

Abstract
We describe an efficient technique for the selective chemical and biological manipulation of the contents of individual cells. This technique is based on the electric-field-induced permeabilization (electroporation) in biological membranes using a low-voltage pulse generator and microelectrodes. A spatially highly focused electric field allows introduction of polar cell-impermeant solutes such as fluorescent dyes, fluorogenic reagents, and DNA into single cells. The high spatial resolution of the technique allows for design of, for example, cellular network constructions in which cells in close contact with each other can be made to possess different biochemical, biophysical, and morphological properties. Fluorescein, and fluo-3 (a calcium-sensitive fluorophore), are electroporated into the soma of cultured single progenitor cells derived from adult rat hippocampus. Fluo-3 also is introduced into individual submicrometer diameter processes of thapsigargin-treated progenitor cells, and a plasmid vector cDNA construct (pRAY 1), expressing the green fluorescent protein, is electroporated into cultured single COS 7 cells. At high electric field strengths, observations of dye-transfer into organelles are proposed.
In the last decade, we have witnessed a tremendous growth in high-resolution techniques driven by the need to understand cellular biochemical and biophysical processes in ever greater detail. For example, patch clamp measurements of ion channel activities (1) have profoundly shaped our understanding of ion channel physiology and cellular communication. The use of carbon fiber ultramicroelectrodes for in vivo voltammetry have revealed exciting information on single transmitter-vesicle exocytotic release events (2, 3). Confocal microscopy and two-photon microscopy have provided striking images of the workings of cellular machinery, such as the dynamics of intracellular calcium ion and the localization of single serotonin-containing granulae in RBL cells (4, 5). The Abbe resolution limit even is bypassed in near-field spectroscopic probes, in which an optical resolution as high as 12 nm has been achieved (6). The manipulation of single organelles and even single biomolecules has been made possible by optical trapping, and this has since been applied to a wide range of interesting biological problems (7–10).
Although numerous high-resolution techniques exist to detect, image, and analyze the contents of single cells and subcellular organelles, few methods exist to control and manipulate the biochemical nature of these compartments. In this study, we demonstrate a high-resolution technique to alter the biochemical content of single cells and organelles in situ, based on permeabilization of phospholipid bilayer membranes by pulsed electric fields (electroporation). During the effective pore-open time, cell-impermeant solutes added to the extracellular medium can enter the cell interior by diffusion. In contrast to microinjection techniques for single cells and single nuclei (11), electric field-induced reagent transfer can be applied for biological containers of subfemtoliter (10−15 liters) volumes and can be extremely fast and precisely timed (12, 13), which is of importance in studying fast reaction phenomena.
The membrane voltage Vm, at different loci on phospholipid bilayer spheres during exposure in a homogenous electric field of duration t, can be calculated from
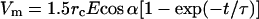 |
1 |
where E is the electric field strength, rc is the cell radius, α is the angle in relation to the direction of the electric field, and τ is the capacitive-resistive time constant. Pore-formation will result at spherical coordinates exposed to a maximal potential shift, which is at the poles facing the electrodes (cosα = 1 for α = 0; cos α = −1 for α = π). Generally, electric field strengths on the order of 1 to 1.5 kV/cm for durations of a few μs to a few ms are sufficient to cause transient permeabilization in 10-μm outer-diameter spherical cells (14–16). A recent study shows that isolated mitochondria, because of their correspondingly smaller size, require 7- to 10-fold higher electric field strengths to incorporate a 7.2-kilobase plasmid DNA (17). Mitochondrial outer-membrane fusion at lower electric field strengths of ≈2.5 kV/cm also has been observed (18).
