

Abstract
Objectives
Obesity and diabetes are strong risk factors that drive the development of type I endometrial cancers. Recent epidemiological evidence suggests that metformin may lower cancer risk and reduce rates of cancer deaths among diabetic patients. In order to better understand metformin's anti-tumorigenic potential, our goal was to assess the effect of metformin on proliferation and expression of key targets of metformin cell signaling in endometrial cancer cell lines.
Methods
The endometrial cancer cell lines, ECC-1 and Ishikawa, were used. Cell proliferation was assessed after exposure to metformin. Cell cycle progression was evaluated by flow cytometry. Apoptosis was assessed by ELISA for caspase-3 activity. hTERT expression was determined by real-time RT-PCR. Western immunoblotting was performed to determine the expression of the downstream targets of metformin.
Results
Metformin potently inhibited growth in a dose-dependent manner in both cell lines (IC50 of 1 mM). Treatment with metformin resulted in G1 arrest, induction of apoptosis and decreased hTERT expression. Western immunoblot analysis demonstrated that metformin induced phosphorylation of AMPK, its immediate downstream mediator, within 24 hours of exposure. In parallel, treatment with metformin decreased phosphorylation of S6 protein, a key target of the mTOR pathway.
Conclusions
We find that metformin is a potent inhibitor of cell proliferation in endometrial cancer cell lines. This effect is partially mediated through AMPK activation and subsequent inhibition of the mTOR pathway. This work should provide the scientific foundation for future investigation of metformin as a strategy for endometrial cancer prevention and treatment.
Keywords: endometrial cancer, metformin, mTOR, telomerase
Introduction
Endometrial cancer is the fourth most common cancer among women in the United States and has been increasing in frequency secondary to an aging female population and changes in dietary and hormonal factors, with obesity being a major culprit [1, 2]. It is estimated that 40,100 new cases will be diagnosed in 2008, and 7,470 women will succumb to this disease [3]. Therefore, there is great interest in identifying novel ways to treat and prevent this disease. Obesity, diabetes and insulin resistance are strong risk factors that drive the development of the more common type I endometrial cancers. Unfortunately, obesity is not only a risk factor for developing endometrial cancer, but it is associated with an increased risk of death [4-6]. Obese women with endometrial cancer have a 6.25 increased risk of death from this disease as compared to their non-obese counterparts [5]. Metformin (dimethylbiguanide) is a biguanide drug that is widely used as the first line pharmacologic treatment of type II diabetes after patients have failed diet and exercise modification. There is recent epidemiological evidence to suggest that metformin use lowers cancer risk and reduces the rate of cancer deaths among diabetic patients as compared to sulfonylureas or insulin use [7, 8]. It is unknown whether the underlying mechanism behind metformin's potential anti-neoplastic effects are related to the systemic action of this drug, by reducing circulating insulin levels, and/or a direct action on cancer cells.
Metformin is commonly thought of as an insulin sensitizer because it enhances signaling through the insulin receptor (IR), leading to an improvement in insulin resistance, followed by a reduction in circulating insulin levels. More recently, evidence suggests that metformin's key target of action is the inhibition of hepatic gluconeogenesis [9], resulting in a secondary decline in insulin levels. Metformin has become the cornerstone of treatment for women with polycystic ovarian syndrome (PCOS), a disease characterized by elevated circulating androgen levels, chronic anovulation, obesity and frequently insulin resistance [10]. Metformin has been found to improve the reproductive abnormalities in women with PCOS, restoring ovulation and improving fertility [10]. Women with PCOS who respond to metformin have generally a decrease in circulating insulin and androgen levels, suggesting that both hyperinsulinemia and hyperandrogenemia contribute to the clinical manifestation of this syndrome which is associated with an increased risk of endometrial cancer. Despite its known benefit in the treatment of women with PCOS, the effect of metformin on the endometrium has yet to be explored.
Although the molecular mechanism of metformin has been well-studied in tissues such as liver, muscle and fat, very little is known about its effects on epithelial tissues. Metformin exerts its beneficial effects through activation of the AMP-activated protein kinase (AMPK) in muscle, adipose tissue and liver [11]. AMPK is a heterotrimeric serine/threonine protein kinase complex, comprising a catalytic α-subunit and regulatory β and γ subunits [11]. AMPK regulates energy metabolism and is activated in response to cellular stresses that deplete cellular energy levels and increase the AMP/ATP ratio [11]. AMPK uniquely detects cellular energy and ensures that cell division only proceeds if there are sufficient metabolic resources to support proliferation. Once activated, AMPK restores cellular energy levels by stimulating catabolic pathways, such as glucose uptake, glycolysis and fatty acid oxidation and halting ATP-consuming processes such as fatty acid, cholesterol and protein synthesis. LKB1 is the kinase responsible for phosphorylating and activating AMPK [9]. Interestingly, LKB1 encodes for a tumor suppressor gene that is lost in Peutz-Jeghers syndrome, a disease characterized by colonic polyps and a predisposition to epithelial malignancies such as breast, colon and lung cancer [12]. Loss of LKB1 expression has also been documented in up to 65% of human endometrial cancers and has been correlated with tumors of higher grade and more advanced stage [13, 14].
Activation of AMPK through LKB1 leads to regulation of multiple signaling pathways involved in the control of cellular proliferation, including the mTOR pathway. It is thought that AMPK mediates its effect on cell growth through inhibition of mTOR via phosphorylation of the tuberous sclerosis complex (TSC2), a subunit of the larger TSC1/TSC2 (hamartin/tuberin) complex that negatively regulates mTOR signaling. Alterations in the mTOR pathway have previously been implicated in endometrial cancer carcinogenesis. PTEN is a negative regulator of this pathway, and loss of PTEN expression is one of the most prevalent molecular abnormalities associated with endometrial cancers, occurring in up to 83% of type I endometrial cancers [15-17]. The mTOR pathway is thought to be a promising target for endometrial cancer treatment, with many mTOR inhibitors already in clinical trials for this disease.
