

Abstract
The ability of amiodarone and its putative metabolites to activate unmodified human trace amine associated receptor 1 (hTAAR1) stably expressed in CHO cell lines was evaluated. Receptor activation was monitored by measuring the accumulation of cAMP, the putative hTAAR1 native second messenger, or calcium mobilization in cells where the receptor was coupled to the promiscuous Gq, Gα16. Despite literature reports of activation of rodent TAAR1 by these agents, no response was seen in either cell line although robust activation was obtained with the endogenous ligand β-phenethylamine. These results indicate that TAAR1 activation by amiodarone and its analogs is species specific.
Keywords: Amiodarone, Human Trace amine-associated receptor 1, hTAAR1, G protein-coupled receptors
The discovery and characterization of several families and subfamilies of mammalian trace amine associated receptors (TAARs)1–3 has prompted much speculation4–8 along with investigations9–13 regarding the role of these G protein-coupled receptors (GPCRs). One line of investigation has demonstrated that iodothyronamines are potent agonists at rodent TAAR1 and this has been linked with the pharmacological effects of exogenously administered thyroid hormone metabolites such as T1AM in rodents.9 Based on the known cardiac effects of thyronamines and their structural similarities with the clinical antiarrythmic agent amiodarone (1), it has been speculated that TAAR1 may be involved in the actions of amiodarone.14 Evaluation of amiodarone (1) along with a series of known and putative amiodarone metabolism products (2–9) (Figure 1) in rat, mouse and rat-human chimera TAAR114 showed some agonist activity in rodent TAAR1, but not in the rat-human chimera cell line. The authors suggested that although amiodarone failed to demonstrate any activity in r-hTAAR1, some of the amiodarones may be active in unmodified hTAAR1. Since we had stably expressed wild-type hTAAR1 in two CHO cell lines, and had developed a rapid assay for evaluation of agonist activity of hTAAR1,15 we were well positioned to carry out such studies.
Figure 1.
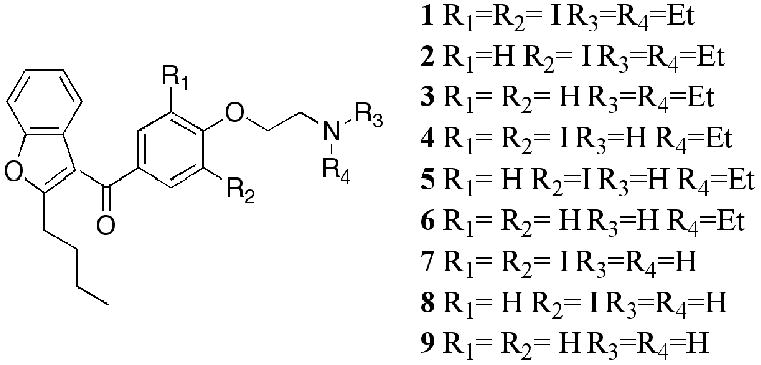
Structures of amiodarones 1–9.
Materials and Methods
The amiodarones 1–9 (in dimethylsulfoxide) were a gift from Professor Scanlan; β–Phenethylamine hydrochloride was purchased from Sigma Aldrich (St. Louis, MO).
hTAAR 1 Activation Assays
Receptor activation was assayed in CHO cells stably expressing Gα16 (RD-HGA16 cells; Molecular Devices Corporation, Sunnyvale, CA) and hTAAR1 as well as in CHO-K1 cells (ATCC) stably expressing only the hTAAR1, as previously described.15 The ability of the test compounds to activate hTAAR1 was assessed in the RD-HGA16-derived cell line using the Calcium 3 Assay Kit (Molecular Devices; Sunnyvale, CA) per manufacturer’s specifications except that the Calcium 3 dye was used at 1/3 the suggested concentration. In the CHO-K1 cell line, stimulation of hTAAR1-coupled adenylyl cyclase activity (cAMP Direct Biotrak EIA Kit; Amersham Biosciences, Piscataway, NJ) was used to evaluate the compounds. In each assay, the test compounds were assayed in duplicate at 0.1, 1.0 and 10 μM (in 100% DMSO). Data are reported as percent maximal stimulation by 1 μM β-PEA. The data represent the mean ± SE from three experiments.
