

Abstract
Human respiratory syncytial virus (RSV) is a major cause of severe lower respiratory tract infection in infants, immunocompromised patients, and the elderly. The RSV fusion (F) protein mediates fusion of the viral envelope with the target cell membrane during virus entry and is a primary target for antiviral drug and vaccine development. The F protein contains two heptad repeat regions, HR1 and HR2. Peptides corresponding to these regions form a six-helix bundle structure that is thought to play a critical role in membrane fusion. However, characterization of six-helix bundle formation in native RSV F protein has been hindered by the fact that a trigger for F protein conformational change has yet to be identified. Here we demonstrate that RSV F protein on the surface of infected cells undergoes a conformational change following exposure to elevated temperature, resulting in the formation of the six-helix bundle structure. We first generated and characterized six-helix-bundle-specific antibodies raised against recombinant peptides modeling the RSV F protein six-helix bundle structure. We then used these antibodies as probes to monitor RSV F protein six-helix bundle formation in response to a diverse array of potential triggers of conformational changes. We found that exposure of ‘membrane-anchored’ RSV F protein to elevated temperature (45-55°C) was sufficient to trigger six-helix bundle formation. Antibody binding to the six-helix bundle conformation was detected by both flow cytometry and cell-surface immunoprecipitation of the RSV F protein. None of the other treatments, including interaction with a number of potential receptors, resulted in significant binding by six-helix bundle-specific antibodies. We conclude that native, untriggered RSV F protein exists in a metastable state that can be converted in vitro to the more stable, fusogenic six-helix bundle conformation by an increase in thermal energy. These findings help to better define the mechanism of RSV F-mediated membrane fusion and have important implications for the identification of therapeutic strategies and vaccines targeting RSV F protein conformational changes.
Keywords: respiratory syncytial virus, paramyxovirus, fusion protein, cell fusion, six-helix bundle, heptad repeat, receptor
INTRODUCTION
Human respiratory syncytial virus (RSV) is an enveloped negative-sense ssRNA virus of the pneumovirus group in the paramyxoviridae family (Collins et al., 2001). RSV is a major cause of pediatric respiratory disease, resulting in severe lower respiratory tract infection in infants, immunocompromised patients, and the elderly (Collins et al., 2001). There are no effective vaccines available for RSV. Currently approved antiviral therapy for RSV infection is limited to ribavirin, a broad-spectrum antiviral drug with limited efficacy. Prophylactic treatment with the anti-RSV monoclonal antibody, palivizumab, is effective in reducing the severity of disease but is only recommended for high-risk patients (Johnson et al., 1997; Groothuis et al., 1993; Pollack and Groothuis, 2002). Therefore development of new therapies for the treatment of RSV infection is a high priority.
RSV enters cells by binding to the target cell surface and fusing its envelope with the plasma membrane of the target cell. There are three viral glycoproteins on the envelope surface: the attachment glycoprotein (G), the fusion (F) protein, and the small hydrophobic (SH) protein (Heminway et al., 1994). However, several lines of evidence indicate that only F, the fusion protein, is both necessary and sufficient for virus entry and cell fusion (Karron et al., 1997; Bukreyev et al., 1997; Kahn et al., 1999; Teecharpornkul et al., 2001). Thus, the critical role of the F protein in virus entry, along with the fact that F is highly conserved and is a major antigen for neutralizing antibodies, makes it an attractive therapeutic target (Johnson et al., 1987; Eckert et al., 1999; Collins et al., 2001, Zhao et al., 2000).
The RSV F protein is synthesized as a 70 kDa precursor (F0) that is processed by proteolytic cleavage in the trans-Golgi complex to yield two disulphide-linked subunits: F1 and F2 (Fig. 1A; Anderson et al., 1982; Collins and Mottet 1991; Collins et al., 1984). The mature F protein is expressed as a homotrimer on the cell-surface (Calder et al., 2000). The ectodomain of the F1 transmembrane subunit contains two heptad repeat regions, HR1 and HR2, and an N-terminal fusion peptide (Fig. 1A) (Collins et al., 2001). Processing of the F0 precursor has been proposed to generate a “metastable” F1/F2 complex (Zhao et al., 2000). In one proposed model, receptor binding triggers conformational changes in the metastable F protein, resulting in insertion of the N-terminal fusion peptide into the target cell membrane. The HR1 and HR2 heptad repeat regions then interact to form a six-helix bundle structure (Carr et al., 1997; Eckert and Kim, 2001). According to this model, formation of the six-helix bundle structure facilitates membrane fusion by bringing the viral and cellular membranes into close apposition (Chan and Kim, 1998; Eckert and Kim, 2001; Russell et al., 2001). The presence of heptad repeat regions that are capable of associating to form a six-helix bundle structure is a feature of several enveloped virus fusion proteins, suggesting a common membrane fusion mechanism (Lamb, 1993; Hernandez et al., 1996; Melikyan et al., 2000; Skehel and Wiley, 2000; Lamb and Kolakofsky, 2001). In support of this model, peptides corresponding to the RSV F protein HR1 and HR2 regions (N and C peptides, respectively) have been shown to form a six-helix coiled-coil bundle structure, wherein three C peptides pack in an antiparallel fashion into the outer grooves of a central N peptide trimeric coiled-coil (Fig. 3B) (Zhao et al., 2000; Matthews et al., 2000; Lawless-Delmedico et al., 2000). This model is also supported in part by the fact that C peptides are often potent inhibitors of fusion mediated by viral fusion proteins, including the RSV F protein (Lambert et al., 1996; Wang et al., 2003). In the case of HIV, C peptide inhibitors have been shown to interact with the HR1 region of the HIV gp41 fusion protein during an intermediate stage of conformational change to form a dead-end peptide-gp41 hybrid six-helix bundle structure (Kilgore et al., 2003). One HIV C peptide inhibitor, enfuvirtide, has been approved for the treatment of HIV infection. Thus, inhibition of viral fusion protein six-helix bundle formation is a validated therapeutic approach for the treatment of viral disease (Wild et al., 1992, 1994; Chan and Kim, 1998; Eckert et al., 1999; Zhao et al., 2000).
FIG. 1. Construction of RSV Fusion (F) protein heptad repeat peptide immunogens.



A) Heptad repeat (HR) domains and other structural features in the RSV Fusion protein. Key domains of the F1 subunit are shown, including the heptad repeats, HR1 (or N region) and HR2 (or C region), the fusion peptide (FP), and the transmembrane domain (TM). In the F2 subunit, the location of an additional predicted heptad repeat, HR3 is shown. Cleavage sites designate the location of furin cleavage sites in F2 and the F2-F1 junction, respectively. The location of conserved glycosylation sites are denoted by lollipop-shaped structures in F2 and F1.
B) Codon optimization of HR1/HR2 cDNA in an RSV peptide expression construct. The nucleotide sequence of the codon-optimized HR1/HR2 cDNA is shown. The wild type RSV A2 strain F gene nucleotides that were replaced to permit optimal expression in E.coli are italicized. The HR1 region (nt 13-187, encoding F residues 153-209) and HR2 region (nt 232-366, encoding F residues 476-520) are joined by a linker region (underlined sequence). Also shown are the locations of restriction enzyme cloning sites and the IEGR-Factor Xa cleavage sequence. The individual HR1 and HR2 cDNA clones have sequences identical to the corresponding regions shown here. The cDNAs expressing RSV peptides NC, N, or C were cloned into the Bam HI-Hind III window of the pET-32a protein expression plasmid.
C) Sequence of recombinant RSV peptides (N, C or single chain NC peptide) generated in the study. The lengths and predicted molecular weights of the peptides are shown.
FIG. 3. Assessment of immune response against the immunogen using a peptide ELISA.



A) ELISA titers demonstrate the immunogenicity of peptide antigen NC or N+C. Antibody response of individual guinea pigs immunized with immunogen NC (guinea pig #82, #83, and #84) or N+C (guinea pig #85, #86, and #87) indicated that antisera #84(NC) and #87 (N+C) displayed the highest titers.
B) Antisera αNC (#84) and αN+C (#87) are qualitatively identical in that they display the highest reactivity to peptide-complexes, NC or N+C, rather than to either of the free peptides N or C. The αNC antiserum displayed the lowest cross-reactivity to the individual N or C free peptides. Data are representative of two experiments. The dilution of αNC antibody used was 1:10,000.
C) Peptide-adsorption ELISA demonstrates the specificity of αNC antiserum to RSV 6HB epitope. Adsorption of αNC antiserum with peptide complex NC or N+C abolishes the reactivity to NC (6HB) peptide. Adsorption with individual free peptide N or C does not significantly affect the reactivity to NC (6HB) peptide. The dilution of αNC antibody used was 1:10,000.
The description of a six-helix bundle structure formed by N and C peptides derived from RSV F protein, along with the fact that RSV C peptides potently inhibit fusion, suggest that RSV F protein undergoes conformation changes during fusion that are analogous to those described in the HIV, SV5, influenza, and related viral systems. Electron microscopy studies of RSV F protein have revealed that trypsin treatment causes a change in conformation from a lollipop-shaped structure to a cone-shaped structure, which are thought to represent the native and fusion-active states, respectively (Calder et al., 2000). However, it has yet to be demonstrated directly that native, membrane-anchored RSV F protein can be triggered to undergo conformational changes leading to the formation of a six-helix bundle structure. Indeed, it is not yet known what triggers these conformational changes. Since, like HIV, RSV fusion takes place at the cell surface and is pH independent (Srinivasakumar et al., 1991; Collins et al., 2001), it is presumed that binding of F protein to a cellular receptor triggers conformational changes. Although several ligands have been identified that bind to RSV F protein, it remains unclear whether any one of these ligands is a bona fide receptor for RSV (Pastey et al., 2000; Behera et al., 2001; Sano et al., 2003; Hallak et al., 2000). The lack of a system in which to study native RSV F protein six-helix bundle formation hinders the development of therapeutics targeting F protein conformational changes.