Given the relationship between the AMPK and mTOR signaling pathways, we hypothesize that metformin may behave like a novel mTOR inhibitor, with important chemotherapeutic implications for endometrial cancer, a disease which is strongly influenced by obesity and insulin resistance. Thus, in order to better understand metformin's anti-tumorigenic potential, our goal was to assess the in vitro effect of metformin on proliferation, apoptosis and expression of key targets of metformin cell signaling in endometrial cancer cell lines.
Materials and Methods
Cell Culture and Reagents
Two endometrial cancer cell lines, ECC-1 and Ishikawa, were used for these experiments. Ishikawa cells were grown in MEM supplemented with 5% fetal bovine serum, 300 mM L-glutamine, 5 μg/ml bovine insulin, 10,000 units/ml penicillin, and 10,000 μg/ml streptomycin under 5% CO2. ECC-1 cells were maintained in RPMI 1640 containing 5% fetal bovine serum, 300 mM L-glutamine, 5 μg/ml bovine insulin, 10,000 units/ml penicillin, and 10,000 μg/ml streptomycin under 5% CO2. Metformin, MTT (3-(,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) dye, RNase A, anti-PTEN antibody and anti-β-actin antibody were purchased from Sigma (St. Louis, MO). The anti-phospho-AMPK, anti-pan-AMPK, anti-phospho-S6, anti-pan-S6 antibodies and the caspase-3 ELISA kit were purchased from Cell Signaling (Beverly, MA). Enhanced chemiluminescence Western blotting detection reagents were purchased from Amersham (Arlington Heights, IL). All other chemicals were purchased from Sigma.
Cell Proliferation Assays
The effects of metformin treatment on cell proliferation was examined by MTT assay. The ECC-1 and Ishikawa cells were plated and grown in 96-well plates at a concentration of 6000 cells/μL and 8000 cells/μL respectively for 24 hours. Cells were then treated with varying doses of metformin for 24-72 hours. Viable cell densities were determined by metabolic conversion of the dye MTT. MTT (5 mg/ml) was added to the 96 well plates at 10 μL/well, and the plates were then incubated for an additional hour. The MTT reaction was terminated by the addition of 100 μl of DMSO. Subsequently, the MTT assay results were read by measuring absorption at 595 nm. The effect of metformin was calculated as a percentage of control cell growth obtained from PBS treated cells grown in the same 96 well plates. Each experiment was performed in triplicate and repeated three times to assess for consistency of results.
Flow Cytometry
To evaluate the mechanism of growth inhibition by metformin, the cell cycle profile was analyzed after treatment with metformin. Both cell lines were plated at 2.5 × 105 cells/well in 6 well plates in their corresponding media for 24 hours. Subsequently, the cells were starved overnight and then treated with 15% serum and metformin at varying concentrations for 36 hours. Cells were collected, washed with PBS, fixed in a 90% methanol solution and then stored at -20 °C until flow cytometric analysis was performed. On the day of analysis, cells were washed and centrifuged using cold PBS, suspended in 100μL PBS and 10uL of RNase A solution (250 μg/ml) followed by incubation for 30 minutes at 37°C. After incubation, 110 μL of PI stain (100 μg/ml) were added to each tube and incubated at 4°C for at least 30 minutes prior to analysis. Flow cytometric analysis was performed on a CyAn machine (Beckman Coulter, Miami, FL). ModFit (Verity Software House, Topsham, ME) was utilized for the analysis to control for dead cells and debris. All experiments were performed in triplicate and repeated twice to assess for consistency of response.
Apoptosis Assay
To evaluate the mechanism of growth inhibition by metformin, the induction of apoptosis was analyzed after exposure to metformin. Both cell lines were cultured in 6 well plates to concentrations of 2-4 × 105 cells/well for 24 hours and then treated with metformin at indicated doses in 0.5% stripped serum for an additional 24 hours. ELISA analysis with a Caspase-3 kit was performed according to manufacturer instructions. Briefly, the cells were lysed and protein concentrations measured to confirm equal loading onto an ELISA plate. Reagents were added as described by the manufacturer, and the ELISA plate was read by measuring absorption at 450 nm. All experiments were performed in triplicate and repeated twice to assess for consistency of response.
Real-time RT-PCR
The hTERT gene encodes the catalytic subunit of telomerase. hTERT expression is the rate-limiting determinant of the enzymatic activity of human telomerase and is thought to be a sensitive marker of telomerase function and cell proliferation. Thus, we examined the effect of metformin treatment on hTERT expression in the endometrial cancer cell lines by real-time RT-PCR. Total RNA was extracted using the AIQshredder kit (Qiagen, Valencia, CA) and further purified by the RNeasy Mini-kit (Qiagen, Valencia, CA). The reverse transcription and PCR reactions were performed using the TaqMan Gold one-step RT-PCR kit in the ABI Prism 7700 Sequence Detection System (Applied Biosystems, Foster City, CA). Reverse transcription was carried out at 48°C for 30 min. The PCR conditions consisted of a 10 min step at 95°C, 40 cycles at 95°C for 15 sec each and 1 min at 65°C. A housekeeping control gene acidic ribosomal phosphoprotein P0 (RPLP0, also known as 36B4) was used as an internal control to correct for differences in the amount of RNA in each sample. Primers and fluorogenic probes for hTERT and RPLP0 have been described previously [18]. The standard curve for hTERT was generated by using dilutions of a known amount of cRNA synthesized by in vitro transcription of a cloned fragment. The normalized level of hTERT in each sample was estimated by a ratio of the hTERT level to the RPLP0 level, as described previously [18]. Each experiment was performed in triplicate and repeated twice to assess for consistency of results.
