

Abstract
Proteins of the plasminogen activation system are broadly expressed throughout the nervous system, and key roles for these proteins in neuronal function have been demonstrated. Recent reports have established that plasminogen is synthesized in neuroendocrine tissues, making this protein and the proteolytic activity of the product of its activation, plasmin, available at sites separated anatomically from circulating, hepatocyte-derived plasminogen. Results with plasminogen-deficient humans and mice suggest a role for plasminogen in neuritogenesis. To elucidate the role of the plasminogen activation system in these processes, the function of plasminogen during neuritogenesis and neurite outgrowth was studied. It is shown here that plasminogen participates in neuritogenesis, as plasmin inhibitors reduced both neurite outgrowth and neurite length in PC-12 cells. The addition of exogenous plasminogen enhanced neurite outgrowth and neurite length in both PC-12 cells and primary cortical neurons. The proteolytic activity of plasmin was required, since mutation of the catalytic serine residue completely abolished the stimulatory activity. Furthermore, mutation of the lysine binding site within kringle 5 of the plasminogen molecule also reduced the neuritogenic activity of plasminogen. Additionally, we demonstrate that plasminogen specifically bound to laminin-1, the interaction resulted in increased plasminogen activation by tissue-type plasminogen activator, and was dependent on a functional lysine binding site within plasminogen kringle 5. Moreover, during NGF-induced neuritogenesis, laminin-1 was degraded, and this cleavage was catalyzed by plasmin. This study provides the first direct evidence that plasminogen participates in neurite outgrowth and also suggests that laminin-1 degradation by plasmin contributes to the process of neuritogenesis.
Introduction
A key feature of the developing nervous system is the growth of neurites along specific pathways. This process requires intracellular signaling, cell migration, interactions with the extracellular matrix (ECM), and proteolytic cleavage of specific substrates. In recent decades, growth factors and growth factor receptors that regulate the development of the nervous system have been identified (Guan and Rao, 2003; Tran and Miller, 2003; Britto et al., 2004). More recently, the molecular components implicated in cell migration and interaction with the ECM have been shown to be key elements in this process (Kiryushko et al., 2004; Pietri et al., 2004). However, the proteolytic enzymes that participate in different steps of axon growth, and the specific substrates they modify, have not been investigated in detail.
The plasminogen (Plg) activation system is composed of a group of serine proteases that play an important role in the remodeling of the ECM during different physiological and pathological processes. Although most studies with this system have focused on hepatic-derived Plg, recent reports have demonstrated that Plg and its activator, tissue plasminogen activator (t-PA), are synthesized throughout neuroendocrine tissues (Qian et al., 1993; Sappino et al., 1993; Tsirka et al., 1997; Basham and Seeds, 2001; Zhang et al., 2002). As a result, there is increasing evidence for an important role of the Plg activation system in neuronal development and recovery (Seeds et al., 1999; Akassoglou et al., 2000; Siconolfi and Seeds, 2001a). For example, patients with severe type I Plg deficiency (Plg activity and antigen both extremely low) exhibit symmetric internal hydrocephalus with a Dandy-Walker malformation, hypoplasia of the cerebellum, and a hypoplastic corpus callosum (Schott et al., 1998). Hydrocephalus has also been recognized in Plg-deficient mice (Drew et al., 1998). These syndromes are consistent with defects in neuronal growth and differentiation that are modulated by proteolysis. Proteins of the Plg activation system are induced during axonal outgrowth in murine embryonic dorsal root ganglia (Hayden and Seeds, 1996; Hoover-Plow et al., 2001). Mice lacking Plg or its activators show delayed functional recovery after sciatic nerve crush in the peripheral nervous system (Siconolfi and Seeds, 2001b). Additionally, a series of studies has documented a requirement for Plg activators in neurite outgrowth induced by nerve growth factor (NGF) (Pittman and DiBenedetto, 1995; Farias-Eisner et al., 2001). Furthermore, synthesis of several proteins of the fibrinolytic system, including the urokinase receptor, Plg activator inhibitor-1 and Plg, is regulated by NGF (Farias-Eisner et al., 2000; Takahashi et al., 2000; Gutierrez-Fernandez et al., 2007). Together, these studies strongly support the participation of Plg in neurologic functions.
The importance of the ECM during neuronal development, and the evidence for a role of Plg in neuronal functions, led us to investigate the role of Plg in neurite outgrowth and determine whether this process was dependent on the interaction of Plg with laminin-1 (LN-1), a major laminin expressed in brain and associated with neuronal growth and development (Lander et al., 1985; Colognato and Yurchenco, 2000). The results of these studies are summarized herein.
Materials and Methods
Cell culture.
Rat pheochromocytoma PC-12 cells, a well established model of NGF-induced neuritogenesis (Greene and Tischler, 1976), were grown in high-glucose DMEM (Invitrogen) containing 2 mm l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin supplemented with 5% heat-inactivated calf serum and 10% heat-inactivated horse serum and maintained in a humidified chamber (37°C, 5% CO2). Primary rat cortical neurons were provided as microsurgically dissected regions of day 18 embryonic Sprague Dawley rat brain from Genlantis, digested with 20 U/mg papain (Sigma), and grown in Neuro culture medium (Neurobasal medium; Invitrogen) supplemented with B27 serum-free supplement and 500 μm l-glutamine (Sigma).
For neurite outgrowth experiments, PC-12 cells were grown in serum-free DMEM for 16 h, harvested, washed with serum-free DMEM, and plated on LN-1, the latter of which was coated onto six-well plates in serum-free DMEM. Neurite outgrowth was induced by treatment with 100 ng/ml NGF (Invitrogen) in serum-free medium. Culture media, inhibitors, and NGF were replaced daily. For neurite outgrowth in cortical neurons, cells were cultured on poly-l-lysine-coated coverslips in serum-free B27 medium in the presence or absence of 25 nm Plg, and media were replaced as described above.
Proteins.
Human Plg was purified from fresh plasma by affinity chromatography on lysine-Sepharose as described previously (Deutsch and Mertz, 1970) and modified in our laboratory (Miles et al., 1988). Plg mutants [D(139)N]Plg, [D(218)N]Plg, [D(413)N]Plg, [D(518)N]Plg, and [S(646)E]Plg were generated by primer-directed mutagenesis and purified from insect conditioned medium as described previously (Menhart et al., 1995). Human plasmin and α2-antiplasmin were purchased from American Diagnostica. Monoclonal antibody (mAb) 51 was raised against kringle domains 1–3 of Plg (Pozzi et al., 2000), and mAb 2D1 was raised against mini-Plg (kringle 5 plus protease chain) (Kenagy et al., 1996).