Conventional electroporation using high-voltage pulse generators is made in a batch mode in relatively large containers, which typically permeabilize the membrane of millions of cells simultaneously (14–16, 19). Instrumentation that can be used for electroporation of a small number of cells in suspension (20, 21) and for a small number of adherent cells grown on a substratum (22, 23) also has been described. In the present work, electroporation with subcellular spatial resolution is accomplished by applying the electric field through carbon fiber ultramicroelectrodes (≈5 μm in diameter). In addition to the high spatial resolution achieved by using microelectrodes, this technique avoids the use of expensive high-voltage pulse generators and complicated microchamber mounts. The method can in principle be battery-operated because the spacing between the electrodes is small, typically 20 μm or less, which results in a high electric field strength with a small amplitude voltage pulse.
MATERIALS AND METHODS
Electroporation Instrumentation.
For electroporation experiments, cell dishes were mounted in a circular polycarbonate holder by using vacuum grease and were transferred to the stage of an inverted microscope (Leica DM IRB, Wetzlar, Germany) equipped with a Leica PL Fluotar 40× objective. A cell was selected, and two carbon fiber microelectrodes (ProCFE, Axon Instruments, Foster City, CA) with an outer diameter of 5 μm were positioned close at each side of the cell by high-graduation micromanipulators (Narishige MWH-3, Tokyo). The two carbon fiber electrode tips (anode and cathode) were positioned 2 to 5 μm from the boundaries of a cell soma or cellular process at an angle of 0–20° and 160–180° with respect to the object plane. The planar electrode arrangement, as schematically shown in Fig. 1, can be used because the electrode tips are flexible and sustain the mechanical stress of being forced against the coverslip. This configuration was, however, only used on sparse cultures of newly seeded cells essentially lacking processes. It is important to not place the electrodes in direct contact with the cell to prevent electrochemical transformation of proteins and other membrane-associated structures. Cells were permeabilized with multiple 1-ms pulses (except in the case of plasmid introduction in which single pulse protocols were used) by using a pulse generator (Digitimer Stimulator DS9A, Welwyn Garden City, U.K.). The voltage output from the pulse generator was calibrated by using high-impedance electrodes and a patch-clamp amplifier. Because the cells sometimes had complex geometries, the membrane voltages reported were obtained by multiplying the applied electric field strength with the average radius from the boundary of the anode-facing cell membrane to the boundary of the cathode-facing membrane at the center of the electrodes.
Figure 1.

Schematic picture showing the positioning of the electrodes for single-cell electroporation. (A) Before electroporation. The electrodes are positioned close to the cell with a distance of ≈2 to 5 μm from the cell surface. The buffer contains the solute (dots) to be introduced into the cytosol. (B) Electroporation by application of a rectangular low-voltage pulse. The applied electric field, highly focused over the selected cell, causes membrane-pore formation, allowing the solute in the extracellular solution to freely diffuse into the cell. (C) After electroporation, the pores are resealed, and the solute is trapped inside the cell. After exchange of the extracellular medium, solute molecules are present in the cell but not in the extracellular solution.
The electroporation buffer in the fluorescein experiments was a Hanks’–Hepes solution (137 mM NaCl/5.4 mM KCl/0.41 mM MgSO4/0.4 mM MgCl2/1.26 mM CaCl2/0.64 mM KH2PO4/3.0 mM NaHCO3/5.5 mM d-glucose/20 mM Hepes, pH adjusted to 7.4 with NaOH) supplemented with 5 to 10 μM fluorescein. A Ca2+-free Hanks’–Hepes solution containing 10 μM fluo-3 and 70 μM EDTA was used in the fluo-3 experiments. For cDNA transfection experiments, a Hepes buffer solution (140 mM NaCl/0.75% dimethyl sulfoxide/10 mM Hepes, pH adjusted to 7.4 with NaOH) containing 10 μg plasmid/ml was used. For electroporation of individual cellular processes, cells first were incubated for 10–30 s in a thapsigargin-supplemented (500 nM) Hanks’–Hepes solution, and then electroporation was performed in a Ca2+-free Hanks’–Hepes solution containing 10 μM fluo-3 and 70 μM EDTA. For selective staining of mitochondria, cells were incubated for 20 min in a rhodamine 123-supplemented (10 μM) Hanks’–Hepes solution and were rinsed with buffer solution before viewed in the microscope.