Results
The amiodarone series consisted of the parent compound 1, its N-nor (4) and N.N-dinor (7) analogs, the monoiodo (2) and desiodo (3) analogs, the monoiodo-N-nor (5) and N,N-dinor (8), and the des iodo-N-nor (6) and N,N-dinor (9) analogs. At concentrations of 0.1 and 1 μM compounds 1–9 elicited small negative responses in the cAMP assay. At 10 μM very small (<5% of maximal activation by 1 μM μ-phenethylamine, μ-PEA) positive effects were seen for compounds 6, 7, and 8. (Fig. 2). In the calcium mobilization assays amiodarone (1) failed to activate hTAAR1 at any of the tested concentrations. The analogs 2–9 were inactive at 0.1 and 1 μM concentrations (data not shown) but elicited calcium responses ranging in magnitude from 5–100% relative to β-PEA at 10 μM. However, with the exception of compound 4, similar responses (within the margin of the experimental error) were seen in the parent Gα16, non-hTAAR1-expressing, cell line. For N-noramiodarone (4) a net (RFUexpressing − RFUparent) stimulation of 44±4% relative to β-PEA was observed. Neither amiodarone (1), nor μ-PEA, affected calcium mobilization in CHO cells stably expressing Gα16 (no hTAAR1); a robust response was obtained for μ-PEA in hTAAR1-Gα16 cells while 1, amiodarone, failed to affect calcium flux in this cell line.
Figure 2.

cAMP accumulation(% of maximal β-PEA response) resulting from interaction of compounds 1–9 with hTAAR1 stably expressed in CHO-K1 cells.
Discussion
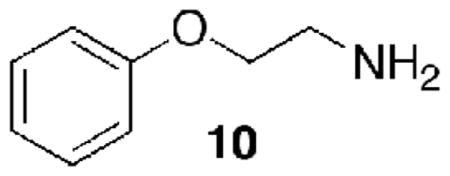
It had been suggested that some of the known side effects of amiodarone (1) may be associated with its, or its metabolism products’, biological activity at TAAR1 and that the lack of any activity in the rat-human TAAR1 might be explained by the inherent difference between this chimeric r-hTAAR1 and wild type hTAAR1.14 Using our hTAAR1-Gα16 cell line we had determined that replacement of the ethylene group (CH2CH2) in μ-PEA by an oxyethylene group (OCH2CH2) (to give 10), reduced potency by an order of magnitude and reduced efficacy by a factor of two (EC50 1,792±430 nM; Emax 41±5, unpublished).
Moreover, our CoMFA of a series of systematically substituted β-PEA analogs, assayed using our hTAAR1-Gα16 cell line, had led to the conclusion that β-PEA analogs with bulky substituents are poorly accommodated by the receptor due to steric constraints, resulting in decreased potency and efficacy.16 In particular, it appeared that N,N-dialkylation and para, or multiple, aryl substitutions resulted in a profound decrease in both potency and efficacy. In addition, it was observed that ortho substitution, particularly by halogens, enhanced potency. Based on these observations and conclusions, amiodarone (1) was not expected to be a potent agonist, but the N,N-dinor analogs 7 and 8 could have had some agonist activity. In fact, 7 and 8 had been found to be the most potent agonists in this series in the rTAAR1 cell line, with 7 exhibiting full agonist activity at 10 μM (relative to T1AM).14
Characterization of the action of the amiodarones on hTAAR1 by measuring the accumulation of cAMP, the putative hTAAR1 native second messenger, in CHO cells stably expressing hTAAR1 showed that none of the amiodarones 1–9 activated hTAAR1 at 0.1–10 μM (Fig. 2). This result was confirmed for 0.1–10 μM concentrations of amiodarone (1) by examining the effect on calcium mobilization in CHO cells stably expressing hTAAR1 coupled to the promiscuous Gq, Gα16 and for 0.1–1.0 μM concentrations of the analogs 2–9. At 10 μM, compound 4 increased calcium mobilization by 44% relative to β-PEA, but the lack of a concentration response relationship does not support a hTAAR1 mediated event. The reason for the responses observed for compounds 2–9 in CHO cells expressing Gα16 but no hTAAR1, is not known. It should be noted that neither amiodarone (1), nor β-PEA, exhibited such behavior. These observations provide yet one more example of the lack of interspecies correspondence of TAAR1 responses.17 Even in the rodent data it was noted14 that while the desiodo analog 3 was a full agonist in rTAAR1 at 10 μM, it was totally inactive in mTAAR1. The reasons for these differences are not known. While both mTAAR1 and rTAAR1 consist of 999 base-pairs, several discrepancies have been noted for rTAAR1. By comparison, hTAAR1 contains 1020 base-pairs with one discrepancy, possibly accounting for the difference in responses between rodent TAAR1 and hTAAR1 to this class of compounds. Whether the accumulation of amiodarone (1), and/or of its possible metabolites 2–9, in adipose tissue could lead to concentrations high enough (>10 μM) to activate TAAR1 in humans, and thus be clinically relevant, as has been suggested,14 is open to question. In rodents, the highest density of TAAR1 was found in the stomach, followed by the brain.1 It would not be surprising to find significant differences in both the receptor distribution of TAAR1 and the metabolic fate of amiodarone between rodent and human, leaving the question of whether the side effects of amiodarone are mediated by hTAAR1 unresolved.