Determination of neurite outgrowth.
Cells were plated at low cell densities to minimize neurite contact, and phase-contrast micrographs were captured every 24 h. Images were acquired with a Zeiss Axioplan microscope equipped with a Zeiss 10× objective and analyzed using the image analysis software Image Pro Plus 4.0 (Media Cybernetics). The percentage of neurite-positive cells was determined by counting the number of cells bearing neurite-like processes greater than the length of the cell soma after treatment and dividing by the total number of cells. A minimum of 150 cells per well in duplicate cultures were counted. For neurite length measurements, data were collected from neurites that did not contact other neurites or cell bodies.
Plg binding assays.
Ninety-six well plates (Nalge Nunc International) were coated with LN-1 (Millipore Bioscience Research Reagents) at 1 μg/ml for 2 h at 37°C in PBS containing Ca2+ and Mg2+. The wells were blocked with 5% bovine serum albumin (BSA) at 37°C for 1 h and washed three times before incubation with 0.25 μm recombinant Plg (rPlg), plasma Plg, or [D(518)N]Plg for 1 h. The wells were then washed three times with PBS to remove unbound Plg and incubated with primary rabbit anti-human Plg antibody (1:5000 dilution). The wells were rinsed with PBS, incubated with an anti-rabbit antibody–HRP conjugate (1:30,000 dilution; Pierce), washed three times with PBS, and developed using 1,2-phenylene diamine-dihydrochloride (Dako). Color development was measured at 450 nm in a microtiter plate reader (Molecular Devices). Each experiment was performed in triplicate.
Plg activation.
Purified LN-1 or BSA was coated onto 96-well plates at 1 μg/ml as above, and wells were blocked with 5% BSA and washed three times with 20 mm HEPES, pH 7.4. Plg was added at a final concentration of 0.3 μm and incubated at 37°C for 1 h, followed by the addition of 0.3 mm of the plasmin substrate VLK-pNA (Val-Leu-Lys- p-nitroanilide) (Sigma). Activation of Plg was initiated by the addition of 0.55 nm t-PA (American Diagnostica). Hydrolysis of VLK-pNA by plasmin was measured as the change in absorbance at 405 nm over time using a microplate reader. Plg activation was determined as the increment in Vo (mol of Pm/min) using the following equation: Vo = b(1 + Km/So)/ε kcat.
Western blotting.
Proteins were subjected to SDS-PAGE on 8% gels under reducing conditions, transferred to nitrocellulose (GE Healthcare), and incubated with a rabbit polyclonal antibody against murine LN-1 (Sigma). The membrane was incubated with an anti-rabbit antibody–HRP conjugate, developed using an ECL substrate (Pierce), and subjected to autoradiography using Kodak Biomax MR Film (Thermo Fisher Scientific).
Statistics.
Data are presented as means ± SEM. Results were analyzed by ANOVA followed by Student-Newman-Keuls post hoc tests for multiple comparisons.
Results
Plasminogen enhances neurite outgrowth in PC-12 cells and primary cortical neurons
A series of studies has supported a requirement for an intact Plg activation system in neurite outgrowth induced by NGF (Pittman and DiBenedetto, 1995; Farias-Eisner et al., 2001; Jacovina et al., 2001). We have recently shown that Plg gene expression is induced by NGF in PC-12 cells (Gutierrez-Fernandez et al., 2007). Therefore, we examined the role of plasmin in NGF-dependent neurite outgrowth. PC-12 cells were grown on LN-1 and treated with NGF in the presence or absence of the plasmin inhibitor α2-antiplasmin, an anticatalytic mAb against the serine protease domain of Plg (mAb 2D1) (Kenagy et al., 1996); ε-aminocaproic acid (EACA), which blocks the interaction of Plg with cells (Miles et al., 2005); or mAb51, an anti-Plg antibody that inhibits binding of Plg to cellular receptors (Pozzi et al., 2000; Tarui et al., 2001). The effects of these reagents on neuritogenesis and neurite outgrowth were evaluated (Fig. 1). Treatment with either α2-antiplasmin or mAb 2D1 resulted in a significant reduction of neurite-positive cells (90 and 78%, respectively, after 48 h of treatment) compared with control cells treated with an isotype control (Fig. 1). Neurite length was also reduced by these inhibitors (data not shown). These data support a requirement for plasmin activity for optimal neurite outgrowth. These data also suggest that Plg is synthesized by the cells and activated to plasmin, consistent with the synthesis of Plg and t-PA by the adrenal gland and by PC-12 cells (Kristensen et al., 1986; Parmer et al., 1997; Zhang et al., 2002; Gutierrez-Fernandez et al., 2007). In addition, treatment with reagents that inhibit binding of Plg/plasmin to cells, including mAb51 or EACA, also reduced NGF-induced neuritogenesis by 38 and 71%, respectively, consistent with a requirement for the interaction of plasmin with the cell surface and/or the ECM. [The percentage of inhibition by MAb51 is consistent with its effect in our previous publication (Pozzi et al., 1998).] Similar results were obtained with rat primary cortical neurons in which EACA treatment reduced the percentage of neurite-positive cells from 62 + 2% to 28 + 7%.
Figure 1.

Plasmin participates in NGF-induced neuritogenesis. PC-12 cells were cultured in serum-free medium on LN-1-coated plates for 12 h and stimulated with NGF in the absence or presence of α2-antiplasmin (α2-AP; 25 nm), EACA (100 mM), mAb51, mAb 2D1, or isotype control (40 μg/ml), and neurite-positive cells were determined after 24 h (filled bars) or 48 h (open bars) as described in Materials and Methods. Data represent the mean of three independent experiments ± SEM (*p < 0.05; **p < 0.01).