Fluorescence imaging was achieved by sending the output of an argon ion laser (Spectra-Physics 2025–05, 488 nm) through a 488-nm line interference filter followed by a spinning disk to break the coherence and scatter the laser light. The laser light was collected by a lens and was sent through a fluorescein filter cube (Leica I-3) into the objective to excite the fluorophores. The resulting fluorescence was collected by the same objective, and the image was detected by a three-chip color charge-coupled device camera (Hamamatsu, Kista, Sweden) and was recorded at a 25-Hz frame collection rate by a Super VHS (Panasonic S-VHS AG-5700, Stockholm, Sweden). The charge-coupled device images were digitized from tape and were processed for presentation. RGB (Red Green Blue) files were converted to CMYK (Cyano Magenta Yellow Black) file format for printing. Occasionally, the charge-coupled device camera was fed directly into a frame grabber.
Cell Culture.
Progenitor cells derived from adult rat brain hippocampus were cultured according to procedures described by Palmer et al. (24) and were plated onto no. 1, 1-inch circular coverslips coated with polyornithine and laminin as described by Ray et al. (25). COS 7 cells derived from African green monkey kidneys, and 293 cells were cultured according to standard procedures.
Chemicals and Materials.
Hepes (>99%), sodium chloride, potassium chloride, and sodium hydroxide (all Suprapur), calcium dichloride, magnesium dichloride, magnesium sulfate, potassium dihydrogen phosphate, sodium hydrocarbonate, and EGTA (Titriplex VI) (all pro analysi) were purchased from Merck. d-glucose (AnalaR) was from BDH and fluorescein (GC-grade) and dimethyl sulfoxide were obtained from Sigma. Fluo-3 (pentaammonium salt), rhodamine 123 and thapsigargin were from Molecular Probes, and the plasmid pRAY 1 was from GIBCO/BRL. Deionized water from a Milli-Q system (Millipore) was used.
RESULTS AND DISCUSSION
Introduction of Membrane-Impermeant Dyes into Single Cells by Electroporation.
Fig. 1 is a schematic setup of the experiment, which depicts the geometrical arrangement of the electrodes with respect to a cell undergoing permeabilization. Because the electrodes are movable in steps of 0.2 μm in three-dimensional space, a high degree of freedom to focus in on a single cellular structure of choice in a complex multicellular network is offered. This is in contrast to fixed-electrode poration chambers in which the dimensions of the electrodes typically extend the size of a cell severalfold. Ideally, a cell is approached with the electrodes at an angle from above, and electrode contact with the cell membrane is avoided, which otherwise can lead to electrochemical transformation of membrane-bound proteins and other structures. Highly focused electrical fields were produced by application of single or multiple rectangular waveform voltage pulses by using a low-voltage pulse-generator.
The high spatial resolution achieved with this technique is demonstrated in Fig. 2, in which fluorescein, a largely cell-impermeant highly fluorescent dye, is electroporated into single cells. The bright-field images in Fig. 2 A and C shows examples of populations with a small number of cells in which one cell was electroporated in a fluorescein-supplemented Hanks’–Hepes buffer. Application of voltage pulses results in formation of pores and diffusion of extracellular fluorescein into the cell. Resealing of the pores after electroporation then traps the dye inside the cell. Fig. 2 B and D shows fluorescence images of the same fields as in Fig. 2 A and C. The electroporated cells exhibit strong fluorescence from fluorescein entrapped in the cytosol.
Figure 2.