Western Immunoblotting
To investigate the mechanisms underlying the anti-proliferative effect of metformin, we characterized the effect of metformin on relevant cell signaling targets, including AMPK and phosphorylated S6. The PTEN status of these two cell lines was also characterized. The Ishikawa and ECC-1 cells were plated at 2 × 105 cells/well in 6 well plates in their corresponding media and then treated for 16 hours with metformin in 0.5% stripped serum. Cell lysates were prepared in RIPA buffer (1% NP40, 0.5 sodium deoxycholate and 0.1% SDS). Equal amounts of protein were separated by gel electrophoresis and transferred onto a nitrocellulose membrane. The membrane was blocked with 5% nonfat dry milk and then incubated with a 1:1000 dilution of primary antibody overnight at 4°C. The membrane was then washed and incubated with a secondary peroxidase-conjugated antibody for 1 hour after washing. Antibody binding was detected using an enhanced chemiluminescence detection system (GE Healthcare Life Sciences, Piscataway, NJ). After developing, the membrane was stripped and re-probed using antibody against β-actin and either pan-S6 or pan-AMPK to confirm equal loading. Each experiment was repeated three times to assess for consistency of results.
Statistical Analysis
Results for experiments were normalized to the mean of the control and analyzed using the student t-test. Differences were considered significant if the p value was less than 0.05 (p<0.05) with a confidence interval of 95%. STATA software (StataCorp, College Station, TX) was used to perform statistical analyses.
Results
Sensitivity of Endometrial Cancer Cells to Metformin
We examined the effect of metformin on proliferation in two endometrial cancer cell lines. As shown in Figure 1, metformin potently inhibited growth in a dose-dependent manner in both endometrial cancer cell lines at 24-72 hours (p=0.0198-0.0348 for ECC-1 and p=0.0056-0.0166 for Ishikawa). The mean IC50 value for each of these cell lines was approximately 1 mM.
Figure 1.
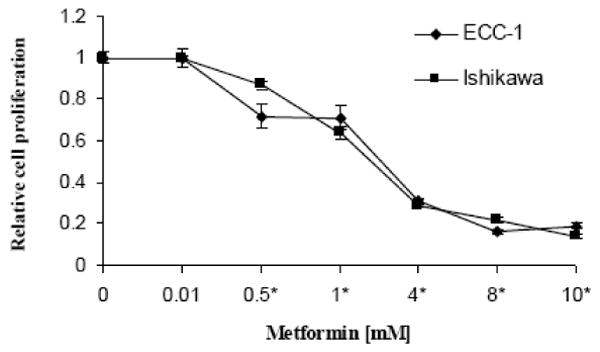
Effect of metformin on proliferation of endometrial carcinoma cells. Ishikawa and ECC-1 cells were cultured in the presence of varying concentrations of metformin for 72 hours. Relative growth of cells were determined by MTT. The results are shown as the mean ± SE of triplicate samples and are representative of three independent experiments. (* indicates statistically significant difference)
Effect of Metformin on Cell Cycle and Apoptosis
To evaluate the mechanism of growth inhibition by metformin, the cell cycle profile and induction of apoptosis was analyzed after treatment with metformin. As expected, serum stimulation resulted in transition of cells from G1 to S phase by 24 hr, with a concomitant decrease in G1 phase. Metformin significantly blocked serum-induced entry to S phase, resulting in G1 cell cycle arrest (Figure 2). This effect was seen in both endometrial cancer cell lines and appeared to be dose-dependent (p= 0.0165-0.0482 for ECC-1 and p=0.0239-0.0586 for Ishikawa). The percent change ranged from 13.4 – 14.1%, depending on the cell line.
Figure 2.
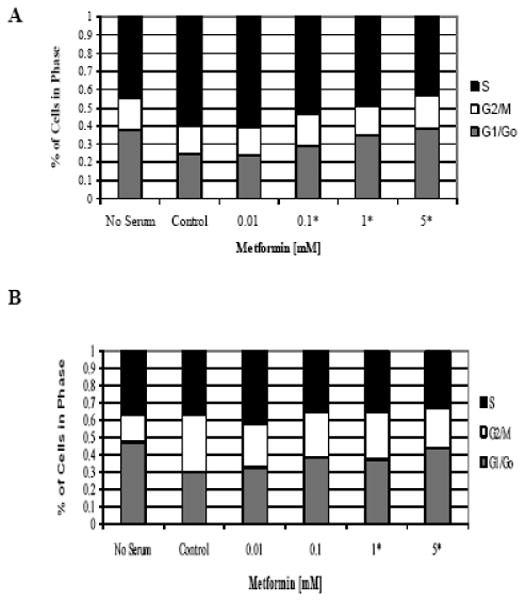
Metformin inhibited cell cycle progression by arrest in G1 phase. The endometrial cancer cell lines, ECC-1 (A) and Ishikawa (B), were starved overnight and then stimulated with 15% serum and metformin at the noted concentrations for 36 hours. Cell cycle analysis was performed by flow cytometry. Results shown are representative of two independent experiments. (* indicates statistically significant difference)
An apoptosis assay using an antibody to caspase-3 was performed after exposure to metformin. Caspases are a family of cysteine proteases that act in a cascade to trigger apoptosis. Caspase-9 is an initiator caspase that is thought to activate the effector caspases (caspase-3 and -6) involved in actual cell disassembly. Caspase-3 is considered to be a specific marker for epithelial apoptosis. Metformin induced apoptosis but only at high doses of treatment (2-5 mM) as demonstrated by increased caspase-3 activity (p=0.008-0.0032 for ECC-1 and p=0.0108-0.0218 for Ishikawa) (Figure 3). Lower doses of metformin had little effect on caspase-3 activity.