To further explore the role of Plg/plasmin in neurite outgrowth, we examined the effect of the addition of exogenous Plg on neurite outgrowth in both PC-12 cells and primary rat cortical cells. [We have previously compared Plg mRNA levels in murine liver and adrenal gland and found that, although adrenal has the highest level of extrahepatic Plg mRNA, this level is only 1.3% of the level of hepatic mRNA and Plg mRNA levels in brain are 0.03% of hepatic levels (Zhang et al., 2002). Thus, we have not been able to detect Plg by ELISA in these cells and so cannot directly compare the levels of exogenous and endogenous Plg. By extrapolation, we added exogenous Plg at 25 nm (12.5% of the plasma Plg concentration) (Collen and Verstraete, 1975), so the ratio of exogenous Plg to endogenous Plg is in the range of 10:1.]
PC-12 cells were plated on wells coated with LN-1, cultured in either the presence or absence of 25 nm Plg, and examined for neurite outgrowth, induced by the addition of NGF. Plg enhanced neurite outgrowth, resulting in a twofold increase in neurite-positive cells (Fig. 2A) and neurite length (Fig. 2B). In the absence of NGF, Plg did not stimulate neurite outgrowth (data not shown). Plg treatment also enhanced neuritogenesis of primary rat cortical neurons. The percentage of neurite-positive cells (Fig. 2C,E) and neurite length (Fig. 2D,E) were increased 1.8-fold and 2.1-fold, respectively, by the addition of Plg. Together, these results suggest that Plg/plasmin enhances neurite outgrowth and the proteolytic activity of plasmin is required for this process.
Figure 2.

Plg enhances neurite outgrowth. A, B, PC-12 cells were cultured in serum-free medium on LN-1-coated plates for 12 h before stimulation with NGF. C–E, Cortical cells were cultured on poly-l-lysine-coated coverslips in serum-free B27 medium in the presence (■) or absence (□) of Plg (25 nm). Neurite-positive cells (A, C) were counted as described in Materials and Methods, and neurite length (B, D) was measured every 24 h for the duration of the experiment from neurites that did not contact other neurites or other cell bodies. E, Representative phase-contrast micrographs of cortical cells incubated with Plg (25 nm). Images were taken using an inverted microscope at 40× magnification. All statistical analyses were performed using the one-way ANOVA (*p < 0.05). Data represent the mean of three independent experiments ± SEM.
Structural requirements for Plg-dependent enhancement of neurite outgrowth
The lysine analog EACA blocks the interactions of Plg with macromolecular cofactors and inhibitors, as well as cell-surface receptors, by interacting with lysine binding sites contained within the kringle structures of Plg (Menhart et al., 1995; Castellino and McCance, 1997; Miles et al., 2005). Therefore, the specific Plg kringles participating in the enhancement in NGF-dependent neurite outgrowth were examined. Plg variants, in which the lysine binding function of kringles 1, 2, 4, and 5 were inactivated individually, were studied: [D(139)N]Plg, [D(218)N]Plg, [D(413)N]Plg, and [D(518)N]Plg (McCance and Castellino, 1995; Menhart et al., 1995). [Kringle 3 does not exhibit significant lysine binding ability (Menhart et al., 1995).] Each of these Plg mutants, wild-type rPlg, or buffer was added, separately, to PC-12 cells grown on LN-1 in the presence of NGF. After 48 h, neurite outgrowth was compared with that observed in the presence of plasma-derived Plg (Fig. 3). Similar to the results in Fig. 2, the number of neurite-positive cells was increased in the presence of either plasma-derived Plg (1.8-fold) or rPlg (1.7-fold) compared with cells treated with NGF without the addition of Plg (Fig. 3A,B). The addition of either the Plg mutants, [D(139)N]Plg, [D(218)N]Plg, or [D(413)N]Plg resulted in an increase in the number of neurite-positive cells similar to that observed with rPlg (Fig. 3A,B), suggesting that a functional lysine binding site in either kringle 1, 2, or 4 is not required for enhancement of neurite outgrowth. In contrast, the Plg mutant ([D(518)N]Plg) exhibited only 15% of the effect of rPlg on the percentage of neurite-positive cells. Similarly, when the cells were treated with NGF plus the mutants [D(139)N]Plg, [D(218)N]Plg, or [D(413)N]Plg, neurite length was increased and was not significantly different than in the presence of rPlg (Fig. 3A,C). However, in the presence of [D(518)N]Plg, neurite length was increased by only 17% compared with rPlg (Fig. 3A,C). These data support a requirement for the lysine binding site within Plg kringle 5 in the ability of Plg to enhance neurite outgrowth.
Figure 3.

Identification of Plg domains responsible for neurite outgrowth. A, Representative phase-contrast micrographs of NGF-treated PC-12 cells stimulated with 25 nm of the indicated Plg variants. Images were taken using an inverted microscope at 10× magnification. B, C, The percentage of neurite-positive cells (B) and the average length of NGF-stimulated cells (C) in the presence of each Plg variant were determined after 48 h of treatment as described in Materials and Methods. Data represent the mean of three independent experiments ± SEM (*p < 0.05) compared with cells incubated without Plg.
The data of Fig. 1, in which anticatalytic anti-Plg mAb 2D1 blocked neurite outgrowth in response to NGF, led to further investigation of the requirement for the plasmin active site in neurite outgrowth. Accordingly, we used a Plg variant in which the Asp residue of the catalytic triad of the serine protease domain was replaced by a Glu residue to generate [D(646)E]Plg. This variant can be cleaved by Plg activators to the two-chain plasmin form but does not exhibit detectable proteolytic activity (Grella and Castellino, 1997). When PC-12 cells were grown on LN-1 and stimulated with NGF in the presence of [D(646)E]Plg, the percentage of neurite-positive cells and neurite length were not significantly increased compared with cells grown in medium alone. These results suggest that the proteolytic activity of plasmin is required for enhancement of neurite outgrowth by Plg, consistent with activation by PC-12-derived t-PA (Parmer et al., 1997).