Photomicrographs demonstrating electroporation of single progenitor cells from the adult rat brain to incorporate fluorescein (10 μM). (A and C) Bright-field images of small groups of progenitor cells in which one was electroporated (arrow). The cells were electroporated with 10 pulses of ≈1 V at 0.5-Hz repetition rate. The false-color coded fluorescence images (fluorescence intensity proportional to color wavelength) of the respective images are shown in B and D. The fluorescence from fluorescein is localized preferentially to the electroporated cells. The fluorescence intensity (measured as pixel intensity per square micrometer over the total cross sectional area of a single cell at the same laser intensity and detector settings) of electroporated cells (n = 10) was 4.3-fold higher (P < 0.001, using an unpaired one-tailed Student’s t test) than the background fluorescence intensity from untreated cells (n = 6).
The success rate for electroporation of progenitor cells as well as individual cells in a kidney 293 cell line for the introduction of fluorescein (n = 28) by using 3 to 10 pulses of 0.99 ± 0.1 V at 0.5 Hz repetition rate in flawless experiments was 86%. We found that, below these plasma membrane threshold potentials, it was impossible to detect any dye accumulation in the cells. Reversible membrane breakdown can nonetheless occur at lower potentials but would require more sensitive detection means to observe than those used in the present study.
Electroporation of Intracellular Organelles and Cellular Processes.
Further experiments were performed with fluorescein to see whether organelles of single progenitor cells could be electroporated in situ. Specifically, fluorescein was introduced into single cells by 10 0.5-Hz superthreshold field applications, resulting in potential shifts at the cell membrane of 1.6 V ± 0.07 V (range 1.5 to 2.4 V). After this, the extracellular dye-containing media was replaced by a Hanks’–Hepes solution. The result of this electroporation scheme is shown in Fig. 3A, in which punctuate fluorescence is observed in the exonuclear cytoplasmic region but not in the nuclear region, which contains a smaller number of organelles. In total, 18 of 20 cells electroporated in this protocol displayed a similar staining pattern, reminiscent of what is observed by using an organelle-specific dye such as rhodamine 123, which labels the mitochondrial fraction of organelles (Fig. 3B). In comparison, cells electroporated at the plasma membrane threshold potential generally displayed a diffuse fluorescence from fluorescein entrapped in the cytosol (Fig. 3C). This observation of a differential localization of fluorescein comparing application of low and high electric-field strengths is consistent with the higher breakdown potential of intracellular organelles as predicted by Eq. 1 because of their smaller dimensions. It has been shown that 2.5- to 10-fold higher electric-field strengths are required for electroporation and fusion of isolated mitochondria compared with 10-μm diameter cells (17, 18). The results are also in accord with morphological observations in giant squid axons following mild, moderate, and severe electroporation protocols (26). It should be stressed, however, that it is unclear what organelles accumulated fluorescein and that electroporation at the plasma membrane threshold potential, in particular by using high-frequency multiple-pulse protocols, might result in permeabilization of organelles. These issues were not dealt with in the present study but warrant further investigation.
Figure 3.
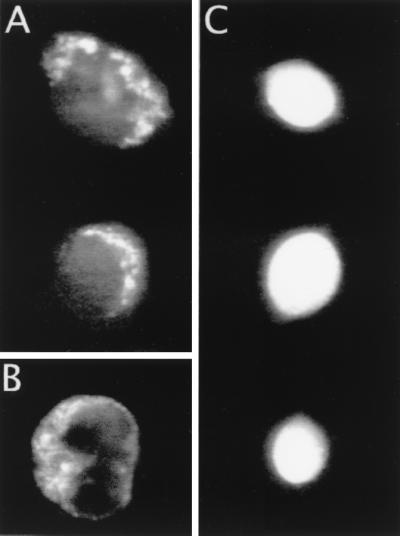
Photomicrographs demonstrating differential fluorescence staining resulting from electroporation of fluorescein into single progenitor cells at high and low electric field strengths, respectively. (A) Two cells electroporated at plasma membrane superthreshold potentials of ≈2 V (10 pulses at 0.5-Hz repetition rate), both of which display a punctuate cytoplasmic fluorescence pattern. (B) Single progenitor cell after incubation with rhodamine 123, a mitochondria-specific dye. (C) Images of three cells after electroporation at the plasma membrane threshold potential of ≈1 V (10 pulses at 0.5-Hz repetition rate) in which the fluorescence is diffuse and evenly distributed over the entire cell.