Figure 3.
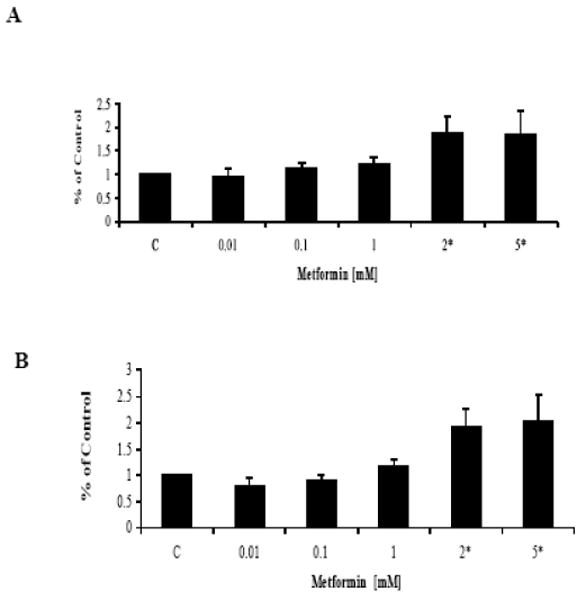
Metformin induced apoptosis but only at high concentrations. The endometrial cancer cell lines (A) ECC-1 and (B) Ishikawa were grown for 24 hours and then treated with metformin at the indicated concentrations for an additional 24 hours. Apoptosis was assessed using an antibody to caspase-3. The results are shown as the mean +/- SD and are representative of two independent experiments. (* indicates statistically significant difference)
These results suggest that metformin predominantly inhibits cell growth via cell cycle arrest and not by induction of apoptosis in these endometrial cancer cell lines. Induction of apoptosis may contribute to metformin's anti-proliferative effect but only in the presence of high concentrations of this drug.
Effect of Metformin on hTERT mRNA Level
The maintenance of telomere length via the expression of telomerase is vital to the ability of cancer cells to remain proliferative. hTERT expression is the rate-limiting determinant of the enzymatic activity of human telomerase; and thus, real time RT-PCR was used to quantify hTERT mRNA expression in the endometrial cancer cell lines. Treatment with metformin decreased hTERT mRNA expression in a dose-dependent manner in both cell lines within 24 hours (p=0.004-0.05 for ECC-1 and p=0.0008-0.0255 for Ishikawa) (Figure 4) and 48 hours (data not shown). This data suggests that metformin may inhibit telomerase activity by rapidly decreasing hTERT mRNA levels.
Figure 4.
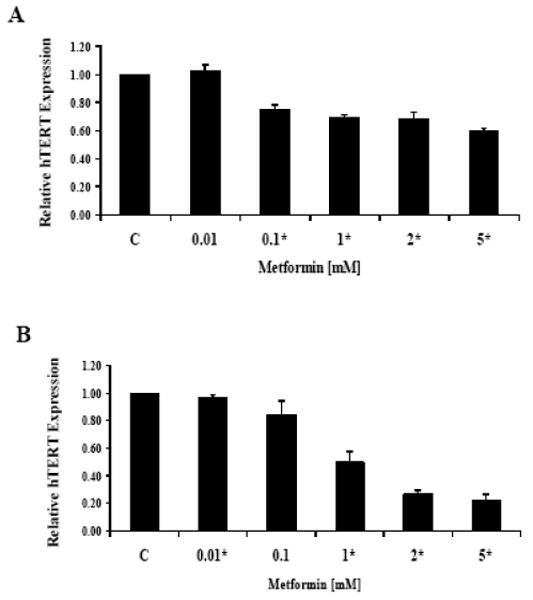
Metformin decreased hTERT mRNA expression in a dose-dependent manner in the (A) ECC-1 and (B) Ishikawa cell lines. Both cell lines were cultured for 24 hours and then treated with the indicated concentrations of metformin for an additional 24 hours. hTERT expression was determined by real-time RT-PCR. The results are shown as the mean ± SE of two independent experiments. (* indicates statistically significant difference)
Effect of Metformin on the mTOR Pathway
To investigate the mechanisms underlying the anti-proliferative effect of metformin, we characterized the effect of metformin on relevant cell signaling targets. In both endometrial cancer cell lines, Western immunoblot analysis demonstrated that metformin induced phosphorylation in a dose-dependent fashion of AMPK, its immediate downstream mediator, within 16 hours of exposure (Figure 5A & 5B). Previous studies suggest that p70S6K is a downstream target of the mTOR pathway [19]. p70S6K kinase directly phosphorylates the 40S ribosomal protein S6, which results in enhanced translation of proteins that contain a polypyrimidine tract in the 5′-untranslated region [19]. Therefore, we studied the effect of metformin on the phosphorylation of the S6 ribosomal protein in both cell lines. Within 16 hours of treatment, metformin dramatically decreased the phosphorylation of S6 (Figure 5C & 5D). Expression of pan-AMPK and pan-S6 were not affected by metformin. This suggests that metformin may exert its anti-proliferative effect through activation of AMPK and subsequent decreased phosphorylation of the S6 protein, resulting in inhibition of the mTOR pathway.