LN-1 is proteolytically cleaved by plasmin
The ability of plasmin to cleave laminins within the ECM (Chen and Strickland, 1997; Indyk et al., 2003) prompted us to investigate whether LN-1 degradation occurred during NGF-induced neurite outgrowth and whether this cleavage was plasmin dependent. PC-12 cells were grown on LN-1 and either untreated or treated with 100 ng/ml NGF in the presence or absence of α2-antiplasmin or t-PA. To investigate the status of LN-1, the cells were detached, and the ECM was harvested, subjected to electrophoresis and Western blotting with polyclonal anti-murine LN-1. LN-1 degradation was observed in ECM harvested from cells treated with NGF, as shown by the decreased intensity of both the α1 chain (migrating with a Mrapp of 440 kDa) and the β1γ1 chain (migrating with a Mrapp of 220 kDa) compared with ECM harvested from cells cultured in the absence of NGF (Fig. 4A). In addition, LN-1 proteolytic fragments in the Mrapp range of 48–140 kDa were detected in the culture medium of NGF-treated cells, but not untreated cells (Fig. 4B). Furthermore, the NGF-dependent LN-1 degradation was consistent with a plasmin-dependent mechanism because α2-antiplasmin reduced the extent of degradation by 68%, compared with ECM harvested from cells treated with NGF alone (Fig. 4A), and also reduced the presence of LN-1 degradation fragments in the conditioned medium (Fig. 4B). The addition of t-PA resulted in enhanced degradation of LN-1 by 160% compared with the ECM harvested from cells treated with NGF alone (Fig. 4A) and also increased the intensity of LN-1 degradation fragments in the conditioned medium (Fig. 4B), consistent with a role for cellular plasmin in degradation of LN-1. These data suggest that LN-1 degradation is increased during NGF-induced neuritogenesis and that the degradation is plasmin dependent.
Figure 4.

LN-1 is cleaved during neuritogenesis. PC-12 cells (1 × 106) were plated on 24-well plates coated with LN-1 (1 μg/ml) in serum-free medium in the absence (buffer) or presence of 100 ng/ml NGF and in the absence or presence of either α2-antiplasmin (α2-AP; 25 nm) or t-PA (10 nm), and cultured for 20 h at 37°C. Conditioned media (B) was harvested, and the ECM (A, C) was collected after detaching the cells by incubation with 5 mm EDTA. Samples were subjected to Western blotting using a polyclonal antibody against LN-1 (A, B) or stained with Coomassie Blue as a loading control (C). Arrowheads denote the α1 chain (migrating with a Mrapp of 440 kDa) and the β1γ1 chain (migrating with a Mrapp of 220 kDa) of LN-1. The relative intensities of the two major chains of LN-1 were determined relative to the Coomassie Blue-stained gel and normalized to the results of cells treated with buffer alone. Representative gels of experiments performed at least three times each are shown.
Plg binding to LN-1 enhances activation by t-PA
A characteristic feature of plasmin substrates, including fibrin and laminin-5, is their ability to interact with Plg and promote its activation by Plg (Goldfinger et al., 2000; Waisman, 2003). Whether Plg could also bind LN-1 and facilitate its activation to plasmin was tested. Microtiter plates coated with LN-1 or BSA were incubated in the presence or absence of rPlg or plasma Plg, and bound protein was detected by ELISA using a mAb against human Plg. Plg binding to LN-1 was detected, whereas no significant binding to BSA was observed (Fig. 5A). Furthermore, this interaction was specific because it was inhibited by 66 ± 7% (n = 3) in the presence of 100 mm EACA. This suggests that the interaction of Plg with LN-1 requires an interaction with the lysine binding sites in Plg. In contrast, the ability of [D(518)N]Plg to bind to LN-1 was decreased by 70% compared with rPlg or plasma Plg, suggesting that an intact lysine binding site in the kringle 5 domain of Plg is required for binding to LN-1 (Fig. 5A), as well as for stimulation of neuritogenesis (Fig. 3).
Figure 5.
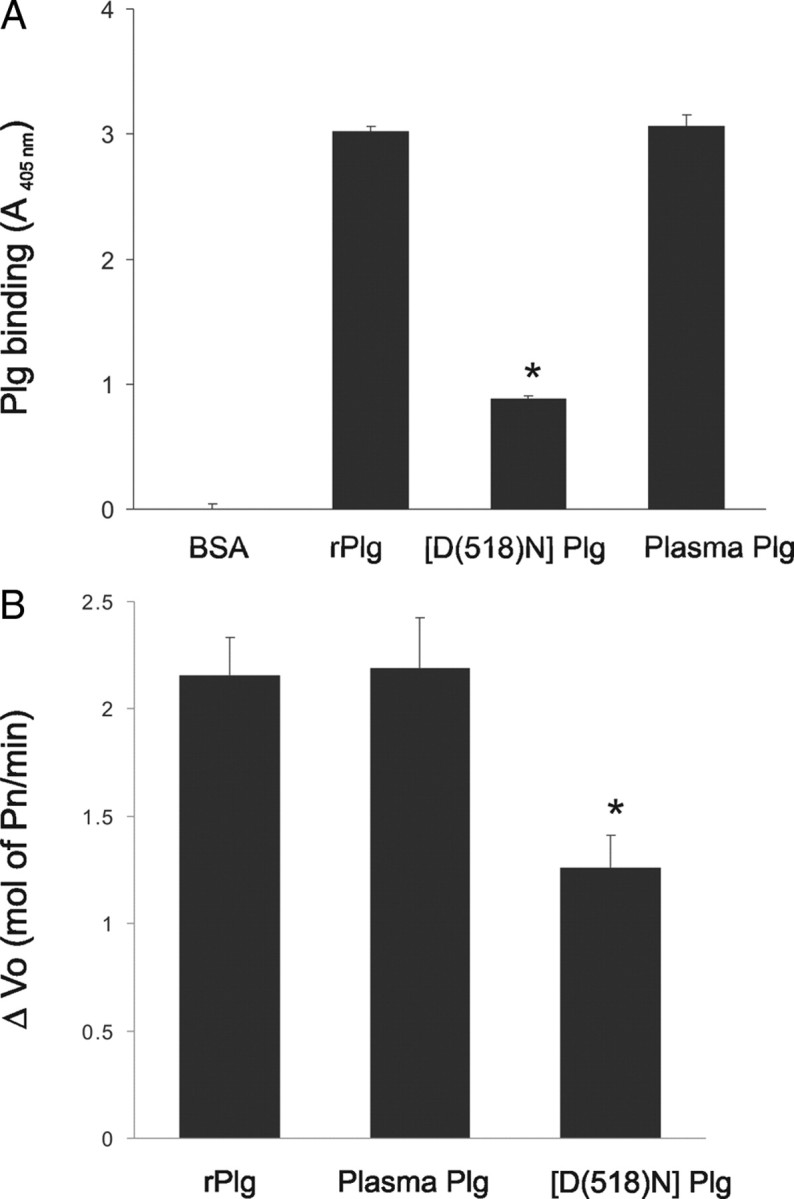
Plasminogen binding to LN-1 enhances its activation by t-PA. A, Ninety-six-well plates coated with LN-1 or BSA were incubated with rPlg, plasma Plg, or [D(518)N]Plg for 1 h at 37°C, and bound Plg was detected by ELISA as described in Materials and Methods. B, The effect of Plg binding to LN-1 on Plg activation was tested as described in Materials and Methods. Plg activation by t-PA when bound to LN-1 was determined as hydrolysis of the chromogenic substrate VLK-pNA after the addition of t-PA and expressed as the increase in Vo (mol of Pm/min) compared with the rate of activation of soluble plasminogen in plates coated with BSA. Data shown are the mean of three independent experiments ± SEM (*p < 0.05).