Acknowledgments
The authors are grateful to Professor Scanlan for providing amidarones 1–9, to K. Warner for technical assistance, and to NIDA (Grant No. R01-DA016327) for financial support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Borowsky B, Adham N, Jones KA, Raddatz R, Arymshym R, Ogozalek KL, Durkin MM, Laklani PP, Bonini JA, Pathirana S, Boyle N, Po X, Kouranova E, Lichtblau H, Ochoa FY, Branchek TA, Gerald C. Proc Natl Acad Sci USA. 2001;98:8966–8971. doi: 10.1073/pnas.151105198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bunzow JR, Sonders MS, Arttmagangkul S, Harrison lM, Zhamg G, Quigley DI, Darland T, Suchland KL, Pasumamula S, Kennedy JL, Olson SB, Magenis RE, Amara SG, Grandy DK. Mol Pharmacol. 2001;60:1181–1188. doi: 10.1124/mol.60.6.1181. [DOI] [PubMed] [Google Scholar]
- 3.Lindemann L, Ebeling M, Kratochwil NA, Bunzow JR, Grandy DK, Hoener MC. Genomics. 2005;85:372–385. doi: 10.1016/j.ygeno.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 4.Premont RT, Gainedtinov RR, Caron MG. Proc Natl Acad Sci USA. 2001;98:9474–9475. doi: 10.1073/pnas.181356198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berry MD. J Neurochem. 2004;90:257–271. doi: 10.1111/j.1471-4159.2004.02501.x. [DOI] [PubMed] [Google Scholar]
- 6.Berry MD. Rev Recnt Clin Trials. 2007;2:3–19. doi: 10.2174/157488707779318107. [DOI] [PubMed] [Google Scholar]
- 7.Branchek TA, Blackburn TP. Cur Opin Pharmacol. 2003;3:90–97. doi: 10.1016/s1471-4892(02)00028-0. [DOI] [PubMed] [Google Scholar]
- 8.Grandy DK. Pharmacol Ther. 2007;116:355–390. doi: 10.1016/j.pharmthera.2007.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scanlan TS, Suchland KL, Hart ME, Chiellini G, Huang Y, Kruzich PJ, Frascarelli S, Crossley DA, Bunzow JR, Ronca-Testoni S, Lin ET, Hatton D, Zucchi R, Grandy DK. Nat Med. 2004;10:638–642. doi: 10.1038/nm1051. [DOI] [PubMed] [Google Scholar]
- 10.Miller GM, Verrico C, Jassen A, Konar M, Yang H, Panas H, Mary B, Johnson R, Madras BK. J Pharmacol Exp Ther. 2005;313:983–994. doi: 10.1124/jpet.105.084459. [DOI] [PubMed] [Google Scholar]
- 11.Xie Z, Miller GM. J Pharmacol Exp Ther. 2007;321:128–136. doi: 10.1124/jpet.106.117382. [DOI] [PubMed] [Google Scholar]
- 12.Xie Z, Westmoreland SV, Bahn ME, Chen GL, Yang H, Vallender EJ, Yao WD, Madras BK, Miller GM. J Pharmacol Exp Ther. 2007;321:116–127. doi: 10.1124/jpet.106.116863. [DOI] [PubMed] [Google Scholar]
- 13.Wollinsky TD, Swanson CJ, Smith KE, Zhong H, Borowsky S, Seeman P, Branhek T, Gerald CP. Genes Brain Behav. 2007;6:628–639. doi: 10.1111/j.1601-183X.2006.00292.x. [DOI] [PubMed] [Google Scholar]
- 14.Snead AN, Miyakawa M, Tan ES, Scanlan TS. Bioorg & Med Chem Lett. 2008;18:5920–5922. doi: 10.1016/j.bmcl.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Navarro HA, Gilmour BP, Lewin AH. J Biomol Screen. 2006;11:688–693. doi: 10.1177/1087057106289891. [DOI] [PubMed] [Google Scholar]
- 16.Lewin AH, Navarro HA, Mascarella SW. Bioorg Med Chem. 2008;16:7415–7423. doi: 10.1016/j.bmc.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lewin AH. In: Drug Addiction from Basic Research to Therapy. Rapaka RS, Sadee W, editors. Springer; New York: 2008. pp. 327–339. [Google Scholar]