In the present study, we generated conformation-specific antibodies to recombinant peptide immunogens modeling the RSV F protein six-helix bundle structure. We show that a low level of RSV F protein in the six-helix bundle conformation exists on the surface of RSV-infected cells in a culture that is actively undergoing fusion. We utilized the six-helix-bundle-specific antibodies as probes to characterize potential triggers of F protein conformational change. We determined that elevated temperature serves as an efficient in vitro trigger of F protein conformation change leading to six-helix bundle formation. The implications of these findings for the development of antiviral therapeutics or vaccines targeting the RSV F protein are discussed.
RESULTS
Expression of immunogens modeling the RSV F protein six-helix bundle
In order to generate antibodies against RSV 6HB, peptides corresponding to the HR regions of RSV F protein were expressed to generate immunogens that mimic the RSV-F fusion core 6HB structure.
We first engineered codon-optimized synthetic bacterial expression constructs expressing a linked N and C single-chain peptide (NC), the N peptide, or the C peptide using sequences derived from the work of Zhao et al. (Zhao et al., 2000) (Figure 1). In each synthetic cDNA construct, selected nucleotide residues in the HR1 and HR2 coding sequences from wild-type RSV F cDNA (Accession # VGNZA2) were replaced with residues that favor optimal expression in E. coli (Fig. 1B). The recombinant RSV peptides (N, C, and NC single-chain) were expressed as histidine (His)-tagged fusion peptides in E. coli (Fig 1B). The His-tag was cleaved off with factor-Xa, the peptides were desalted, lyophilized and resuspended at a concentration of ~0.5 mg/ml in water.
The linked single-chain NC peptide or a 1:1 mixture of individual N and C peptides (N+C) are hypothesized to form a complex in vitro that mimics the authentic 6HB structure formed due to coiled-coil interactions of RSV-F HR α-helical domains during the fusion process. The 14-amino-acid-long glycine-rich peptide inserted between the N and C domains allows for folding and interaction between the arms of the NC peptide (Fig. 1C).
Characterization of recombinant RSV peptides
The recombinant RSV peptides were analyzed by SDS-PAGE before and after factor-Xa cleavage to determine their approximate molecular weights. The larger size of the His-tagged peptides is due to the presence of uncleaved TRX, Thrombin, and S-Tags encoded in the pET-32a leader sequence that precedes the engineered IEGR (Factor Xa)-RSV peptides (N, C, and NC) (Fig. 1C). Factor-Xa enzyme-mediated cleavage of the His-tagged peptides releases free-N, C, or NC peptide, respectively (Fig 1C). The predicted molecular weights of the N (57aa), C (45aa), and NC (116 aa) peptides based on their amino acid (aa) sequences were 6.13, 5.11, and 12.21 kDa, respectively (Fig. 1C). The migration pattern of the RSV peptides relative to a 36-amino-acid (4.5 kDa) control peptide (DP-178) and molecular weight markers coincided well with their predicted molecular weights (compare Fig 2A with Fig 1C).
FIG. 2. Characterization of the recombinant RSV HR peptides.
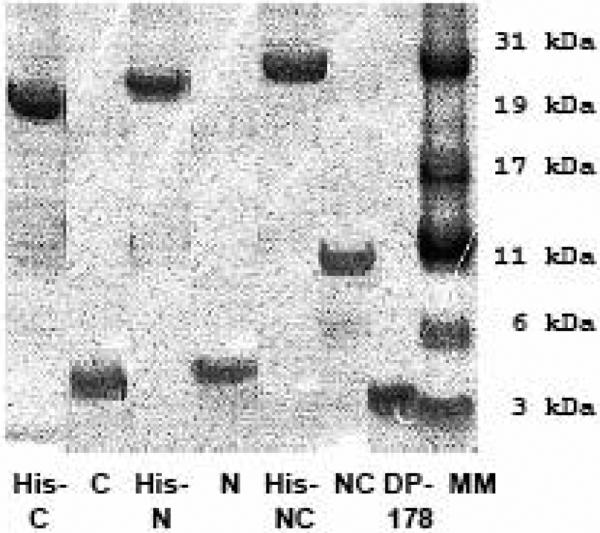

A) Coomasie stained polyacrylamide gel showing the molecular weights of the histidine-tagged fusion peptide and purified RSV peptides (N, C, or NC) released by factor Xa cleavage of the corresponding histidine-tagged fusion proteins. A commercially synthesized HIV peptide, DP-178 (~4.5kd in size), was included for comparison.
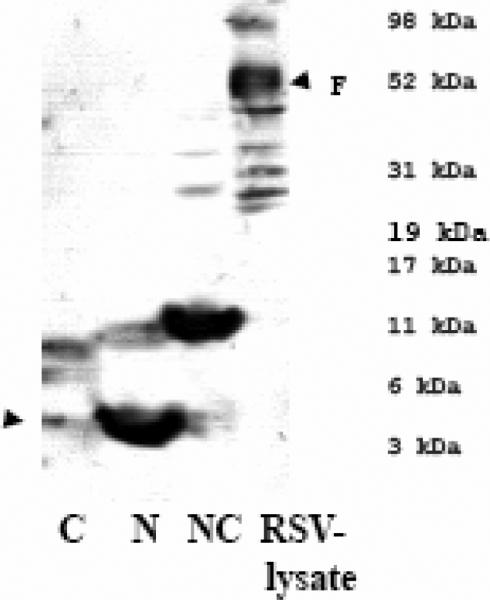
B) αRSV serum recognizes the peptides, N and NC but not the C peptide in an immunoblot. The arrow indicates the predicted migration for the C-peptide that was not recognized by αRSV serum.
C) Circular Dichroism spectra of the NC single-chain peptide are consistent with a 6HB solution structure. The double minima at 222 and 208 nm are characteristic of the high level of helical secondary structure found in a 6HB.
To determine whether antibodies to RSV could recognize the recombinant peptides, an immunoblot of the recombinant peptides was probed using RSV neutralizing polyclonal serum. The αRSV polyclonal serum recognized the N and NC peptides thus confirming that these peptides contained authentic F protein epitopes (Fig. 2B). Surprisingly, the C peptide did not appear to react with αRSV serum; only faint background bands were seen, likely attributed to cross-reactivity with contaminating bacterial proteins. One possible explanation for this observation is the presence of a conserved N-linked glycosylation site (aa: 500NQS502) within the HR2 (C-peptide region) domain in the Fusion protein (Figs. 1A and 1C, Collins et al., 2001, Zimmer et al., 2001). Perhaps the presence of the carbohydrate side chain makes this region poorly antigenic. Alternatively, the HR2-C region may be buried within the native, untriggered RSV F protein oligomer and inaccessible to circulating antibodies. Studies of soluble F proteins suggest that this latter explanation is unlikely, since the HR2 region is proposed to form a stalk structure in the pre-fusion state (Lamb and Jardetzky, 2007).
Antiviral activity of RSV peptides
The C (HR2) and N (HR1) peptides have been reported to potently inhibit RSV fusion and infection (Lambert et al 1994; Gao et al. 2003). To determine whether the peptides that we generated had antiviral activity, we tested each peptide (N, C, NC, or an N+C mixture) for its ability to inhibit RSV infection and syncytia formation in vitro in a dose-response assay. Consistent with previous observations (Lambert et al., 1996; Wang et al., 2003), both the N and C peptides were found to inhibit RSV infection (data not shown). Also consistent with previous reports, the C peptide was found to be more potent than the N peptide (IC50 = 8.0 μg/ml [1.6 μM] and 62.5 μg/ml [10.2 μM], respectively). The existence of a prehairpin intermediate conformation for RSV F1 is strongly supported by our observation and by others that RSV infection can be potently inhibited by HR-C peptides (Lambert et al., 1996; Wang et al., 2003).
We further found that the linked peptide NC did not exhibit any noticeable antiviral activity (data not shown). Similarly, mixing N- and C-peptides together in equimolar ratio (N+C) resulted in the loss of antiviral activity of either peptide, presumably due to 6HB-like structure formation (data not shown).
Circular Dichroism reveals that the RSV peptide NC or an equimolar mixture of N and C peptides (N+C) assumes a helicity characteristic of a six-helix bundle structure
Circular dichroism (CD) was performed to determine whether the peptides NC or N+C have the solution structures similar to those predicted for the RSV fusion core from previous studies (Matthews et al., 2000; Lawless-Delmedico et al., 2000; Zhao 2000). The formation of the 6HB structure is marked by a signature-increase in helicity that can be detected by CD spectroscopy upon mixing of individual N and C peptides (Zhao et al., 2000; Matthews et al., 2000; Lawless-Delmedico et al., 2000; Wild et al., 1992). We therefore analyzed the helicity of the NC peptide (or a 1:1 mixture of N and C, [N+C]). The CD spectra for a 10μM sample of peptide NC in PBS at 4 °C, displayed a double minima at 222 and 208nm that is characteristic of the high degree of α-helical secondary structure found in the 6HB complex (Fig 2C). The mean molar elipticity value of −27,000 at 222nm indicates that under the conditions of the experiment the protein is ~85% helical. Similar results were obtained with a 1:1 mixture of the individual N- and C-peptide constructs (data not shown). These data indicate that the complex formed by the recombinant RSV peptides, NC or N+C, models the 6HB structure in a manner similar to peptides utilized in previous studies (Lawless-Delmedico et al., 2000; Matthews et al., 2000, Zhao et al., 2000).
Peptide ELISA demonstrates that the antibodies generated against recombinant peptide complexes are specific for 6HB epitopes
Having established that these peptides possess the characteristics of a 6HB structure, the recombinant peptide complexes NC or N+C were used as immunogens to raise antibodies that would recognize the 6HB epitope generated during the RSV F-mediated fusion process. Three guinea pigs were immunized with each immunogen (NC or N+C mixed peptides). We have used this approach previously to generate antibodies against the HIV gp41 6HB structure that forms following triggering with CD4 (deRosny et al., 2001; Furuta et al., 1998).