To determine whether Plg binding to LN-1 enhanced its activation by t-PA, microtiter plates coated with either LN-1 or BSA were incubated with rPlg or plasma for 30 min to allow binding, and then t-PA was added. The activation rate of rPlg or plasma Plg bound to LN-1 was fourfold greater than the activation rate of soluble Plg (incubated with plates coated with BSA; data not shown). In contrast, the activation rate of [D(518)N]Plg bound to LN-1 was only 50% of the activation rates of rPlg and plasma Plg (Fig. 5B). These results demonstrate that Plg binding to LN enhances its activation and that a functional kringle 5 domain within the Plg molecule is required for the enhanced activation. No significant differences where observed in the activation rates of soluble rPlg, plasma Plg, and soluble [D(518)N]Plg. Together, these results suggest that the neurite outgrowth promoting capacity of Plg is dependent on its direct effect on laminin degradation.
Plg binding to LN-1 enhances neuritogenesis
The above data suggested that plasmin activity was required for neurite outgrowth and that the interaction of Plg with LN-1 promoted Plg activation. Therefore, to examine whether Plg binding to LN-1 could influence NGF-induced neurite outgrowth, LN-1-coated plates were either preincubated with 25 nm Plg (Fig. 6, open bars) or buffer (Fig. 6, filled bars) for 1 h to allow binding of Plg to LN-1 before the addition of the cells. Next, PC-12 cells were added simultaneously with soluble Plg(+) or buffer(−), at the initiation of culture. NGF was added to the culture medium, and neurite outgrowth was quantified at 16 h after plating. Pretreatment of LN-1 with Plg enhanced both the percentage of neurite-positive cells and neurite length compared with cells that had been exposed to Plg at the initiation of the cultures (Fig. 6, compare open and filled bars).
Figure 6.

Neurite outgrowth enhancement by plasminogen binding to LN-1. LN-1-coated plates were preincubated in the presence of Plg 25 nm (open bars) or buffer (filled bars) at 37°C for 1 h before the addition of the cells. Then PC-12 cells were added simultaneously at the initiation of the culture with soluble Plg(+) or buffer(−). NGF was then added, and percentage of neurite-positive cells and neurite length were quantified at 16 h. Neurite-positive cells (A) and neurite length (B) were quantified as described in Materials and Methods. Data shown are the mean of three independent experiments ± SEM (*p < 0.05; **p < 0.01).
Discussion
The importance of proteolysis in the nervous system has been demonstrated by the identification of numerous neurological diseases caused by alterations in the activity of specific proteases, including neurotrypsin in mental retardation; UCHL-1 in Parkinson's disease; and α-, β-, and γ-secretases in Alzheimer's disease (Esler and Wolfe, 2001; Liu et al., 2002; Molinari et al., 2002; Vassar, 2002). However, the physiological role for most proteases expressed in the nervous system remains primarily unknown. A series of previous studies has documented a requirement for Plg activators in neurite outgrowth induced by NGF (Pittman and DiBenedetto, 1995; Farias-Eisner et al., 2001). In the current study, we investigated the function of plasmin in these processes. We found that (1) Plg enhanced neurite outgrowth and neuritogenesis; (2) the proteolytic activity of plasmin and the lysine binding sites in the kringle 5 domain of Plg were necessary for the enhancement; (3) LN-1 was cleaved during neurite outgrowth; and (4) Plg bound to LN-1, resulting in increased Plg activation and the enhancement of neurite outgrowth and neuritogenesis. As a whole, these data suggest a major mechanism by which proteolysis of a major component of the neuronal ECM promotes neurite outgrowth and neuritogenesis.
Although the fibrinolytic system has been classically associated with vascular processes, recent reports have shown that some of its proteins participate in neurological functions (Hayden and Seeds, 1996; Drew et al., 1998; Schott et al., 1998; Seeds et al., 1999; Akassoglou et al., 2000; Hoover-Plow et al., 2001; Siconolfi and Seeds, 2001b). The presence of the hematoencephalic barrier prevents the diffusion of hepatic-derived Plg into neural tissues. However, recent reports have demonstrated that proteins of the fibrinolytic system (i.e., Plg and t-PA) are broadly expressed throughout the neuroendocrine system (Kristensen et al., 1986; Parmer et al., 1997; Zhang et al., 2002; Gutierrez-Fernandez et al., 2003). In fact, our studies have shown that NGF upregulates the expression of the Plg gene (Gutierrez-Fernandez et al., 2007). In the current study, we found that blockade of the proteolytic activity of plasmin delayed NGF-dependent neuritogenesis and neurite outgrowth.
These data are consistent with a requirement for cellular Plg during neuritogenesis and suggest that Plg, activated by t-PA, is necessary for this process. We suggest that the direct participation of Plg in neuritogenesis contributes to the neurological abnormalities associated with defects in this protein. In this regard, it has been shown that patients with severe type I Plg deficiency exhibit neuropathologies (Schott et al., 1998). These syndromes are consistent with defects in neuronal growth and differentiation that are modulated by proteolysis, and these data support a major role for proteins of the fibrinolytic system in neuritogenesis.
The activity of Plg during neuritogenesis not only depends on its proteolytic activity but also requires additional interactions with proteins. In fact, as shown here, the lysine-binding properties of Plg are necessary for neuritogenic activity.
