.05.085. [DOI] [PubMed] [Google Scholar]
- Uddin S, Siraj AK, Al-Rasheed M, Ahmed M, Bu R, Myers JN, Al-Nuaim A, Al-Sobhi S, Al-Dayel F, Bavi P, Hussain AR, Al-Kuraya KS. Fatty acid synthase and AKT pathway signaling in a subset of papillary thyroid cancers. J Clin Endocrinol Metab. 2008;93:4088–4097. doi: 10.1210/jc.2008-0503. [DOI] [PubMed] [Google Scholar]
- Kridel SJ, Axelrod F, Rozenkrantz N, Smith JW. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res. 2004;64:2070–2075. doi: 10.1158/0008-5472.can-03-3645. [DOI] [PubMed] [Google Scholar]
- Kuhajda FP, Landree LE, Ronnett GV. The connections between C75 and obesity drug-target pathways. Trends Pharmacol Sci. 2005;26:541–544. doi: 10.1016/j.tips.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Knowles LM, Axelrod F, Browne CD, Smith JW. A fatty acid synthase blockade induces tumor cell-cycle arrest by down-regulating Skp2. J Biol Chem. 2004;279:30540–30545. doi: 10.1074/jbc.M405061200. [DOI] [PubMed] [Google Scholar]
- Orita H, Coulter J, Lemmon C, Tully E, Vadlamudi A, Medghalchi SM, Kuhajda FP, Gabrielson E. Selective inhibition of fatty acid synthase for lung cancer treatment. Clin Cancer Res. 2007;13:7139–7145. doi: 10.1158/1078-0432.CCR-07-1186. [DOI] [PubMed] [Google Scholar]
- Fong PY, Xue WC, Ngan HY, Chiu PM, Chan KY, Tsao SW, Cheung AN. Caspase activity is downregulated in choriocarcinoma: a cDNA array differential expression study. J Clin Pathol. 2006;59:179–183. doi: 10.1136/jcp.2005.028027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiguchi A, Asano T, Asano T, Ito K, Sumitomo M, Hayakawa M. Pharmacological inhibitor of fatty acid synthase suppresses growth and invasiveness of renal cancer cells. J Urol. 2008;180:729–736. doi: 10.1016/j.juro.2008.03.186. [DOI] [PubMed] [Google Scholar]
- Zhao W, Kridel S, Thorburn A, Kooshki M, Little J, Hebbar S, Robbins M. Fatty acid synthase: a novel target for antiglioma therapy. Br J Cancer. 2006;95:869–878. doi: 10.1038/sj.bjc.6603350. [DOI] [PMC free article] [PubMed] [Google Scholar]