A peptide-based indirect ELISA was used for initial characterization of the antiserum. The peptides NC or N+C were highly immunogenic and yielded relatively high-titer antiserum (Fig 3A), which we designated as α-NC or α-N+C. The antibody responses were directed primarily against peptide complexes NC or N+C rather than individual peptides N or C (Fig. 3B). Furthermore the reactivity of antiserum α-NC (or α-N+C) to peptides NC or N+C could be completely adsorbed out by either immunogens (NC or N+C) but not by free peptides N or C (Fig 3C). These observations demonstrate that α-NC antibodies predominantly recognize 6HB-specific epitopes. The αNC or α-N+C antiserum did not react with a peptide complex corresponding to HIV 6HB (P15+P16) indicating its specificity to RSV 6HB (data not shown). Our results are also supported by findings in the HIV system, wherein a polyclonal 6HB-specific antiserum displayed similar specificity to a gp41 6HB peptide complex (deRosny et al., 2001). Collectively, these results demonstrate that the antiserum generated against the NC or N+C peptides was predominantly complex-specific and therefore suitable to probe for RSV-F 6HB structure. Since the αNC antibodies displayed a higher titer (Fig 3A) as well as lower cross reactivity to free peptides N or C (Fig 3B), this antiserum (84F = αNC; Fig 3A) was used for further characterization.
αNC recognizes RSV F protein in lysate strips as well as lysate immunoprecipitation
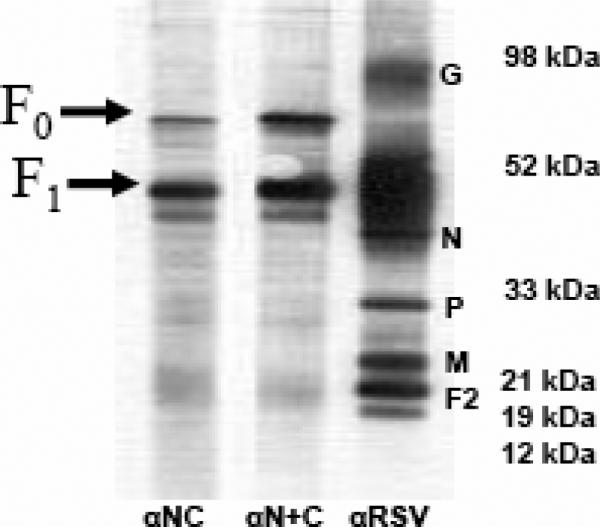
The antiserum αNC (or αN+C), when used to probe lysate strips prepared from RSV infected cells, reacted to protein with molecular weight ~48-50 kDa (F1 subunit) predominantly and to an ~70 kDa (F0) protein (Fig 4A), suggesting that these antisera recognized epitopes in the RSV F protein.
FIG. 4. αNC antiserum recognizes epitopes located in the F1 subunit.

A) αNC antiserum recognizes RSV F protein in RSV-lysate western blot strips. The reactivity of αRSV serum is shown as a control.
B) αNC antiserum selectively immunoprecipitates F1 subunit from RSV-infected cell lysate. Pre-immunization serum (PI) from guinea pig #84 is run as a negative control. RSV proteins immunoprecipitated by two RSV αF monoclonal antibodies (858-1-Sigma and 3216-Serotec) and polyclonal αRSV serum were run as controls.
In immunoprecipitation (IP) experiments, the αNC (or αN+C) antiserum immunoprecipitated F1 subunit from RSV-infected cell lysate, but no F0 form whatsoever was observed (Fig.4B). These results are consistent with the αNC antibody epitopes being located in the F1 subunit (Fig. 4). The lack of binding to F0 was interesting, since studies of soluble hPIV3 F protein suggest that cleavage of the F precursor is not required for adoption of a post-fusion conformation (Lamb and Jardetzky, 2007). It may be that the requirements for 6HB formation differ between soluble and native, membrane-bound F proteins (Ruiz-Arguello et al., 2004; Yin et al., 2005, 2006). Conversely, the reactivity of the αNC antibody to the F0 form of RSV F protein in the western blot (Fig 4A) could either be non-specific or result from small amounts of antibodies against uncomplexed N and C components that bind to denatured F0 (and, potentially, F1). Collectively, the above findings, along with the results of the peptide ELISA, strongly suggest that the F1 epitope recognized by the αNC antiserum is the RSV 6HB structure (Fig. 3 and Fig. 4).
Cell-surface IP adsorption experiment demonstrates the specificity of antibodies for the RSV F six-helix bundle
We next employed cell-surface immunoprecipitation to determine if 6HB epitopes could be found on the surface of RSV-infected cells that are actively undergoing cell fusion. Using the αNC serum, F1 subunit was immunoprecipitated from the surface of RSV infected cells (Fig 5B). In order to determine if the F1 being immunoprecipitated from the cell-surface was indeed a 6HB conformation of F1, we adsorbed the αNC antiserum with peptides N, C, NC, or N+C, respectively, and used this serum in cell-surface IP experiments. Our results show that adsorption of the antiserum with NC (or N+C; data not shown) peptides that mimic the RSV F 6HB structure completely abolished the capacity of αNC antiserum to immunoprecipitate F1 protein. Whereas, adsorption with peptides N or C alone did not prevent F1 binding (Fig. 5B).
FIG. 5. Immunoprecipitation of F protein from the surface of RSV-infected cells using αNC antibodies.

Cell-surface IP demonstrates the presence of 6HB epitopes on the surface of RSV-infected cells actively undergoing fusion. Cell-surface reactivity to these epitopes can be adsorbed out with NC or N+C peptide mixtures but not with individual N or C peptides.
The peptide-adsorption cell-surface IP results, along with peptide-ELISA binding data, demonstrate that α-NC antibodies are conformation-specific for 6HB epitopes and, therefore, can be used to detect the final conformation state of the RSV F protein. Our results provide proof-of-concept that, starting with synthetic-peptide immunogens that mimic the RSV F 6HB structure, it is possible to generate antibodies that specifically bind the 6HB epitopes being created on the surface of RSV-infected cells undergoing fusion.
Search for a method to trigger six-helix bundle formation
Using flow cytometry, the staining of RSV-infected cells with αNC antiserum detected the presence of a low level of F protein in the 6HB conformation on the cell-surface (Fig 6A). The mean fluorescence due to antibody binding to 6HB epitopes was significantly lower in comparison with the total F protein detected using an α-F monoclonal antibody (Fig. 6B). These data generated using flow cytometry correlated well with cell-surface IP results and suggested that a low level of F protein 6HB exists on the surface of RSV-infected cells.
FIG. 6. Detection of F protein 6HB epitopes on the surface of RSV-infected cells.

Flow cytometry of αNC antibody-stained intact cells demonstrates:
A) A relatively low amount of F protein is recognized by anti-6HB (αNC) antiserum on the surface of RSV-infected cells.
B) By comparison, a large amount of total F protein is detected on RSV-infected cells using an α-F monoclonal antibody.
Using flow cytometry, we rapidly ruled out the possibility that the following conditions / reagents would trigger a conformation change in cell-surface expressed RSV F into its fusion active six-helix bundle formation: 1) addition of reported F-interacting proteins such as RhoA or ICAM-1 or glycosaminoglycans (such as heparin, heparan sulphate, dextran sulphate or Chondroitin B); 2) human or bovine lactoferrin, reported to interact with G or F glycoprotein; 3) exposure to different pH (3.2-11.0) or trypsin or disulphide bond reducing agents (β-Mercaptoethanol or dTT) or denaturing agent (SDS) (data not shown).
Temperature as an efficient trigger of RSV F protein six-helix bundle formation
Previous work using a soluble form of the F protein from parainfluenza virus type 5 (PIV5) demonstrated an effect of temperature on the conformational state of the protein (Connolly et al., 2006). To determine if elevated temperature could trigger 6HB formation in native RSV F protein, we incubated RSV-infected cells for 5 min at 4 °C or 37 °C or a range of temperature from 37 °C to 65 °C prior to staining with 6HB antibody. Following incubation at temperatures between 45 °C - 55 °C, a specific increase in staining of RSV-infected cells, but not uninfected control cells, was observed (Fig. 7A and data not shown). Repeated testing of the effects of temperature on triggering during a 5 min incubation using a thermocycler revealed that triggering reaches a plateau between 50 °C and 55 °C (Fig. 7A). At 55 °C the level of staining by 6HB-specific antibody was almost equivalent to that observed with an anti-F monoclonal antibody positive control (data not shown), suggesting that a large proportion of the total population of F protein expressed on the cell surface was triggered under these conditions.
FIG. 7. Identification of temperature as a trigger of 6HB formational change in a flow cytometry-based triggering assay.



A) Temperature course (5 min incubation) @ 37 °C, 40 °C, 42.5 °C, 45 °C, 47.5 °C, 50 °C, 52.5 °C, 55 °C, 57.5 °C, 60 °C. Temperature dependent triggering starts around 42.5 °C and plateaus between 47.5 °C and 55 °C, where a maximum signal to noise ratio of 2.4-2.5 times that observed at 37 °C (1.4) is achieved.
B) Time course @ 55 °C for 5 min, 15 min, 30 min, 45 min, 60 min. 6HB formation occurs rapidly within the first 5 minutes of exposure to 55 °C.
C) Loss of staining of α-NC antiserum upon adsorption with a mixture of N+C peptides but not with N or C peptide alone. Triggering was performed for 15 min at 55 °C.
In order to identify the optimal time for triggering RSV infected Vero cells were incubated at 55 °C for various lengths of time up to an hour prior to staining. Exposure to 55 °C for a 15 min duration resulted in the most dramatic/rapid increase in triggering (Fig. 7B). Only small and gradual increases in triggering were observed beyond 15 min exposure up to 45 minutes, and then this effect subsided. These data, along with the results of 5-min temperature-course experiments (Fig. 7A), demonstrated that a short incubation of 5-15 minutes at 50 – 55 °C was sufficient to trigger optimal 6HB formation on the surface of RSV-infected cells.
Triggering was compared for four different cell lines: Vero, Hep2, HeLa and Hela-S3. The results demonstrated that the best combination of F expression and triggering signal-to-noise was observed with Vero cells (data not shown).