Figure 5.

In both endometrial cancer cell Lines ECC-1 (A & C) and Ishikawa (B & D), metformin increased phosphorylation of AMPK and decreased phosphorylation of S6 in a dose-dependent manner within 16 hours of exposure as determined by Western immunoblotting. Little effect was seen on total AMPK (Pan-AMPK) or total S6 (Pan-S6). Both cell lines expressed wild-type PTEN (E).
The expression of wild-type PTEN in the endometrial cancer cell lines was determined by Western immunoblotting (Figure 5E). Loss of PTEN expression is thought to enhance sensitivity of tumor cells to the effects of mTOR inhibitors [20-23]. The Ishikawa and ECC-1 cell lines were both strongly positive for PTEN. Thus, we were unable to assess the effect of PTEN status on response to treatment with metformin. However, we can conclude that PTEN does not need to be absent for metformin to be a potent inhibitor of cell proliferation.
Discussion
We have demonstrated that metformin is a potent inhibitor of cell proliferation in two endometrial cancer cell lines, predominantly through G1 cell cycle arrest. Metformin was capable of inducing apoptosis but only at high concentrations of treatment. In addition, metformin suppressed hTERT mRNA expression in both of these cell lines. Treatment with metformin resulted in activation of AMPK, its immediate downstream target, followed by decreased phosphorylation of S6, signifying inhibition of the mTOR pathway. These findings suggest that metformin may function as a novel mTOR inhibitor and emerge as a targeted chemotherapeutic strategy for both the treatment and prevention of endometrial cancer. Although metformin has been shown to inhibit proliferation in vitro in breast, prostate, colon and ovarian cancer cell lines [24-26], this is the first time that this has been demonstrated for endometrial cancer, which of all of these cancers is the one most impacted by obesity and insulin resistance.
Metformin's immediate downstream target is AMPK, and AMPK activation leads to regulation of multiple downstream pathways involved in the control of cellular proliferation, including the mTOR pathway (Figure 6). AMPK mediates its effect on cell growth through inhibition of mTOR via phosphorylation of the tuberous sclerosis complex (TSC2), a subunit of the larger TSC1/TSC2 (hamartin/tuberin) complex that negatively regulates mTOR signaling. In breast cancer cell lines, metformin-mediated AMPK activation was shown to decrease cell growth through inhibition of mTOR and a decrease in phosphorylation of its downstream targets, S6 and eIF-4E-binding proteins [25, 27]. This ultimately resulted in a significant reduction in initiation of translation. Similar results have been found in prostate, colon and ovarian cancer cell lines [24, 26] and confirmed by our findings in endometrial cancer cell lines (Figure 5).
Figure 6.
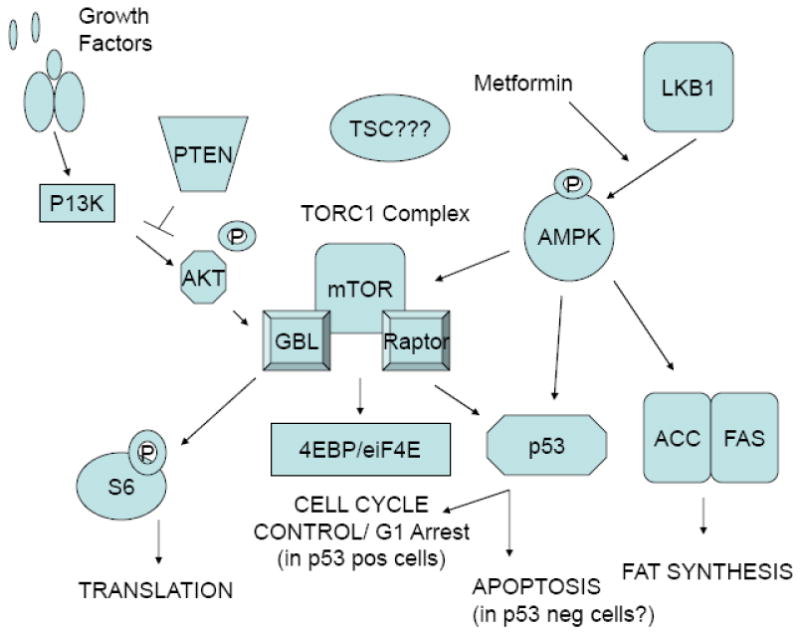
Interaction between the metformin and mTOR pathways.
AMPK activation is thought to be a possible therapeutic target for cancers with activated Akt signaling, since AMPK inhibits mTOR signaling downstream of Akt. Given the high prevalence of PTEN mutations in endometrial cancer which leads to constitutive Akt expression, AMPK activation through metformin may be a particularly compelling anti-cancer strategy for this disease. Both of the endometrial cancer cell lines examined in these studies expressed wild-type PTEN, demonstrating that PTEN does not need to be absent for metformin to be an effective inhibitor of cell proliferation. Other researchers have shown that loss of PTEN function in PTEN-null cancer cells confers increased sensitivity to mTOR inhibition by rapamycin [20-23]. Our future plans include further evaluation of PTEN status on response to treatment with metformin through either comparison with other PTEN-null endometrial cancer cell lines or selective silencing of PTEN via small interfering RNA (siRNA). However, given that both wild-type PTEN and PTEN-null tumors may potentially respond to metformin treatment, this would increase the generalizability of this therapy for endometrial cancer patients and may also have a bearing on type II endometrial cancers, which less frequently have PTEN mutations.