We found that binding of Plg to LN-1 enhanced Plg activation and neurite outgrowth, and this binding, enhancement of Plg activation, and enhancement of neurite outgrowth were dependent on a functional kringle 5 domain. Furthermore, LN-1 was found to be a plasmin substrate that was cleaved during NGF-induced neurite outgrowth. Furthermore, inhibition of plasmin activity inhibited neurite outgrowth as well as LN-1 degradation. Our results indicate that LN-1 is a plasmin substrate and also suggest that LN-1 degradation by plasmin is a major mechanism facilitating neurite outgrowth. Our studies suggest that binding to LN-1 may provide a general mechanism to achieve Plg activation in neuronal tissues, similar to the activation of fibrin-bound Plg during fibrinolysis. This interaction would not only contribute to the spatial localization of Plg, but plasmin generated at the cell surface would be available to cleave LN-1 or other ECM substrates, thus facilitating neurite outgrowth. Thus, during neural development and neuritogenesis, plasmin activity is likely to be required to cleave LN-1 and may activate other proteinases, facilitating migration and perhaps promoting different signaling events by exposing cryptic sites or liberating proteolytic fragments with biological activity. Cleavage of LN-5 by MMP-2 has been shown to expose a cryptic site that induces migration of epithelial cells (Giannelli et al., 1997), demonstrating that processing of laminins by specific proteases may contribute to processes requiring cell migration. Interestingly, although LN-1 (α1β1γ1) is one of the major laminins in neural tissues, other components of this family, including LN-2 (α2β1γ1) and LN-10 (α5β1γ1), are also expressed in neural tissues and have been associated with neurite outgrowth (Vuolteenaho et al., 1994; Powell et al., 1998; Colognato and Yurchenco, 2000; Fried et al., 2005), suggesting that cleavage of these proteins by plasmin could also contribute to neuritogenesis.
The results presented here suggest that the Plg activation system upregulates neuritogenesis. Consistent with our findings, several studies have shown that Plg and t-PA protect against axonal degeneration after sciatic nerve injury in the peripheral nervous system by cleaving fibrin (Akassoglou et al., 2000). However, the role of the fibrinolytic system in degradation of LN-1 in nervous tissue may be multifaceted. Plasmin-catalyzed degradation of LN-1 or LN-10 in the hippocampus mediates excitotoxic-induced neuronal cell anoikis attributable to loss of cell attachment (Chen and Strickland, 1997; Indyk et al., 2003). Excitotoxic glutamate agonists stimulate synthesis of Plg that is then activated to plasmin, contributing to neuronal cell death (Chen and Strickland, 1997). Thus, Plg expression and activation within neuronal tissues must be strictly regulated because failure to control this balance can have pathologic consequences.
In summary, our studies suggest that proteolysis by plasmin regulates neurite outgrowth by cleavage of LN-1. The interaction of this protease with LN-1 may represent a general mechanism by which neuronal Plg can be efficiently activated to facilitate ECM remodeling and neurite outgrowth, suggesting a previously unrecognized role of the fibrinolytic system in neurological functions.
Footnotes
This work was supported by National Institutes of Health Grants HL045934 and HL081046 (L.A.M.), HL050398 (R.J.P.), and HL013423 (F.J.C.) and by the Department of Veterans Affairs (R.J.P.). We thank Dr. X. S. Puente for helpful revisions and comments and N. Baik for excellent technical assistance. This is publication number 19764 from The Scripps Research Institute.
References
- Akassoglou K, Kombrinck KW, Degen JL, Strickland S. Tissue plasminogen activator-mediated fibrinolysis protects against axonal degeneration and demyelination after sciatic nerve injury. J Cell Biol. 2000;149:1157–1166. doi: 10.1083/jcb.149.5.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basham ME, Seeds NW. Plasminogen expression in the neonatal and adult mouse brain. J Neurochem. 2001;77:318–325. doi: 10.1046/j.1471-4159.2001.t01-1-00239.x. [DOI] [PubMed] [Google Scholar]
- Britto JM, Lukehurst S, Weller R, Fraser C, Qiu Y, Hertzog P, Busfield SJ. Generation and characterization of neuregulin-2-deficient mice. Mol Cell Biol. 2004;24:8221–8226. doi: 10.1128/MCB.24.18.8221-8226.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellino FJ, McCance SG. The kringle domains of human plasminogen. Ciba Found Symp. 1997;212:46–60. doi: 10.1002/9780470515457.ch4. [DOI] [PubMed] [Google Scholar]
- Chen LW, Strickland S. Neuronal death in the hippocampus is promoted byplasmin-catalyzed degradation of laminin. Cell. 1997;91:917–925. doi: 10.1016/s0092-8674(00)80483-3. [DOI] [PubMed] [Google Scholar]
- Collen D, Verstraete M. Molecular biology of human plasminogen. II. Metabolism in physiological and some pathophysiological conditions in man. Thromb Diath Haemorrh Suppl. 1975;34:403–408. [PubMed] [Google Scholar]
- Colognato H, Yurchenco PD. Form and function: the laminin family of heterotrimers. Dev Dyn. 2000;218:213–234. doi: 10.1002/(SICI)1097-0177(200006)218:2<213::AID-DVDY1>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Deutsch DG, Mertz ET. Plasminogen: purification from human plasma by affinity chromatography. Science. 1970;170:1995–1996. doi: 10.1126/science.170.3962.1095. [DOI] [PubMed] [Google Scholar]
- Drew AF, Kaufman AH, Kombrinck KW, Danton MJS, Daugherty CC, Degen JL, Bugge TH. Ligneous conjunctivitis in plasminogen-deficient mice. Blood. 1998;91:1616–1624. [PubMed] [Google Scholar]
- Esler WP, Wolfe MS. A portrait of Alzheimer secretases–new features and familiar faces. Science. 2001;293:1449–1454. doi: 10.1126/science.1064638. [DOI] [PubMed] [Google Scholar]