In order to confirm that the 55 °C incubation was not having deleterious effects on cell-membranes, RSV-infected Vero cells treated at 55 °C for ½ hour were stained with antibodies that recognize two intracellular constituents: 1) Rho A (found on the intracellular face of the plasma membrane) or 2) the Golgi-complex (Sigma Golgi-specific antibody #2404). We observed that flow cytometry staining to RhoA or the Golgi-complex was dramatically increased in uninfected Vero cells following treatment with detergent-containing buffer (Perm 2, BD Biosciences); whereas, no increase was observed following incubation at 55 °C for ½ hr in the absence of detergent (data not shown). These results demonstrated that significant cell-permeabilization was not occurring at 55 °C, and the triggering detected at that temperature was, instead, due to 6HB formation in cell-surface expressed F protein. The specificity of the staining following a 55 °C trigger was further confirmed in experiments which showed that no significant staining above background was observed following adsorption of antibodies to 6HB complexes in an N+C peptide mixture (Fig 7C).
Cell-surface IP demonstrates a temperature-dependent increase in triggering
A cell-surface immunoprecipitation experiment was performed in which 6HB-specific αNC antiserum was incubated with RSV-infected cells at increasing temperatures for 15 min. Following this triggering step, unbound antibody was washed away, and the immune complexes were then immunoprecipitated. As is shown in Fig. 8A, the amount of RSV F protein on the cell surface that was recognized by αNC antiserum was significantly increased at temperatures above 37° C, with the highest level being pulled down at 50-55° C. These results were consistent with the results presented in Fig. 7 from flow cytometry experiments and confirmed that the increased antibody binding detected at elevated temperature was specific for the RSV F protein.
FIG. 8. Cell-surface IP demonstrates temperature-induced triggering of six-helix bundle formation in cell-surface-expressed F protein.


A) 6HB formation increases as a function of increasing temperature. A qualitative increase in F1 subunit immunoprecipitation was observed in the samples triggered at 55 or 50 °C for 15 min (lanes 6 & 7) as compared to those triggered at 37 °C (lane 5). The amount of F1 subunit immunoprecipitated following 45 °C triggering appeared to be somewhat lower than that observed at 50-55 °C, however, it still appeared to be higher than that observed at 37 °C or 40 °C (compare lane 8 with lanes 5 and 9, respectively). Lane 1 is a control showing F expression in RSV-infected cells immunoprecipitated with an anti-F monoclonal antibody (858-1). The pre-immunization serum (PI) did not immunoprecipitate any relevant proteins either in uninfected or RSV-infected cells (lanes 2 and 3).
B) Specificity of the α-NC antiserum binding at elevated temperature for 6HB epitope. Increased amount of 6HB formed during elevated temperature can be adsorbed out with corresponding 6HB peptides. The α-NC antiserum was preadsorbed with 5 μg each of peptides NC, N, C, or N+C, respectively or PBS (-) overnight at 4 °C and used in triggering experiments at 55 °C for 15min as described above. The NC and N+C peptides appeared to almost completely adsorb out the reactivity of α-NC antiserum at 55 °C (compare lanes 3 and 6 with lane 2). The adsorption of α-NC antiserum with N peptide alone (lane 4) or C peptide alone (lane 5) did not appear to significantly impair their ability to immunoprecipitate F1 subunit.
To confirm the specificity of the antibody binding to the six-helix bundle conformation of RSV F following temperature triggering, a cell-surface IP experiment was performed in which 6HB-specific NC antiserum was pre-incubated with either the N, C, or NC single-chain peptide or a mixture of the N and C peptides (N+C) prior to incubation with RSV-infected cells at 55° C for 15 min. As is shown in Fig.8B, virtually all of the reactivity of the NC antiserum for temperature-triggered RSV F protein was adsorbed out by the NC single-chain 6HB peptide or an equimolar mixture of N+C peptide complexes; whereas, the N or C peptide alone failed to adsorb out all of the antibodies. These results confirm that elevated temperature triggers RSV F protein 6HB formation on the surface of infected cells.
DISCUSSION
In this study, we describe the generation of reagents and identification of a system that permits the study of 6HB formation directly in native, membrane-anchored F protein expressed on the surface of RSV-infected cells. Using conformation-specific antibodies to the RSV F 6HB structure, we directly demonstrate that elevated temperature is an efficient trigger of F protein conformational change. The flow cytometry-based assay described in this report is an attractive starting point for the development of novel strategies for discovery of RSV fusion inhibitors. We have previously used a similar approach successfully to study HIV-1 gp41 6HB formation following triggering with CD4 and to identify small molecule inhibitors of HIV-1 fusion (Salzwedel et al., unpublished results, Furuta et al., 1998; deRosny et al., 2001).
The physiologic trigger(s) that activate conformational changes in RSV F are not known. The activation of most paramyxovirus F proteins at neutral pH is triggered by a three-step process including 1) binding of a viral receptor-binding protein such as HN or H to its cellular receptor; 2) interaction of HN with F, and 3) conformational changes in F that mediate membrane fusion (Crennell et al., 2000; Russell et al., 2001; Takimoto et al., 2002; Colman and Lawrence, 2003). Coexpression of homotypic receptor binding protein is required for efficient fusion activity of F (review see Lamb & Kolakofsky, 2001). In the case of RSV, G protein mediates attachment; however G is not absolutely essential for fusion (Karron et al., 1997; Bukreyev et al., 1997; Kahn et al., 1999; Teecharpornkul et al., 2001). The RSV F protein alone can mediate infection and syncytia formation entirely in the absence of G, suggesting that the RSV-F1 + F2 complex, like HIV gp120-gp41 carries all the domains necessary to mediate membrane fusion (Collins et al., 2001; Karron et al., 1997; Bukreyev et al., 1997; Kahn et al., 1999; Teecharpornkul et al., 2001). In the case of HIV, the fusion protein, gp41, is triggered by binding of its associated subunit (gp120) to CD4 receptor on the target cell surface (Hernandez et al., 1996). Since, like HIV, RSV fusion takes place at the cell surface and is pH independent (Collins et al., 2001), it is likely that F protein also interacts with a cell-surface receptor, although such a receptor has not yet been identified.
The conformational change in HIV-1 gp41 leading to 6HB formation can be triggered in vitro in HIV-1-infected cells by addition of soluble CD4 and detected with 6HB-specific antibodies generated by peptide immunization (Furuta et al., 1998; de Rosny et al., 2001; Kilgore et al., 2003, Salzwedel et al., unpublished results). In the absence of a known receptor that can trigger RSV F, we looked at other viral systems with similar mechanism of fusion and tested several possible 6HB triggers in a flow cytometry assay. After a thorough literature search we selected the following approaches with some rationale for testing in the flow cytometry assay: Rho A (Pastey et al., 2000); ICAM-1 (Behera et al., 2001); human/bovine lactoferrin (Sano et al., 2003); Iduronic acid containing glycosaminoglycans (GAG) including dextran sulfate, chondroitin sulfate, heparin sulfate and heparin (Hallak et al., 2000).; trypsin treatment to enhance F2 internal cleavage (Gonzalez-Reyes et al., 2001; Zimmer et al., 2001); pH range 3.2 -11.0 (Hernandez et al., 2001; Mothes et al., 2000; Sturman et al., 1990; Zelus et al., 2003; Hsu et al., 1982; Holmes and Choppin 1966; Plemper et al., 2003); Destabilizing F2-F1 arrangement with dithiothreitol(dTT) and β-mercaptoethanol (β-ME) (Schlender et al., 2003; Plemper et al., 2003; Barbouche et al., 2003; Gallina et al., 2002) and mild denaturant, 0.05% sodium dodecyl sulfate (SDS). None of these approaches resulted in a clear, consistent triggering effect (data not shown).
We could not test hydrostatic pressure for its reported effect to induce fusion active state due to the need for specialized equipment (Gaspar et al., 2002). Similarly we could not test urea or guanidine HCl in our system due to their deleterious effect on cells (Ruigrok et al., 1986; Paterson et al., 2000; Wharton, et al., 2000).
A few early studies discovered that the low pH triggering effect for influenza hemagglutinin (HA) could be substituted by elevated temperature in the range of 58-62 °C (Wharton, 1986; Haywood and Boyer, 1986; Ruigrok, 1987). The heat-triggered conformational change in HA coincided with the formation of a fusogenic 6HB conformation that is biochemically indistinguishable from the low pH-triggered conformation (Carr et al., 1997). These studies suggested to us that elevated temperature could potentially serve as a trigger for RSV F protein. Using the αNC antibody as a probe, we observed in flow cytometry and cell-surface IP experiments that exposure of RSV infected cells to a temperature of 45-55 °C for 5-15 min was sufficient to cause a 3-5-fold increase in the formation of 6HB conformation of F (Figs. 7B and 7C). We concluded that elevated temperature could serve as an efficient in vitro trigger of 6HB formation in RSV-F.
One caveat of this study is that we cannot completely rule out the possibility that the α-NC antibodies may recognize a conformation of the HR2 domain that is not represented by the free C peptide. Such an epitope could potentially be unmasked by partial denaturation during incubation at elevated temperature and may not faithfully represent a conformation of the F protein that is related to authentic conformational changes. Alternatively, it is possible that the antibodies may detect an intermediate conformation of the F protein rather than the post-fusion 6HB structure. However, we consider these scenarios to be unlikely given the results obtained.
Our finding is consistent with similar observations in the other paramyxoviruses (Wharton et al., 2000; Patterson et al., 2000; Seth et al., 2003; Russell et al., 2001). Notably, Wharton et al. (2000) observed that following exposure to 55 °C, Sendai virus Fusion protein assumes the 6HB conformation. Pre-incubation of Sendai virus at that temperature (55 °C) blocked fusion by inactivating the F protein, presumably by pre-triggering 6HB formation in the absence of the target membrane (Wharton et al., 2000). Patterson and co-workers working with SV5 –F mutants that required coexpression of homotypic HN protein to promote membrane fusion, demonstrated that the requirement of HN for F triggering can be replaced by elevated temperature (Patterson et al., 2000). More recently, for a newly identified paramyxovirus – SER virus (a rubula virus) – elevated temperature in the range of 55-65 °C could enhance hemifusion, cytoplasmic content mixing and syncytium formation in dye-transfer experiments between SER F-expressing cells and dye-labeled guinea pig RBC (Seth et al., 2003).