In addition, we have demonstrated that metformin suppressed proliferation in the endometrial cancer cell lines via G1 cell cycle arrest (Figure 2). This result is not surprising, given that mTOR inhibitors have also been shown to arrest cells in the G1 phase in a number of different cancer cell types, including our work in endometrial, ovarian and cervical cancer cell lines [28, 29]. It is controversial whether metformin may also induce apoptosis. In the endometrial cancer cells, metformin was able to induce apoptosis but only at high concentrations of treatment (Figure 3), suggesting that cell cycle arrest as opposed to cell death is most likely the major contributor to metformin's anti-proliferative effect. In contrast, metformin failed to induce apoptosis in prostate and breast cancer cell lines at similar doses of treatment but did block cell cycle progression in G1 phase [30, 31]. However, metformin was found to induce apoptosis in vitro in colon cancer cells but only in those cells that lacked the tumor suppressor p53 [32]. Both endometrial cancer cell lines used in this work express wild-type p53 (data not shown); and thus, it is possible that metformin may selectively induce apoptosis at more physiologic doses in p53-deficient endometrial cancer cells. This may have possible implications for type II endometrial cancers which more often have mutated p53.
In most normal somatic cell types, telomerase activity is usually undetectable; however, the endometrium is one exception [33]. It is thought that telomerase plays a critical role in the ability of normal endometrium to repeatedly proliferate from the onset of menarche to menopause. Furthermore, activation of telomerase has also been implicated as a fundamental step in cellular immortality and oncogenesis in many cancers [33], including endometrial cancers [34, 35]. Ninety percent of endometrial cancers have been found to express telomerase. Telomerase is comprised of an RNA template (hTR) and the catalytic protein hTERT which has reverse transcriptase activity. hTERT is considered to be the rate-limiting factor in the formation of functional telomerase. In this study, metformin was found to decrease hTERT expression in the endometrial cancer cell lines, which to our knowledge is the first time metformin has been linked to regulation of telomerase activity.
Regulation of hTERT transcription during the cell cycle is controversial with some investigators reporting high activity in S phase and undetectable levels in G2-M phase and others demonstrating no change throughout the cell cycle [36, 37]. If hTERT transcription is cell cycle dependent and non-cycling cells do not express hTERT, then the effect of metformin on hTERT expression may potentially occur as an indirect consequence of cell cycle arrest rather than a direct effect on hTERT transcription. We have previously demonstrated that the mTOR inhibitor, rapamycin, profoundly suppresses telomerase activity via inhibition of hTERT mRNA expression in endometrial, ovarian and cervical cancer cell lines [28, 29]. However, in cell lines that were resistant to rapamycin's anti-proliferative effects and failed to undergo G1 arrest, rapamycin still decreased hTERT expression, suggesting that rapamycin's effect on regulation of hTERT expression was independent of its ability to induce cell cycle arrest [29]. Thus, we concluded that rapamycin may regulate hTERT mRNA expression through an alternative pathway downstream from mTOR, unrelated to cell cycle control. This concept may also apply to metformin, given its known interaction with the mTOR pathway, but this remains to be proven.
We should acknowledge that the doses of metformin (0.01 – 5 mM) used in these in vitro studies are supratherapeutic compared to those doses used in diabetic patients. This has been a previously raised criticism of similar work in other cancer cell types, such as breast, prostate and colon cancer cell lines [24-27]. The maximum recommended clinical metformin dose is 2250 mg/day. In clinical pharmacokinetic studies, therapeutic levels measured in healthy volunteers range from 0.5 to 2.0 mg/L (peak plasma levels 0.6 to 1.8 ug/mL), and other investigators have calculated 20 uM as a clinically equivalent dose in vitro [38, 39]. However, it is important to consider that cells in culture are grown under hyperglycemic conditions in an environment of overabundant nutrients. Tissue culture media alone contains high concentrations of glucose, and 5-10% fetal bovine serum is typically added, resulting in excessive growth stimulation. This, of course, is an inherent limitation to in vitro studies using cell lines and may be a viable explanation for why higher doses are needed to see the effects of metformin in cell culture than what is typically used in diabetic patients.
Metformin has also been shown to accumulate in tissues at higher concentrations than in blood, suggesting that increased concentrations of metformin may be achieved in target organs at lower, more pharmacologic doses [40]. Thus, despite the discrepancy in dosing between these in vitro studies and clinical metformin use for diabetic treatment, metformin's anti-tumorigenic potential more than deserves a thorough assessment for endometrial cancer prevention and treatment. These issues in regards to dosing may be more accurately addressed in vivo using animal models prior to validation in clinical trials, which is one of our future research goals.
A well-established phenomenon of tumor cells is a shift in glucose metabolism from oxidative phosphorylation to aerobic glycolysis, termed the “Warburg” effect [41]. The PI3K/Akt/mTOR pathway is known to play a crucial role in both growth control and glucose metabolism. PI3K signaling through Akt can modulate glucose transporter expression, stimulate glucose capture by hexokinase and increase phosphofructokinase activity [41]. In support of this theory, response to anti-cancer therapies, such as tyrosine kinase inhibitors, can often be predicted by the decreased metabolism of glucose by tumors as measured by FDG-PET [41]. Thus, it is intriguing to postulate that an additional anti-tumorigenic benefit of metformin may be through regulation of glucose uptake and utilization by endometrial cancer cells. We plan to investigate this relationship between metformin cell signaling and glucose metabolism as part of our future work.