To find functional evidence of organellar electroporation, single progenitor cells were electroporated with 10 μM fluo-3, a dye that becomes fluorescent when chelating Ca2+ (27). Because the concentration of free intracellular Ca2+ is extremely low (≈100 nM), the cytosol only becomes weakly fluorescent on addition of fluo-3. Specifically, fluo-3 supplemented to a Ca2+-free Hanks’–Hepes buffer is first introduced into the cells with a sequence of 1- to 5-, 0.5- to 10-ms rectangular waveform pulses at the plasma membrane threshold potential. This pulse protocol only induced a slight increase in cytoplasmic fluorescence. After this, the fluo-3-containing buffer was replaced with a Ca2+-free Hanks’–Hepes buffer, and additional pulses of 1.3 to 2.8 V (1 ms) were applied. Of a total of 47 cells, this protocol resulted in increased cytoplasmic fluorescence in 31 cells, most likely because of membrane breakdown of intracellular calcium stores.
In experiments using multiple pulse protocols with plasma membrane superthreshold field applications, cells frequently displayed a swollen and damaged appearance. Presumably, such cells enter a hyper-leakage state with deterioration of many cellular functions (26). Of importance, high-voltage applications appear to affect organellar structures to a lesser extent (17) and were found in single-pulse applications for gene transfer not to affect considerably cell viability in COS 7 cells (see below). Electroporation of organelles in situ might become useful for probing and selective labeling of organelles for functional studies and for recently emerging single-organelle analysis schemes in conjunction with chemical separations (10).
Experiments were conducted to electroporate individual submicrometer diameter cellular processes as a further demonstration of the high spatial resolution of this technique. Specifically, fluo-3 was electroporated into individual processes of thapsigargin-treated neuronal progenitor cells. Thapsigargin releases ≈50% of total Ca2+ from intracellular stores in Jurkat cells (28) to the cytoplasm by inhibition of a Ca2+-ATPase of the endoplasmic reticulum (29). Fig. 4 shows a time sequence of the spreading of a fluorescence wave emanating from the electroporation loci in two cells. These experiments were performed in quadruplicate and consistently showed the same pattern.
Figure 4.

Images showing electroporation of fluo-3 into individual cellular processes of thapsigargin-treated progenitor cells. (A) Bright-field image of two progenitor cells. The position of the microelectrodes in relation to the cellular process undergoing permeabilization is shown. (B) The same image as A in false-color coded fluorescence (fluorescence intensity proportional to color wavelength) before application of the electric field. (C–E) Time sequences starting at 70 s after electroporation (C) and ending 90 s after electroporation (E). In D and E, the background fluorescence was subtracted from the images. The arrows in C indicate the location of electroporation along the cellular process. In C, right, the process loosened from the substratum after electroporation. The processes were electroporated with three 1-ms pulses of ≈0.5 V.
Transfection of Single COS 7 Cells.
In a third category of experiments, individual COS 7 cells were electroporated to investigate the feasibility of selective gene transfer by using the presented technique as well as to investigate the survival rate of cells after electroporation. The plasmid vector pRAY 1 was used as a reporter gene and was electroporated into individual COS 7 cells. A Hepes buffer with 10 μg plasmid/ml and 0.75% dimethyl sulfoxide was used throughout the experiments. In initial experiments, using a plasmid-supplemented Hanks’–Hepes buffer, it was noted that it was difficult to achieve a high transfection yield. Addition of dimethyl sulfoxide at low concentrations to the electroporation buffer, however, facilitated the electroporation-induced uptake of plasmid into cells. This is in accordance with observations made by other researchers (30). For experiments, single 1-ms rectangular waveform voltage pulses of ≈1.9 V were applied followed by careful washing of the cells with Hepes buffer. Transfected cells (n = 50, five cell dishes) were detected 36 hours after electroporation by using the same laser-induced fluorescence microscope setup as for the other dyes. The transfection yield and survival rate was 97%. Individual electroporated cells generally exhibited strong fluorescence from expression of green fluorescent protein (Fig. 5). Control cells (n > 50, five cell dishes) exposed to plasmid at the same concentration and time did not exhibit any fluorescence over background level under the same detection conditions (Fig. 5).