More recently, Connolly et al. (2006) engineered a soluble form of the parainfluenza virus 5 Fusion protein in its pre-fusion state, wherein the function of the transmembrane anchor domain (TM) was preserved by replacing it with a trimerization domain (PIV5 F-GCNt). The authors demonstrated that this form of the protein could be triggered to undergo conformational change to its more stable post-fusion state by heating to 50 °C (range: 45 °C - 60 °C). Upon triggering at 50 °C, PIV5-F GCNt associated with liposome membranes, formed rosettes, and lost or gained reactivity to conformation-specific antibodies that were predicted to recognize pre- or post-fusion conformational states, respectively. A change in the shape of the soluble protein from a prefusion “ball-and-stem” to a more elongated post-fusion “golf-tee” shape was also observed by electron microscopy (Connolly et al., 2006). Thus, our findings that elevated temperature serves as a surrogate trigger of RSV F conformational changes is consistent with previous studies of F protein conformational change in other paramyxovirus systems (Lamb and Jardetzky, 2007).
Study of the process of conformation change in fusion proteins from a diverse group of enveloped viruses including SV5, Influenza HA and HIV have lead to the development of a ‘spring-loaded trap’ model (Carr, 1993; Russell, 2003). In this model it is suggested that the non-fusogenic neutral pH conformation of viral fusion glycoproteins (vFGps), including the native F1-F2 complex in paramyxoviruses such as RSV, exist in a kinetically trapped high-energy state, and that energy is released as a result of conformation change in the protein. In agreement with the spring-trap model, at neutral pH, heat, urea, or pressure can ‘loosen’ the native non-fusogenic form, causing it to transition spontaneously into a fusogenic form, suggesting that the native state of vFGps is ‘metastable’ (Carr et al., 1993, 1997; Paterson et al., 2000; Wharton et al., 2000; Gaspar et al., 2000; Epand et al., 2002). These studies also propose that physiological triggers such as pH change or receptor binding fulfill a common energy requirement for performing the work of membrane fusion that could be satisfied directly by an increase in temperature or other destabilizing force such as urea or pressure (Patterson et al., 2000; Wharton et al., 1986, 2000; Carr et al., 1997; Haywood et al., 1986; Ruigrok et al., 1987; Epand et al., 2002; Seth et al., 2003). The energy released during the 6HB formation is used to perform the work of membrane fusion (Melikyan et al., 2000). Our demonstration here that heat can trigger the formation of the 6HB structure in the RSV fusion protein is consistent with the native state of RSV F protein existing in a metastable state. Our findings, therefore, extend the metastability hypothesis to RSV, a member of the pneumovirus genus in the family Paramyxoviridae.
The assay system described in this report has potential applications in the testing of future RSV receptor candidates by measuring their triggering effect on F protein conformational change. Similarly, this approach could be used to further study the requirements for 6HB formation in native RSV F. For example: Do RSV G or SH proteins have any influence on this effect? Does F0 undergo the same conformational change as cleaved F1? The thermal triggering approach could also be adapted to study the conformational changes of fusion proteins from other viruses for which receptors have yet to be identified. The triggering event and the conformational changes in the RSV F protein leading to 6HB formation are attractive antiviral targets, since disruption of these key steps can prevent F protein mediated membrane fusion. For treating HIV infection, a C peptide inhibitor (enfuvirtide; T-20) has been approved that targets the HIV gp41 fusion protein during an intermediate stage of conformational change to form a dead-end peptide-gp41 hybrid six-helix bundle structure (Wild et al., 1994; Kilgore et al., 2003; Matthews et al., 2004). Thus, inhibition of viral fusion protein six-helix bundle formation is a validated therapeutic approach for the treatment of viral disease. The RSV 6HB antibody probe and temperature triggering technique that we describe could be applied towards the development of high-throughput drug screening assays to identify small molecule drugs that specifically block the conformation changes in the fusion protein. Proof-of-concept for this idea has been provided by the identification of an influenza virus fusion inhibitor capable of blocking temperature-induced 6HB formation in influenza virus HA (Yoshimoto et al., 1999).
MATERIALS AND METHODS
Cells, viruses, and antibodies
Vero cells and the A2 strain of human RSV were obtained from the ATCC. Cells were cultured in Dulbecco's modified Eagle's medium with 10% fetal bovine serum containing antibiotics. Virus was propagated and titrated in Vero cells. The following antibodies were used: anti-RSV polyclonal antibodies (B65860G, Biodesign), anti-F protein mAb (RSV3216, Serotec), anti-F protein mAb (858-1, Chemicon), anti-G protein mAb (858-2, Chemicon), anti-RhoA mAb (26C4, sc-418, Santa Cruz Biotechnology), anti-Golgi mAb (G-2404, Sigma), donkey anti-goat IgG-HRP (AP180P, Chemicon).
Construction of synthetic genes encoding RSV F protein heptad repeat peptides
Synthetic genes encoding the heptad repeat regions, HR1 (N) and HR2 (C), of RSV F protein (A2 strain; accession number VGNZA2) were designed based on the work of Zhao et al. (Zhao et al., 2000) and codon optimized for optimal expression in Escherichia coli (Fig. 1). A cDNA construct (HR1/HR2) encoding both heptad repeat regions, HR1 (amino acid residues 153-209 of RSV F) and HR2 (amino acid residues 476-520 of RSV F), linked via a glycine-rich linker was generated (Fig 1, B and C). Briefly, a series of overlapping complementary oligonucleotides spanning the entire length of either the N or C coding sequence was designed. The codons within the oligo sequences were biased for optimal expression in E. coli (Fig. 1B). The oligos were annealed and extended in the appropriate order in successive PCR reactions to form two complete double-stranded DNAs encoding either N peptide (HR1) or C peptide (HR2). The resulting HR1 or HR2 PCR products were then re-amplified in a final PCR reaction using an upstream primer encoding a Bam HI-factor-Xa cleavage sequence (residues IEGR) at the 5’ end and a reverse primer encoding the restriction site Hind III at the 3’ end. The final PCR product from each reaction was cloned into the Bam HI-Hind III sites of the pET32a bacterial expression vector (Novagen) to generate the cDNAs HR1 or HR2, respectively. The linked HR1/HR2 cDNA (Fig 1B) was constructed by first amplifying the individual cDNA fragments, HR1 and HR2, in separate PCR reactions using complementary, overlapping primers that encoded each half of the intervening linker sequence, as well as a unique Kpn I site at their 5’ ends. The resulting PCR fragments encoding HR1 or HR2 were then ligated together at their Kpn I site to yield a fragment encoding both HR1 and HR2, joined via a glycine-rich linker (NC; Fig. 1C). The ligated HR1/HR2 was amplified once more in a PCR reaction using a primer encoding the 5’ end of HR1 and a primer encoding the 3’ end of HR2. The final product was cloned via Bam HI-Hind III into the pET32a vector to yield the HR1/HR2 cDNA that encoded the single-chain NC peptide (Fig. 1, B and C). The constructs HR1, HR2, and HR1/HR2 were sequenced in their entirety. The hexahistidine tag encoded in the pET32a vector allowed for histidine affinity purification of the fusion peptide.
Peptide expression and purification
The recombinant RSV peptides were expressed as histidine-tagged fusion proteins in E coli (Fig. 1, B, C and Fig. 2A). The plasmids HR1, HR2, or HR1/HR2 were transformed into E.coli strain pLysS (Novagen) for protein expression. Individual colonies of transformed bacteria were inoculated into Luria-Bertani (LB) broth and grown to an optical density of 0.6 at 600 nm. Protein expression was induced with 1 mM isopropyl-β-D-thiogalactopyranoside for 3 hr. The cells were harvested by pelleting in a centrifuge. The cell pellet was lysed with Benzonase reagent (Novagen), and lysate was clarified by centrifugation. The histidine-tagged fusion proteins in the lysates were captured on Ni+2 affinity columns. The eluted histidine-tagged fusion proteins were dialyzed against factor-Xa cleavage buffer (50mM Tris-HCl [pH 8.0]-100mM NaCl-2mM CaCl2) in order to set up factor-Xa digestion. The fusion proteins were digested with factor-Xa (1:500 wt/wt ratio of protease:tagged protein) for 24h at room temperature to remove the leader peptide and histidine tag residues. The leader peptide-histidine tag was removed from the cleavage mixture by passing the mixture over a Ni+2 affinity column; the flow-through contained the RSV peptides (N, C, or NC) (Fig. 2A). The N and NC peptides were further desalted by passing through PD-10 Sephadex columns. The peptides were lyophilized and resuspended in ultra pure water. The RSV peptides (N, C, or NC) were each adjusted to 0.5 mg/ml final concentration.
Confirmation of molecular weight and reactivity to RSV polyclonal serum by immunoblot
Purified N, C, or NC peptides, along with their histidine-tagged forms, were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) using 12% polyacrylamide Bis-Tris Novex gels (Invitrogen) alongside Multimark molecular weight marker and a commercially synthesized peptide of known molecular weight (~4.5 kDa). The proteins were detected by Coomassie blue staining.
In order to ascertain if the synthetic peptides could be recognized by anti-RSV (αRSV) antibodies, the purified N, C, or NC peptides were separated by SDS-PAGE as above, transferred to a nitrocellulose membrane, and probed with goat αRSV polyclonal serum. Antibody binding was detected using a horseradish peroxidase-conjugated anti-goat secondary antibody and the ECL chemiluminescent detection system (Amersham Pharmacia Biotech).