Since obesity and diabetes have been linked to an increased risk of mortality from endometrial cancer, patients after diagnosis are strongly encouraged to make lifestyle changes such as diet, weight reduction and exercise. Unfortunately, these lifestyle changes are challenging for patients, and pharmacologic intervention through metformin may be an innovative chemotherapeutic strategy to both treat endometrial cancer and improve outcomes for these “high risk” women by impacting insulin resistance. Furthermore, we postulate that maintenance therapy with metformin after endometrial cancer diagnosis and treatment may prevent recurrences in this unique patient population where obesity and diabetes are such predominant risk factors. This concept is even more intriguing given that metformin has been shown to improve outcomes in diabetic cancer patients as compared to sulfonylureas or insulin use [7, 8]. Thus, metformin may emerge as an effective targeted therapy in the treatment and long-term management of endometrial cancer, with the additional benefits of low cost, oral route of administration, proven safety and very little toxicity. We hope that this work will begin to provide the scientific foundation for future clinical trials of metformin for endometrial cancer treatment and ultimately, prevention.
Acknowledgments
This work was generously supported by the UNC Clinical Translational Science Award - K12 Scholars Program (KL2 RR025746), the V Foundation for Cancer Research and the Steelman Fund. The project described was supported by Award Number KL2RR025746 from the National Center for Research Resources. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Footnotes
This work was presented at the 2009 Annual Meeting of the Society of Gynecologic Oncologists, San Antonio, TX.
Conflict of Interest Statement: We, the authors of this manuscript, have no financial or personal relationships to disclose that could inappropriately influence or bias this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fader AN, Arriba LN, Frasure HE, von Gruenigen VE. Endometrial cancer and obesity: epidemiology, biomarkers, prevention and survivorship. Gynecol Oncol. 2009;114(1):121–7. doi: 10.1016/j.ygyno.2009.03.039. [DOI] [PubMed] [Google Scholar]
- 2.von Gruenigen VE, Gil KM, Frasure HE, Jenison EL, Hopkins MP. The impact of obesity and age on quality of life in gynecologic surgery. Am J Obstet Gynecol. 2005;193(4):1369–75. doi: 10.1016/j.ajog.2005.03.038. [DOI] [PubMed] [Google Scholar]
- 3.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58(2):71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 4.Chia VM, Newcomb PA, Trentham-Dietz A, Hampton JM. Obesity, diabetes, and other factors in relation to survival after endometrial cancer diagnosis. Int J Gynecol Cancer. 2007;17(2):441–6. doi: 10.1111/j.1525-1438.2007.00790.x. [DOI] [PubMed] [Google Scholar]
- 5.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348(17):1625–38. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 6.Steiner E, Plata K, Interthal C, et al. Diabetes mellitus is a multivariate independent prognostic factor in endometrial carcinoma: a clinicopathologic study on 313 patients. Eur J Gynaecol Oncol. 2007;28(2):95–7. [PubMed] [Google Scholar]
- 7.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. Bmj. 2005;330(7503):1304–5. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care. 2006;29(2):254–8. doi: 10.2337/diacare.29.02.06.dc05-1558. [DOI] [PubMed] [Google Scholar]
- 9.Shaw RJ, Lamia KA, Vasquez D, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310(5754):1642–6. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dronavalli S, Ehrmann DA. Pharmacologic therapy of polycystic ovary syndrome. Clin Obstet Gynecol. 2007;50(1):244–54. doi: 10.1097/GRF.0b013e31802f35a0. [DOI] [PubMed] [Google Scholar]
- 11.Hadad SM, Fleming S, Thompson AM, Targeting AMPK. A new therapeutic opportunity in breast cancer. Crit Rev Oncol Hematol. 2008 doi: 10.1016/j.critrevonc.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 12.Carling D. LKB1: a sweet side to Peutz-Jeghers syndrome? Trends Mol Med. 2006;12(4):144–7. doi: 10.1016/j.molmed.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 13.Contreras CM, Gurumurthy S, Haynie JM, et al. Loss of Lkb1 provokes highly invasive endometrial adenocarcinomas. Cancer Res. 2008;68(3):759–66. doi: 10.1158/0008-5472.CAN-07-5014. [DOI] [PubMed] [Google Scholar]
- 14.Lu KH, Wu W, Dave B, et al. Loss of tuberous sclerosis complex-2 function and activation of mammalian target of rapamycin signaling in endometrial carcinoma. Clin Cancer Res. 2008;14(9):2543–50. doi: 10.1158/1078-0432.CCR-07-0321. [DOI] [PubMed] [Google Scholar]
- 15.Mutter GL, Lin MC, Fitzgerald JT, et al. Altered PTEN expression as a diagnostic marker for the earliest endometrial precancers. J Natl Cancer Inst. 2000;92(11):924–30. doi: 10.1093/jnci/92.11.924. [DOI] [PubMed] [Google Scholar]
- 16.An HJ, Lee YH, Cho NH, et al. Alteration of PTEN expression in endometrial carcinoma is associated with down-regulation of cyclin-dependent kinase inhibitor, p27. Histopathology. 2002;41(5):437–45. doi: 10.1046/j.1365-2559.2002.01455.x. [DOI] [PubMed] [Google Scholar]