Figure 5.

Fluorescence images demonstrating selective transfection of single COS 7 cells with the pRAY 1 expression vector containing the cDNA for green fluorescent protein. The images were taken 36 hours after electroporation. Control cells exposed to plasmid but not electroporated are outlined with white contour lines. The location of control cells was identified from bright-field images and superimposed onto the fluorescence images.
CONCLUSIONS
We have demonstrated selective transfer of membrane-impermeant solutes into single cells by using a miniaturized version of electroporation. The technique might become valuable for basic and applied studies of electroporation on the single-cell and organelle level. Further miniaturization of the technique by using nanotip electrodes with insulated shanks that can be inserted into a cell for exclusive electroporation of organellar structures with minimal impairment of cellular physiology would be feasible. The methodology is compatible with a wide range of biochemical and biophysical techniques and chemical microseparation formats that operate on the single cell and single organelle level (see refs. 1–10). It contrasts microinjection techniques for single cells and single nuclei in that it can be timed precisely and applied to biological containers of sizes much smaller than can be resolved in a light microscope.
Acknowledgments
We are grateful to Daniel T. Chiu and Stephen G. Weber for fruitful discussions, T. D. Palmer and F. H. Gage for the generous gift of adult-derived neural progenitor cells, Per-Arne Svensson for enthusiastic help and advice on cell transfections, and Susanne Orwar for digital image-editing and photographic work. This work was made possible through grants by the Swedish Natural Science Research Council (Grants 10481-305, 308, 309) and the Swedish Foundation for Strategic Research. P.S.E. is supported by the Gunvor och Josef Anérs foundation and the Swedish Medical Research Council.
References
- 1.Hamill O P, Marty A, Neher E, Sakman B, Sigworth F J. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 2.Chow R H, von Rüden L, Neher E. Nature (London) 1992;356:60–63. doi: 10.1038/356060a0. [DOI] [PubMed] [Google Scholar]
- 3.Wightman R M, Jankowski J A, Kennedy R T, Kawagoe K T, Schroeder T J, Leszczyszyn D J, Near J A, Diliberto E J, Jr, Viveros O H. Proc Natl Acad Sci USA. 1991;88:10754–10758. doi: 10.1073/pnas.88.23.10754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bacskai B J, Wallen P, Lev-Ram V, Grillner S, Tsien R Y. Neuron. 1995;14:19–28. doi: 10.1016/0896-6273(95)90237-6. [DOI] [PubMed] [Google Scholar]
- 5.Maiti S, Shear J B, Williams R M, Zipfel W R, Webb W W. Science. 1997;275:530–532. doi: 10.1126/science.275.5299.530. [DOI] [PubMed] [Google Scholar]
- 6.Betzig E, Trautman J K, Harris T D, Weiner J S, Kostelak R L. Science. 1991;251:1468. doi: 10.1126/science.251.5000.1468. [DOI] [PubMed] [Google Scholar]
- 7.Ashkin A. Phys Rev Lett. 1970;24:156–160. [Google Scholar]
- 8.Svoboda K, Block S M. Annu Rev Biophys Biomol Struct. 1994;23:247–285. doi: 10.1146/annurev.bb.23.060194.001335. [DOI] [PubMed] [Google Scholar]