Determination of antiviral activity of peptides
The N, C, and NC peptides or a 1:1 equimolar mixture of N and C peptides (N+C) was tested in a dose-response assay for their ability to inhibit RSV-mediated cell fusion and infection in vitro using a modification of an RSV microneutralization assay (Johnson 1997). Briefly, 100 TCID50 of RSV was mixed with various concentrations of each RSV peptide. After a 1-hr pre-incubation at room temperature, the peptide/virus mixture was added to a Vero cell monolayer in a 96-well plate (2 × 104 cells/well). The plates were incubated at 37 °C for 5 days and then scored visually for inhibition of virus-mediated cell killing.
Circular dichroism studies
CD measurements were obtained with an Aviv Associates model 202 spectropolarimeter calibrated with a standard solution of 10-camphorsulfonic acid as per Wild et al., (1992). Samples contained 10-50 μM total concentration of peptide (either NC or the N+C peptide mixture) in 0.1 M NaCl / 10 mM potassium phosphate, adjusted to pH 7.0. Spectra was collected at 1 °C with 1.5 mM bandwidth, a 0.5 nm step size, and a 2.0-sec time constant in either 1- or 10-mm-path-length cells. Blank-corrected data was smoothed with a third-order polynomial function. A thermoelectric cell holder accurate to within 1 °C regulated sample temperature.
Immunization protocol
To generate antibodies against the RSV F protein 6HB structure, two groups of 3 guinea pigs were immunized with either the single-chain peptide, NC, or an equimolar (1:1) mixture of the N and C peptides, N+C. All of the animals were primed subcutaneously with 200 μg of total peptide in complete Freund's adjuvant and boosted three times with 100 μg peptide in incomplete Freund's adjuvant. The pre-immunization (PI) sera and final bleeds post–immunization were stored at –20 °C.
Peptide ELISA
An indirect ELISA was established to determine the reactivity of anti-NC (αNC) or anti-N+C (αN+C) antiserum to RSV peptides (N, C, NC, and the N+C mixture). 96-well Immulon-2 plates (VWR cat. # 62402-972) were coated with 100 ng/well of each peptide antigen. After blocking with 1% BSA, serum samples were added in duplicate and allowed to incubate for 2 hr at 37 °C. After incubation, the plates were washed in PBS-0.05% Tween-20 and a phosphatase-labeled anti-guinea pig secondary antibody was added at a 1:5000 dilution. After a 1-hr incubation at room temperature with the secondary antibody, substrate (fast p-nitrophenyl phosphate, Sigma) was added, and absorbance was detected at 405 nm in a plate reader.
RSV immunoblot
Immunoblot strips prepared from RSV-infected cell-lysate were probed with αNC or αN+C antiserum. Briefly, a lysate was prepared from RSV-infected Vero cells. RSV proteins in the lysate were separated by SDS-PAGE (12%). The proteins were then transferred to a nitrocellulose membrane and strips were prepared for probing individually with different antibodies to RSV proteins. The RSV lysate strips were probed with αN+C or αNC serum at a dilution of 1:500 in 5% milk-PBS. The strip-blots were probed with peroxidase-conjugated secondary antibody, followed by detection using the ECL chemiluminescent detection system.
Lysate Immunoprecipitation
The αNC and αN+C antisera were tested for their ability to immunoprecipitate RSV proteins from the lysates of RSV-infected cells. RSV-infected Vero cells were lysed in 0.2 ml lysis buffer (1% Nonidet P-40-150mM NaCl-100mM Tris [pH 8.0]) and clarified by centrifugation at 13000 rpm for 20 minutes. 2 μl of αNC or αN+C antiserum was added to clarified lysate and incubated for 1 hr at 4 °C. Protein G agarose beads (Invitrogen) were then added and incubated for 1 hr at 4 °C. The antigen-antibody-bead complexes were washed five times in wash buffer (0.1% SDS, 300mM NaCl- 0.5% IGEPAL, 50mM Tris [pH 7.5]) and resuspended in protein-loading buffer (Invitrogen). The immunoprecipitated proteins were separated by SDS-PAGE (10%). The proteins were then transferred to a nitrocellulose membrane, the membrane was blocked in blocking buffer (5% milk-PBS) and probed with a α-RSV polyclonal antibody (Biodesign International cat. # B65860G) at a 1:500 dilution in blocking buffer. The blots were washed and probed with a horseradish peroxidase-conjugated donkey anti-goat secondary antibody (Chemicon International cat. # AP180P) at a dilution of 1:10,000 in blocking buffer. The blots were washed again and the proteins were detected by chemiluminescence using the ECL chemiluminescent detection system.
Cell-surface immunoprecipitation
Cell-surface immunoprecipitation (IP) was used to selectively immunoprecipitate the fusion protein expressed on the cell-surface of RSV infected Vero cells. Vero cells were infected with RSV at 0.03-0.05 MOI. 48h post infection RSV infected Vero monolayer was washed three times with PBS and resuspended into a single-cell suspension using enzyme-free cell-dissociation buffer (Invitrogen). The cells were pelleted and resuspended in stain/wash buffer (SWB: PBS containing 1% BSA-0.1% NaN3) at a concentration of 2 × 107 cells/ml. 100 μl of cell suspension (2 × 106 cells) was used for each cell-surface IP. 2 μl of primary antibody, αNC or α-F monoclonal (Chemicon International # MAB858-1) was used in each IP. The antibody-cell mixture was incubated for 1h at 4 °C. Following this, the cells were washed twice with PBS and lysed in 0.2 ml lysis buffer. The clarified lysate was then processed and analyzed as described in lysate-IP experiment. The immunoprecipitated proteins were separated using SDS-PAGE (10%) and immunoblotting was performed as for the lysate IP described above.
In temperature triggering cell-surface IP experiments, a master mix was prepared wherein αNC antibody was added at the concentration of 2.0 μl /100 μl of RSV infected Vero cells (2 × 106 cells) resuspended in SWB. After thorough mixing, 100 μl aliquots were distributed into 0.5 ml PCR tubes for heat treatment in a thermocycler. The cells were exposed to elevated temperature in the range of 4 °C - 55°C (or the indicated temperature) for 5-15 min (or the indicated time period). Following temperature treatment, all subsequent steps of the cell-surface IP and immunoblotting were performed as described above.
Flow cytometry
Antibody staining and flow cytometry analysis was used to determine the level of F protein 6HB present on the surface of RSV-infected cells. RSV infected Vero cells were resuspended in stain/wash buffer at a concentration of 2 × 107 cells/ml. 50 μl (1 × 106 cells) of the cell-suspension was used for flow cytometry. A 1:50 dilution (2.0 μl) of αNC or control monoclonal antibodies αF or αG were used as primary antibodies for each flow cytometry sample. The cells were incubated with antiserum for 1 hr at 4 °C and then washed twice in stain/wash buffer. A secondary antibody, FITC-labeled α-guinea pig antibody at a final concentration of 1 μg/ml in stain/wash buffer was added to each sample and incubated for 45 min at 4 °C. The cells were washed twice and then resuspended in stain/wash buffer and analyzed using a Becton Dickinson FACSCalibur flow cytometer and CellQuest software.
Flow cytometry in the above format was used as a screening assay to search for potential trigger of RSV 6HB formation. We tested a wide variety of ligands and conditions such as recombinant RhoA protein (Cytoskeleton Inc. # RHO1-B) or recombinant ICAM-1(R&D Systems Inc # ADP4); Glycosaminoglycans such as heparin, dextran sulphate, chondroitin sulphate B (Sigma); Lactoferrin bovine or human (Sigma), pH in the range of 11-3.2, trypsin; disulphide bond reducing agents like dTT or β-Mercaptoethanol; denaturing agent such as SDS (Pastey et al., 2000; Behera et al., 2001; Sano et al., 2003; Hallak et al., 2000; Gonzales-Reyes et al., 2001; Zimmer et al., 2001; Schlender et al., 2003; Plemper et al., 2003; Barbouche 2003; Gallina et al., 2002). We exposed RSV infected Vero cells to elevated temperature (above 37 °C up to 65 °C) for varying time period in course of identifying temperature as a trigger of 6HB formation of RSV F protein
The integrity of the cells following triggering at 55 °C was further confirmed using control antibodies to intracellular proteins Rho A or Golgi complex. In order to compare the level of staining obtained at 55°C with that obtained following a true-cell membrane permeabilization effect, we used a cell-permeabilization buffer (Perm2 FACS-permeabilizing solution 2, Becton Dickinson) to permeabilize the cells prior to flow cytometry staining. 2 × 107 RSV infected Vero cells/ml or uninfected Vero cells were treated with 1× Perm 2 buffer as per manufacturer's recommendation. Briefly, the cells were exposed to 1× Perm 2 solution prepared in water for ½ hr, followed by 2 washes in stain/wash buffer and resuspended back into original cell-concentration (i.e., 2 × 107 cells/ml). A 50 μl (= 1×106 cells) aliquot of cells was used in each flow cytometry experiment as above. The staining to cellular protein ‘RhoA’ or a cell-organelle ‘Golgi complex’ was determined with monoclonal antibodies α-RhoA (Santa Cruz Biotechnology Inc. # SC418) or α-Golgi, (Sigma #2404), respectively.