- 17.Terakawa N, Kanamori Y, Yoshida S. Loss of PTEN expression followed by Akt phosphorylation is a poor prognostic factor for patients with endometrial cancer. Endocr Relat Cancer. 2003;10(2):203–8. doi: 10.1677/erc.0.0100203. [DOI] [PubMed] [Google Scholar]
- 18.Bieche I, Nogues C, Paradis V, et al. Quantitation of hTERT gene expression in sporadic breast tumors with a real-time reverse transcription-polymerase chain reaction assay. Clin Cancer Res. 2000;6(2):452–9. [PubMed] [Google Scholar]
- 19.Jefferies HB, Fumagalli S, Dennis PB, et al. Rapamycin suppresses 5′TOP mRNA translation through inhibition of p70s6k. Embo J. 1997;16(12):3693–704. doi: 10.1093/emboj/16.12.3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Podsypanina K, Lee RT, Politis C, et al. An inhibitor of mTOR reduces neoplasia and normalizes p70/S6 kinase activity in Pten+/- mice. Proc Natl Acad Sci U S A. 2001;98(18):10320–5. doi: 10.1073/pnas.171060098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi Y, Gera J, Hu L, et al. Enhanced sensitivity of multiple myeloma cells containing PTEN mutations to CCI-779. Cancer Res. 2002;62(17):5027–34. [PubMed] [Google Scholar]
- 22.Grunwald V, DeGraffenried L, Russel D, et al. Inhibitors of mTOR reverse doxorubicin resistance conferred by PTEN status in prostate cancer cells. Cancer Res. 2002;62(21):6141–5. [PubMed] [Google Scholar]
- 23.Neshat MS, Mellinghoff IK, Tran C, et al. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc Natl Acad Sci U S A. 2001;98(18):10314–9. doi: 10.1073/pnas.171076798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zakikhani M, Dowling RJ, Sonenberg N, Pollak MN. The effects of adiponectin and metformin on prostate and colon neoplasia involve activation of AMP-activated protein kinase. Cancer Prev Res (Phila Pa) 2008;1(5):369–75. doi: 10.1158/1940-6207.CAPR-08-0081. [DOI] [PubMed] [Google Scholar]
- 25.Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006;66(21):10269–73. doi: 10.1158/0008-5472.CAN-06-1500. [DOI] [PubMed] [Google Scholar]
- 26.Gotlieb WH, Saumet J, Beauchamp MC, et al. In vitro metformin anti-neoplastic activity in epithelial ovarian cancer. Gynecol Oncol. 2008;110(2):246–50. doi: 10.1016/j.ygyno.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 27.Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007;67(22):10804–12. doi: 10.1158/0008-5472.CAN-07-2310. [DOI] [PubMed] [Google Scholar]
- 28.Zhou C, Gehrig PA, Whang YE, Boggess JF. Rapamycin inhibits telomerase activity by decreasing the hTERT mRNA level in endometrial cancer cells. Mol Cancer Ther. 2003;2(8):789–95. [PubMed] [Google Scholar]
- 29.Bae-Jump VL, Zhou C, Gehrig PA, Whang YE, Boggess JF. Rapamycin inhibits hTERT telomerase mRNA expression, independent of cell cycle arrest. Gynecol Oncol. 2006;100(3):487–94. doi: 10.1016/j.ygyno.2005.08.053. [DOI] [PubMed] [Google Scholar]
- 30.Alimova IN, Liu B, Fan Z, et al. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle. 2009;8(6) doi: 10.4161/cc.8.6.7933. [DOI] [PubMed] [Google Scholar]
- 31.Ben Sahra I, Laurent K, Loubat A, et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27(25):3576–86. doi: 10.1038/sj.onc.1211024. [DOI] [PubMed] [Google Scholar]
- 32.Buzzai M, Jones RG, Amaravadi RK, et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67(14):6745–52. doi: 10.1158/0008-5472.CAN-06-4447. [DOI] [PubMed] [Google Scholar]
- 33.Stewart SA, Weinberg RA. Telomerase and human tumorigenesis. Semin Cancer Biol. 2000;10(6):399–406. doi: 10.1006/scbi.2000.0339. [DOI] [PubMed] [Google Scholar]
- 34.Yokoyama Y, Takahashi Y, Shinohara A, Lian Z, Tamaya T. Telomerase activity in the female reproductive tract and neoplasms. Gynecol Oncol. 1998;68(2):145–9. doi: 10.1006/gyno.1997.4921. [DOI] [PubMed] [Google Scholar]
- 35.Zheng PS, Iwasaka T, Yamasaki F, et al. Telomerase activity in gynecologic tumors. Gynecol Oncol. 1997;64(1):171–5. doi: 10.1006/gyno.1996.4523. [DOI] [PubMed] [Google Scholar]
- 36.Zhu X, Kumar R, Mandal M, et al. Cell cycle-dependent modulation of telomerase activity in tumor cells. Proc Natl Acad Sci U S A. 1996;93(12):6091–5. doi: 10.1073/pnas.93.12.6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holt SE, Aisner DL, Shay JW, Wright WE. Lack of cell cycle regulation of telomerase activity in human cells. Proc Natl Acad Sci U S A. 1997;94(20):10687–92. doi: 10.1073/pnas.94.20.10687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scheen A. Clinical pharmacokinetics of metformin. Clin Pharmacokinet. 1996;30(5):359–71. doi: 10.2165/00003088-199630050-00003. [DOI] [PubMed] [Google Scholar]
- 39.Isoda K, Young JL, Zirlik A, et al. Metformin inhibits proinflammatory responses and nuclear factor-kappaB in human vascular wall cells. Arterioscler Thromb Vasc Biol. 2006;26(3):611–7. doi: 10.1161/01.ATV.0000201938.78044.75. [DOI] [PubMed] [Google Scholar]
- 40.Wilcock C, Bailey CJ. Accumulation of metformin by tissues of the normal and diabetic mouse. Xenobiotica. 1994;24(1):49–57. doi: 10.3109/00498259409043220. [DOI] [PubMed] [Google Scholar]
- 41.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]