- 9.Chiu D T, Hsiao A, Gaggar A, Garza-López R A, Orwar O, Zare R N. Anal Chem. 1997;69:1801–1807. doi: 10.1021/ac961226g. [DOI] [PubMed] [Google Scholar]
- 10.Chiu D T, Lillard S J, Scheller R H, Zare R N, Rodriguez-Cruz S E, Williams E R, Orwar O, Sandberg M, Lundqvist J A. Science. 1998;279:1190–1193. doi: 10.1126/science.279.5354.1190. [DOI] [PubMed] [Google Scholar]
- 11.Capecchi M R. Cell. 1980;22:479–488. doi: 10.1016/0092-8674(80)90358-x. [DOI] [PubMed] [Google Scholar]
- 12.Kinosita K, Jr, Ashikawa I, Saita N, Yoshimura H, Itoh H, Nagayama K, Ikegami A. J Biophys. 1988;53:1015–1019. doi: 10.1016/S0006-3495(88)83181-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hibino M, Shigemori M, Itoh H, Nagayama K, Kinosita K., Jr Biophys J. 1991;59:209–220. doi: 10.1016/S0006-3495(91)82212-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zimmermann U. Biochim Biophys Acta. 1982;694:227–277. doi: 10.1016/0304-4157(82)90007-7. [DOI] [PubMed] [Google Scholar]
- 15.Tsong T Y. Biophys J. 1991;60:297–306. doi: 10.1016/S0006-3495(91)82054-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weaver J C. J Cell Biochem. 1993;51:426–435. doi: 10.1002/jcb.2400510407. [DOI] [PubMed] [Google Scholar]
- 17.Collombet J-M, Wheeler V C, Vogel F, Coutelle C. J Biol Chem. 1997;272:5342–5347. doi: 10.1074/jbc.272.8.5342. [DOI] [PubMed] [Google Scholar]
- 18.Reynaud J A, Labbe H, Lequoc K, Lequoc D, Nicolau C. FEBS Lett. 1989;247:106–112. doi: 10.1016/0014-5793(89)81250-5. [DOI] [PubMed] [Google Scholar]
- 19.Templeton N S, Roberts D D, Safer B. Gene Ther. 1997;4:700–709. doi: 10.1038/sj.gt.3300457. [DOI] [PubMed] [Google Scholar]
- 20.Kinosita K, Jr, Tsong T Y. Biochim Biophys Acta. 1979;554:479–497. doi: 10.1016/0005-2736(79)90386-9. [DOI] [PubMed] [Google Scholar]
- 21.Chang D C. Biophys J. 1989;56:641–652. doi: 10.1016/S0006-3495(89)82711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marszalek P E, Farrell B, Verdugo P, Fernandez J M. Biophys J. 1997;73:1160–1168. doi: 10.1016/S0006-3495(97)78148-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Teruel M N, Meyer T. Biophys J. 1997;73:1785–1796. doi: 10.1016/S0006-3495(97)78209-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palmer T D, Takahashi J, Gage F H. Mol Cell Neurosci. 1997;8:389–404. doi: 10.1006/mcne.1996.0595. [DOI] [PubMed] [Google Scholar]
- 25.Ray J, Peterson D A, Schinstine M, Gage F H. Proc Natl Acad Sci USA. 1993;90:3602–3606. doi: 10.1073/pnas.90.8.3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gallant P E, Galbraith J A. J Neurotrauma. 1997;14:811–822. doi: 10.1089/neu.1997.14.811. [DOI] [PubMed] [Google Scholar]
- 27.Minta A, Kao J P Y, Tsien R Y. J Biol Chem. 1989;264:8171–8178. [PubMed] [Google Scholar]
- 28.Guse A H, Roth E, Emmrich F. Biochem J. 1993;291:447–451. doi: 10.1042/bj2910447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thastrup O, Cullen P J, Drobak B K, Hanley M R, Dawson A P. Proc Natl Acad Sci USA. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Melkonyan H, Sorg C, Klemp M. Nucleic Acids Res. 1996;24:4356–4357. doi: 10.1093/nar/24.21.4356. [DOI] [PMC free article] [PubMed] [Google Scholar]