ACKNOWLEDGMENTS
This work was supported by NIH grant AI052668 awarded to K. S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Anderson K, Stott EJ, Wertz GW. Intracellular processing of the human respiratory syncytial virus fusion glycoprotein: amino acid substitutions affecting folding, transport and cleavage. J Gen Virol. 1992;73(Pt 5):1177–88. doi: 10.1099/0022-1317-73-5-1177. [DOI] [PubMed] [Google Scholar]
- 2.Baker KA, Dutch RE, Lamb RA, Jaredtzky TS. Structural basis for paramyxovirus-mediated membrane fusion. Mol. Cell. 1999;3(3):309–319. doi: 10.1016/s1097-2765(00)80458-x. [DOI] [PubMed] [Google Scholar]
- 3.Barretto N, Hallak LK, Peeples ME. Neuraminidase treatment of respiratory syncytial virus-infected cells or virions, but not target cells, enhances cell-cell fusion and infection. Virology. 2003 Aug 15;313(1):33–43. doi: 10.1016/s0042-6822(03)00288-5. 2003. [DOI] [PubMed] [Google Scholar]
- 4.Begona Ruiz-Arguello M, Gonzalez-Reyes L, Calder LJ, Palomo C, Martin D, Saiz MJ, Garcia-Barreno B, Skehel JJ, Melero JA. Effect of proteolytic processing at two distinct sites on shape and aggregation of an anchorless fusion protein of human respiratory syncytial virus and fate of the intervenig segment. Virology. 2002 Jul 5;298(2):317–26. doi: 10.1006/viro.2002.1497. 2002. [DOI] [PubMed] [Google Scholar]
- 5.Behera AK, Matsuse H, Kumar M, Kong X, Lockey RF, Mohapatra SS. Blocking intercellular adhesion molecule-1 on human epithelial cells decreases respiratory syncytial virus infection. Biochem. Biophys. Res. Commun. 2001;280(1):188–95. doi: 10.1006/bbrc.2000.4093. [DOI] [PubMed] [Google Scholar]
- 6.Calder LJ, Gonzalez-Reyes L, Garcia-Barreno B, Wharton SA, Skehel JJ, Wiley DC, Melero JA. Electron microscopy of the human respiratory syncytial virus fusion protein and complexes that it forms with monoclonal antibodies. Virology. 2000;271:122–131. doi: 10.1006/viro.2000.0279. 25. [DOI] [PubMed] [Google Scholar]
- 7.Carr CM, Chaudhry C, Kim PS. Influenza hemagglutinin is spring-loaded by a metastable native conformation. Proc. Natl. Acad. Sci. USA. 1997;94:14306–14313. doi: 10.1073/pnas.94.26.14306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chambers P, Pringle CR, Easton AJ. Heptad repeat sequences are located adjacent to hydrophobic regions in several types of virus fusion glycoproteins. J. Gen. Virol. 1990;71:3075–3080. doi: 10.1099/0022-1317-71-12-3075. [DOI] [PubMed] [Google Scholar]
- 9.Chan DC, Kim PS. HIV entry and its inhibition. Cell. 1998;93:681–684. doi: 10.1016/s0092-8674(00)81430-0. [DOI] [PubMed] [Google Scholar]
- 10.Chen CH, Greenberg ML, Bolognesi DP, Matthews TJ. Monoclonal antibodies that bind to the core of fusion-active glycoprotein 41. J. Virol. 1995;69:1462–1472. doi: 10.1089/088922200750054765. [DOI] [PubMed] [Google Scholar]
- 11.Collins PL, Chanock RM, Murphy BR. Respiratory Syncytial Virus. In: Knipe DM, Howley PM, editors. Fields Virology. 4th edn Lippincot, Williams and Wilkins; Philadelphia, PA: 2001. [Google Scholar]
- 12.Collins PL, Huang YT, Wertz GW. Nucleotide sequence of the gene encoding the fusion (F) glycoprotein of human respiratory syncytial virus. Proc. Natl. Acad. Sci. USA. 1984;81:7683–7687. doi: 10.1073/pnas.81.24.7683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collins PL, Mottet G. Post-translational processing and oligomerization of the fusion glycoprotein of human respiratory syncytial virus. J.Gen.Virol. 1991;72:3095–3101. doi: 10.1099/0022-1317-72-12-3095. [DOI] [PubMed] [Google Scholar]
- 14.Connolly SA, Leser GP, Yin HS, Jardetzky TS, Lamb RA. Refolding of a paramyxovirus F protein from prefusion to postfusion conformations observed by liposome binding and electron microscopy. Proc. Natl. Acad. Sci. U S A. 2006;103:17903–17908. doi: 10.1073/pnas.0608678103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Rosny E, Vassell R, Wingfield PT, Wild CT, Weiss CD. Peptides corresponding to the heptad repeat motifs in the transmembrane protein (gp41) of human immunodeficiency virus type 1 elicit antibodies to receptor-activated conformations of the envelope glycoprotein. J Virol. 2001;75(18):8859–8863. doi: 10.1128/JVI.75.18.8859-8863.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eckert DM, Kim PS. Mechanism of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 2001;70:777–810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- 17.Eckert DM, Malashkevich VN, Hong LH, Carr PA, Kim PS. Inhibiting HIV-1 entry: discovery of D-peptide inhibitors that target the gp41 coiled-coil pocket. Cell. 1999;99:103–115. doi: 10.1016/s0092-8674(00)80066-5. [DOI] [PubMed] [Google Scholar]
- 18.Epand RM, Epand RF. Thermal denaturation of influenza virus and its relationship to membrane fusion. Biochem. J. 2002;365:841–848. doi: 10.1042/BJ20020290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feldman SA, Audet S, Beeler JA. The fusion glycoprotein of human respiratory syncytial virus facilitates virus attachment and infectivity via an interaction with cellular heparan sulfate. J Virol. 2000;74(14):6442–7. doi: 10.1128/jvi.74.14.6442-6447.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frey G, Rits-Volloch S, Zhang XQ, Schooley RT, Chen B, Harrison SC. Small molecules that bind the inner core of gp41 and inhibit HIV envelope-mediated fusion. Proc. Natl. Acad. Sci. U S A. 2006;103:13938–13943. doi: 10.1073/pnas.0601036103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Furuta RA, Wild CT, Weng Y, Weiss CD. Capture of an early fusion-active conformation of HIV-1 gp41. Nat. Struct. Biol. 1998;5(4):276–279. doi: 10.1038/nsb0498-276. [DOI] [PubMed] [Google Scholar]
- 22.Gallina A, Hanley TM, Mandel R, Trahey M, Broder CC, Viglianti GA, Ryser HJ. Inhibitors of protein-disulfide isomerase prevent cleavage of disulfide bonds in receptor-bound glycoprotein 120 and prevent HIV-1 entry. J Biol Chem. 2002;277(52):50579–88. doi: 10.1074/jbc.M204547200. [DOI] [PubMed] [Google Scholar]
- 23.Gaspar LP, Silva ACB, Gomes AMO, Freitas MS, Ano Bom APD, Schwarcz WD, Mestecky J, Novak MJ, Foguel D, Silva JL. Hydrostatic pressure induces the Fusion-active state of enveloped viruses. J. Biol. Chem. 2002;277:8433–8439. doi: 10.1074/jbc.M106096200. [DOI] [PubMed] [Google Scholar]
- 24.Gonzalez-Reyes L, Ruiz-Arguello MB, Garcia-Barreno B, Calder L, Lopez JA, Albar JP, Skehel JJ, Wiley DC, Melero JA. Cleavage of the human respiratory syncytial virus fusion protein at two distinct sites is required for activation of membrane fusion. Proc Natl Acad Sci U S A. 2001;98(17):9859–64. doi: 10.1073/pnas.151098198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gorny MK, Zolla-Pazner S. Recognition by human monoclonal antibodies of free and complexed peptides representing the prefusogenic and fusogenic forms of human immunodeficiency virus type 1 gp41. J. Virol. 2000;74:6186–6192. doi: 10.1128/jvi.74.13.6186-6192.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Groothuis JR, Simoes EA, Levin MJ, Hall CB, Long CE, Rodriguez WJ, Arrobio J, Meissner HC, Fulton DR, Welliver RC, et al. Prophylactic administration of respiratory syncytial virus immune globulin to high-risk infants and young children. The Respiratory Syncytial Virus Immune Globulin Study Group. N. Engl. J. Med. 1993;329:1524–1530. doi: 10.1056/NEJM199311183292102. [DOI] [PubMed] [Google Scholar]
- 27.Hallak LK, Collins PL, Knudson W, Peeples ME. Iduronic acid-containing glycosaminoglycans on target cells are required for efficient respiratory syncytial virus infection. Virology. 2000;271(2):264–75. doi: 10.1006/viro.2000.0293. [DOI] [PubMed] [Google Scholar]
- 28.Haywood AM, Boyer BP. Time and temperature dependence of influenza virus membrane fusion at neutral pH. J.Gen.Virol. 1986;67:2813–2817. doi: 10.1099/0022-1317-67-12-2813. [DOI] [PubMed] [Google Scholar]
- 29.Heminway BR, Yu Y, Tanaka Y, Perrine KG, Gustafson E, Bernstein JM, Galinski MS. Analysis of respiratory syncytial virus F, G, and SH proteins in cell fusion. Virology. 1994;200(2):801–5. doi: 10.1006/viro.1994.1245. [DOI] [PubMed] [Google Scholar]
- 30.Hernandez LD, Hoffman LR, Wolfsberg TG, White TG. Virus-cell and cell-cell fusion. Annu Rev Cell Dev Biol. 1996;12:627–661. doi: 10.1146/annurev.cellbio.12.1.627. [DOI] [PubMed] [Google Scholar]
- 31.Holmes KV, Choppin PW. On the role of the response of the cell membrane in determining virus virulence. Contrasting effects of the parainfluenza virus SV5 in two cell types. J Exp Med. 1966;124(3):501–520. doi: 10.1084/jem.124.3.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsu MC, Scheid A, Choppin PW. Enhancement of membrane-fusing activity of sendai virus by exposure of the virus to basic pH is correlated with a conformational change in the fusion protein. Proc. Natl. Acad. Sci. U S A. 1982;79(19):5862–5866. doi: 10.1073/pnas.79.19.5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson PR, Jr., Olmsted RA, Prince GA, Murphy BR, Alling DW, Walsh EE, Collins PL. Antigenic relatedness between glycoproteins of human respiratory syncytial virus subgroups A and B: evaluation of the contributions of F and G glycoproteins to immunity. J. Virol. 1987;61:3163–3166. doi: 10.1128/jvi.61.10.3163-3166.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson S, Oliver C, Prince GA, Hemming VG, Pfarr DS, Wang SC, Dormitzer M, O'Grady J, Koenig S, Tamura JK, Woods R, Bansal G, Couchenour D, Tsao E, Hall WC, Young JF. Development of a humanized monoclonal antibody (MEDI-493) with potent in vitro and in vivo activity against respiratory syncytial virus. J Infect Dis. 1997;176(5):1215–1224. doi: 10.1086/514115. [DOI] [PubMed] [Google Scholar]