- Farias-Eisner R, Vician L, Silver A, Reddy S, Rabbani SA, Herschman HR. The urokinase plasminogen activator receptor (UPAR) is preferentially induced by nerve growth factor in PC12 pheochromocytoma cells and is required for NGF-driven differentiation. J Neurosci. 2000;20:230–239. doi: 10.1523/JNEUROSCI.20-01-00230.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farias-Eisner R, Vician L, Reddy S, Basconcillo R, Rabbani SA, Wu YY, Bradshaw RA, Herschman HR. Expression of the urokinase plasminogen activator receptor is transiently required during “priming” of PC12 cells in nerve growth factor-directed cellular differentiation. J Neurosci Res. 2001;63:341–346. doi: 10.1002/1097-4547(20010215)63:4<341::AID-JNR1028>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Fried K, Sime W, Lillesaar C, Virtanen I, Tryggvasson K, Patarroyo M. Laminins 2 (alpha2beta1gamma1, Lm-211) and 8 (alpha4beta1gamma1, Lm-411) are synthesized and secreted by tooth pulp fibroblasts and differentially promote neurite outgrowth from trigeminal ganglion sensory neurons. Exp Cell Res. 2005;307:329–341. doi: 10.1016/j.yexcr.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Giannelli G, Falk-Marzillier J, Schiraldi O, Stetler-Stevenson WG, Quaranta V. Induction of cell migration by matrix metalloprotease-2 cleavage of laminin-5. Science. 1997;277:225–228. doi: 10.1126/science.277.5323.225. [DOI] [PubMed] [Google Scholar]
- Goldfinger LE, Jiang L, Hopkinson SB, Stack MS, Jones JC. Spatial regulation and activity modulation of plasmin by high affinity binding to the G domain of the alpha 3 subunit of laminin-5. J Biol Chem. 2000;275:34887–34893. doi: 10.1074/jbc.M006652200. [DOI] [PubMed] [Google Scholar]
- Greene LA, Tischler AS. Establishment of a noradrenergic clonal cell line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci U S A. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grella DK, Castellino FJ. Activation of human plasminogen by staphylokinase. Direct evidence that preformed plasmin is necessary for activation to occur. Blood. 1997;89:1585–1589. [PubMed] [Google Scholar]
- Guan KL, Rao Y. Signalling mechanisms mediating neuronal responses to guidance cues. Nat Rev Neurosci. 2003;4:941–956. doi: 10.1038/nrn1254. [DOI] [PubMed] [Google Scholar]
- Gutierrez-Fernandez A, Gingles NA, Zhang L, Garcia Bannach F, Jenkins GR, Loskutoff DJ, Parmer RJ, Miles LA. Regulation of plasminogen gene expression. In: Waisman DM, editor. Plasminogen regulation. New York: Academic/Plenum Publishers; 2003. pp. 67–80. [Google Scholar]
- Gutierrez-Fernandez A, Parmer RJ, Miles LA. Plasminogen gene expression is regulated by nerve growth factor. J Thromb Haemost. 2007;5:1715–1725. doi: 10.1111/j.1538-7836.2007.02636.x. [DOI] [PubMed] [Google Scholar]
- Hayden SM, Seeds NW. Modulated expression of plasminogen activator system components in cultured cells from dissociated mouse dorsal root ganglia. J Neurosci. 1996;16:2307–2317. doi: 10.1523/JNEUROSCI.16-07-02307.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover-Plow J, Skomorovska-Prokvolit O, Welsh S. Selective behaviors altered in plasminogen-deficient mice are reconstituted with intracerebroventricular injection of plasminogen. Brain Res. 2001b;898:256–264. doi: 10.1016/s0006-8993(01)02191-6. [DOI] [PubMed] [Google Scholar]
- Indyk JA, Chen ZL, Tsirka SE, Strickland S. Laminin chain expression suggests that laminin-10 is a major isoform in the mouse hippocampus and is degraded by the tissue plasminogen activator/plasmin protease cascade during excitotoxic injury. Neuroscience. 2003;116:359–371. doi: 10.1016/s0306-4522(02)00704-2. [DOI] [PubMed] [Google Scholar]
- Jacovina AT, Zhong F, Khazanova E, Lev E, Deora AB, Hajjar KA. Neuritogenesis and the nerve growth factor-induced differentiation of PC-12 cells requires annexin II-mediated plasmin generation. J Biol Chem. 2001;276:49350–49358. doi: 10.1074/jbc.M106289200. [DOI] [PubMed] [Google Scholar]
- Kenagy RD, Vergel S, Mattsson E, Bendeck M, Reidy MA, Clowes AW. The role of plasminogen, plasminogen activators, and matrix metalloproteinases in primate arterial smooth muscle cell migration. Arterioscler Thromb Vasc Biol. 1996;16:1373–1382. doi: 10.1161/01.atv.16.11.1373. [DOI] [PubMed] [Google Scholar]
- Kiryushko D, Berezin V, Bock E. Regulators of neurite outgrowth: role of cell adhesion molecules. Ann N Y Acad Sci. 2004;1014:140–154. doi: 10.1196/annals.1294.015. [DOI] [PubMed] [Google Scholar]
- Kristensen P, Hougaard DM, Nielsen LS, Dano K. Tissue-type plasminogen activator in rat adrenal medulla. Histochemistry. 1986;85:431–436. doi: 10.1007/BF00982674. [DOI] [PubMed] [Google Scholar]
- Lander AD, Fujii DK, Reichardt LF. Laminin is associated with the “neurite outgrowth-promoting factors” found in conditioned media. Proc Natl Acad Sci U S A. 1985;82:2183–2187. doi: 10.1073/pnas.82.7.2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Fallon L, Lashuel HA, Liu Z, Lansbury PT., Jr The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson's disease susceptibility. Cell. 2002;111:209–218. doi: 10.1016/s0092-8674(02)01012-7. [DOI] [PubMed] [Google Scholar]
- McCance SG, Castellino FJ. Contributions of individual kringle domains toward maintenance of the chloride-induced tight conformation of human glutamic acid-1 plasminogen. Biochemistry. 1995;34:9581–9586. doi: 10.1021/bi00029a035. [DOI] [PubMed] [Google Scholar]
- Menhart N, Hoover GJ, McCance SG, Castellino FJ. Roles of individual kringle domains in the functioning of positive and negative effectors of human plasminogen activation. Biochemistry. 1995;34:1482–1488. doi: 10.1021/bi00005a003. [DOI] [PubMed] [Google Scholar]
- Miles LA, Dahlberg CM, Plow EF. The cell-binding domains of plasminogen and their function in plasma. J Biol Chem. 1988;263:11928–11934. [PubMed] [Google Scholar]
- Miles LA, Hawley SB, Baik N, Andronicos NM, Castellino FJ, Parmer RJ. Plasminogen receptors: the sine qua non of cell surface plasminogen activation. Front Biosci. 2005;10:1754–1762. doi: 10.2741/1658. [DOI] [PubMed] [Google Scholar]
- Molinari F, Rio M, Meskenaite V, Encha-Razavi F, Auge J, Bacq D, Briault S, Vekemans M, Munnich A, Attie-Bitach T, Sonderegger P, Colleaux L. Truncating neurotrypsin mutation in autosomal recessive nonsyndromic mental retardation. Science. 2002;298:1779–1781. doi: 10.1126/science.1076521. [DOI] [PubMed] [Google Scholar]
- Parmer RJ, Mahata M, Mahata S, Sebald MT, O'Connor DT, Miles LA. Tissue plasminogen activator (t-PA) is targeted to the regulated secretory pathway: catecholamine storage vesicles as a reservoir for the rapid release of t-PA. J Biol Chem. 1997;272:1976–1982. doi: 10.1074/jbc.272.3.1976. [DOI] [PubMed] [Google Scholar]
- Pietri T, Eder O, Breau MA, Topilko P, Blanche M, Brakebusch C, Fassler R, Thiery JP, Dufour S. Conditional beta1-integrin gene deletion in neural crest cells causes severe developmental alterations of the peripheral nervous system. Development. 2004;131:3871–3883. doi: 10.1242/dev.01264. [DOI] [PubMed] [Google Scholar]
- Pittman RN, DiBenedetto AJ. PC12 cells overexpressing tissue plasminogen activator regenerate neurites to a greater extent and migrate faster than control cells in complex extracellular matrix. J Neurochem. 1995;64:566–575. doi: 10.1046/j.1471-4159.1995.64020566.x. [DOI] [PubMed] [Google Scholar]
- Powell SK, Williams CC, Nomizu M, Yamada Y, Kleinman HK. Laminin-like proteins are differentially regulated during cerebellar development and stimulate granule cell neurite outgrowth in vitro. J Neurosci Res. 1998;54:233–247. doi: 10.1002/(SICI)1097-4547(19981015)54:2<233::AID-JNR11>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Pozzi A, Wary KK, Giancotti FG, Gardner HA. Integrin alpha1beta1 mediates a unique collagen-dependent proliferation pathway in vivo. J Cell Biol. 1998;142:587–594. doi: 10.1083/jcb.142.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzi A, Moberg PE, Miles LA, Wagner S, Soloway P, Gardner HA. Elevated matrix metalloprotease and angiostatin levels in integrin alpha 1 knockout mice cause reduced tumor vascularization. Proc Natl Acad Sci U S A. 2000;97:2202–2207. doi: 10.1073/pnas.040378497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z, Gilbert ME, Colicos MA, Kandel ER, Kuhl D. Tissue-type plasminogen activator is induced as an immediate-early gene during seizure, kindling and long-term potentiation. Nature. 1993;361:453–457. doi: 10.1038/361453a0. [DOI] [PubMed] [Google Scholar]
- Sappino AP, Madani R, huarte J, Belin D, Kiss JZ, Wohlwend A, Vassalli JD. Extracellular proteolysis in the adult murine brain. J Clin Invest. 1993;92:679–685. doi: 10.1172/JCI116637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schott D, Dempfle CE, Beck P, Liermann A, Mohr-Pennert A, Goldner M, Mehlem P, Azuma H, Schuster V, Mingers AM, Schwarz HP, Kramer MD. Therapy with a purified plasminogen concentrate in an infant with ligneous conjunctivitis and homozygous plasminogen deficiency. N Engl J Med. 1998;339:1679–1686. doi: 10.1056/NEJM199812033392305. [DOI] [PubMed] [Google Scholar]
- Seeds NW, Basham ME, Haffke SP. Neuronal migration is retarded in mice lacking the tissue plasminogen activator gene. Proc Natl Acad Sci U S A. 1999;96:14118–14123. doi: 10.1073/pnas.96.24.14118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siconolfi LB, Seeds NW. Induction of the plasminogen activator system accompanies peripheral nerve regeneration after sciatic nerve crush. J Neurosci. 2001a;21:4336–4347. doi: 10.1523/JNEUROSCI.21-12-04336.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siconolfi LB, Seeds NW. Mice lacking tPA, uPA, or plasminogen genes showed delayed functional recovery after sciatic nerve crush. J Neurosci. 2001b;21:4348–4355. doi: 10.1523/JNEUROSCI.21-12-04348.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Uno S, Watanabe Y, Arakawa K, Nakagawa S. Expression of nerve growth factor-induced type 1 plasminogen activator inhibitor (PAI-1) mRNA is inhibited by genistein and wortmannin. Neuroreport. 2000;11:1111–1115. doi: 10.1097/00001756-200004070-00040. [DOI] [PubMed] [Google Scholar]
- Tarui T, Miles LA, Takada Y. Specific interaction of angiostatin with integrin alpha vbeta 3 in endothelial cells. J Biol Chem. 2001;276:39562–39568. doi: 10.1074/jbc.M101815200. [DOI] [PubMed] [Google Scholar]
- Tran PB, Miller RJ. Chemokine receptors: signposts to brain development and disease. Nat Rev Neurosci. 2003;4:444–455. doi: 10.1038/nrn1116. [DOI] [PubMed] [Google Scholar]
- Tsirka SE, Bugge TH, Degen JL, Strickland S. Neuronal death in the central nervous system demonstrates a non-fibrin substrate for plasmin. Proc Natl Acad Sci U S A. 1997;94:9779–9781. doi: 10.1073/pnas.94.18.9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R. Beta-secretase (BACE) as a drug target for Alzheimer's disease. Adv Drug Deliv Rev. 2002;54:1589–1602. doi: 10.1016/s0169-409x(02)00157-6. [DOI] [PubMed] [Google Scholar]
- Vuolteenaho R, Nissinen M, Sainio K, Byers M, Eddy R, Hirvonen H, Shows TB, Sariola H, Engvall E, Tryggvason K. Human laminin M chain (merosin): complete primary structure, chromosomal assignment, and expression of the M and A chain in human fetal tissues. J Cell Biol. 1994;124:381–394. doi: 10.1083/jcb.124.3.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waisman DM. New York: Klewer Academic/Plenum Publishers; 2003. Plasminogen: structure, activation, and regulation. [Google Scholar]
- Zhang L, Seiffert D, Fowler B, Jenkins GR, Thinnes T, Loskutoff DJ, Parmer RJ, Miles LA. Plasminogen has a broad extrahepatic distribution. Thromb Haemost. 2002;87:493–501. [PubMed] [Google Scholar]