- 35.Jiang S, Lin K, Lu M. A conformation-specific monoclonal antibody reacting with fusion-active gp41 from the human immunodeficiency virus type 1 envelope glycoprotein. J. Virol. 1998;72:10213–10217. doi: 10.1128/jvi.72.12.10213-10217.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang S, Lin K, Zhang L, Debnath AK. A screening assay for antiviral compounds targeted to the HIV-1 gp41 core structure using a conformation-specific monoclonal antibody. J Virol Methods. 1999;80(1):85–96. doi: 10.1016/s0166-0934(99)00041-5. [DOI] [PubMed] [Google Scholar]
- 37.Kahn JS, Schnell MJ, Buonocore L, Rose JK. Recombinant vesicular stomatitis virus expressing respiratory syncytial virus (RSV) glycoproteins: RSV fusion protein can mediate infection and cell fusion. Virology. 1999;254(1):81–91. doi: 10.1006/viro.1998.9535. [DOI] [PubMed] [Google Scholar]
- 38.Karron RA, Buonagurio DA, Georgiu AF, Whitehead SS, Adamus JE, Clements-Mann ML, Harris DO, Randolph VB, Udem SA, Murphy BR, Sidhu MS. Respiratory syncytial virus (RSV) SH and G proteins are not essential for viral replication in vitro: clinical evaluation and molecular characterization of a cold-passaged, attenuated RSV subgroup B mutant. Proc. Natl. Acad. Sci. U S A. 1997;94(25):13961–13966. doi: 10.1073/pnas.94.25.13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kilgore NR, Salzwedel K, Reddick M, Allaway GP, Wild CT. Direct evidence that C-peptide inhibitors of human immunodeficiency virus type 1 entry bind to the gp41 N-helical domain in receptor-activated viral envelope. J Virol. 2003;77(13):7669–7672. doi: 10.1128/JVI.77.13.7669-7672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lamb RA. Paramyxovirus fusion: a hypothesis for changes. Virology. 1993;197:1–11. doi: 10.1006/viro.1993.1561. [DOI] [PubMed] [Google Scholar]
- 41.Lamb RA, Kolakofsky D. Paramyxoviridae,: the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. 2nd ed. Lippincott, Williams and Wilkins; Philadelphia: 2001. pp. 1305–1340. [Google Scholar]
- 42.Lamb RA, Jardetzky TS. Structural basis of viral invasion: lessons from paramyxovirus F. Curr Opin Struct Biol. 2007;17(4):427–436. doi: 10.1016/j.sbi.2007.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lambert DM, Barney S, Lambert AL, Guthrie K, Medinas R, Davis DE, Bucy T, Erickson J, Merutka G, Petteway SR., Jr. Peptides from conserved regions of paramyxovirus fusion (F) proteins are potent inhibitors of viral fusion. Proc. Natl. Acad. Sci. U S A. 1996;93(5):2186–2191. doi: 10.1073/pnas.93.5.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lawless-Delmedico MK, Sista P, Sen R, Moore NC, Antczak JB, White JM, Greene RJ, Leanza KC, Matthews TJ, Lambert DM. Heptad-repeat regions of respiratory syncytial virus F1 protein form a six-membered coiled-coil complex. Biochemistry. 2000;39(38):11684–11695. doi: 10.1021/bi000471y. [DOI] [PubMed] [Google Scholar]
- 45.Levine S, Klaiber-Franco R, Paradiso PR. Demonstration that glycoprotein G is the attachment protein of respiratory syncytial virus. J. Gen.Virol. 1987;68:2521–2524. doi: 10.1099/0022-1317-68-9-2521. [DOI] [PubMed] [Google Scholar]
- 46.Matthews JM, Young TF, Tucker SP, Mackay JP. The core of the respiratory syncytial virus fusion protein is a trimeric coiled coil. J. Virol. 2000;74(13):5911–5920. doi: 10.1128/jvi.74.13.5911-5920.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matthews T, Salgo M, Greenburg M, Chung J, DeMasi R, Bolognesi D. Enfuvirtide: the first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat. Rev. Drug Discov. 2004;3(3):215–225. doi: 10.1038/nrd1331. [DOI] [PubMed] [Google Scholar]
- 48.McGinnes LW, Sergel T, Morrison TG. A single amino acid change in the Newcastle disease virus fusion protein alters the requirement for HN protein in fusion. J. Virol. 2000;74:5101–5107. doi: 10.1128/jvi.74.11.5101-5107.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McGinnes LW, Sergel T, Morrison TG. Mutations in the fusion peptide and adjacent heptad repeat inhibit folding or activity of the Newcastle disease virus fusion protein. J.Virol. 2001;75:7934–7943. doi: 10.1128/JVI.75.17.7934-7943.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Melikyan GB, Markosyan RM, Hemmati H, Delmedico MK, Lambert DM, Cohen FS. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J Cell Biol. 2000;151(2):413–423. doi: 10.1083/jcb.151.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mothes W, Boerger AL, Narayan S, Cunningham JM, Young JA. Retroviral entry mediated by receptor priming and low pH triggering of an envelope glycoprotein. Cell. 2000;103(4):679–89. doi: 10.1016/s0092-8674(00)00170-7. [DOI] [PubMed] [Google Scholar]
- 52.Olmsted RA, Elango N, Prince GA, Murphy BR, Johnson PR, Moss B, Chanock RM, Collins PL. Expression of the F glycoprotein of respiratory syncytial virus by a recombinant vaccinia virus: comparison of the individual contributions of the F and G glycoproteins to host immunity. Proc. Natl. Acad. Sci. U S A. 1986;83(19):7462–7466. doi: 10.1073/pnas.83.19.7462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pastey MK, Crowe JE, Jr., Graham BS. RhoA interacts with the fusion glycoprotein of respiratory syncytial virus and facilitates virus-induced syncytium formation. J Virol. 1999;73(9):7262–70. doi: 10.1128/jvi.73.9.7262-7270.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pastey MK, Gower TL, Spearman PW, Crowe JE, Jr., Graham BS. A RhoA-derived peptide inhibits syncytium formation induced by respiratory syncytial virus and parainfluenza virus type 3. Nat. Med. 2000;6(1):35–40. doi: 10.1038/71503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pollack P, Groothius JR. Development and use of palivizumab (Synagis): a passive immunoprophylactic agent for RSV. J. Infect. Chemother. 2002;8(3):201–206. doi: 10.1007/s10156-002-0178-6. [DOI] [PubMed] [Google Scholar]
- 56.Plemper RK, Lakdawala AS, Gernert KM, Snyder JP, Compans RW. Structural features of paramyxovirus F protein required for fusion initiation. Biochemistry. 2003;42(22):6645–55. doi: 10.1021/bi034385k. [DOI] [PubMed] [Google Scholar]
- 57.Ruiz-Arguello MB, Martin D, Wharton SA, Calder LJ, Martin SR, Cano O, Calero M, Garcia-Barreno B, Skehel JJ, Melero JA. Thermostability of the human respiratory syncytial virus fusion protein before and after activation: implications for the membrane-fusion mechanism. J Gen Virol. 2004;85(Pt 12):3677–87. doi: 10.1099/vir.0.80318-0. [DOI] [PubMed] [Google Scholar]
- 58.Ruigrok RW, Martin SR, Wharton SA, Skehel JJ, Bayley PM, Wiley DC. Conformation changes in the hemagglutinin of influenza virus which accompany heat-induced fusion of virus with liposomes. Virology. 1987;155(2):484–497. doi: 10.1016/0042-6822(86)90210-2. [DOI] [PubMed] [Google Scholar]
- 59.Russell CJ, Jardetzky TS, Lamb RA. Membrane fusion machines of paramyxoviruses: capture of intermediates of fusion. EMBO J. 2001;20(15):4024–4034. doi: 10.1093/emboj/20.15.4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sano H, Nagai K, Tsutsumi H, Kuroki Y. Lactoferrin and surfactant protein A exhibit distinct binding specificity to F protein and differently modulate respiratory syncytial virus infection. Eur. J. Immunol. 2003;33(10):2894–902. doi: 10.1002/eji.200324218. [DOI] [PubMed] [Google Scholar]
- 61.Seth S, Vincent A, Compans RW. Activation of fusion by the SER virus F protein: a low-pH-dependent Paramyxovirus entry process. J. Virol. 2003;77:6520–6527. doi: 10.1128/JVI.77.11.6520-6527.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sergel TA, McGinnes LW, Chen H, Hamo L, Schwertz S, Li D, Morrison TG. Mutational analysis of the membrane proximal heptad repeat of the Newcastle disease virus fusion protein. Virology. 2001;289:343–352. doi: 10.1006/viro.2001.1123. [DOI] [PubMed] [Google Scholar]
- 63.Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 2000;69:531–561. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 64.Srinivasakumar N, Ogra PL, Flanagan TD. Characteristics of fusion of respiratory syncytial virus with HEp-2 cells as measured by R18 fluorescence dequenching assay. J. Virol. 1991;65:4063–4069. doi: 10.1128/jvi.65.8.4063-4069.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sturman LS, Richard CS, Holmes KV. Conformational change of the coronavirus peplomer glycoprotein at pH 8.0 and 37 degrees C correlates with virus aggregation and virus-induced cell fusion. J Virol. Jun. 1990;64(6):3042–50. doi: 10.1128/jvi.64.6.3042-3050.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Teecharpornkul S, Barretto N, Peeples ME. Functional analysis of recombinant respiratory syncytial virus deletion mutants lacking the small hydrophobic and/or attachment glycoprotein gene. J Virol. 2001;75(15):6825–6834. doi: 10.1128/JVI.75.15.6825-6834.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Techaarpornkul S, Collins PL, Peeples ME. Respiratory syncytial virus with the fusion protein as its only viral glycoprotein is less dependent on cellular glycosaminoglycans for attachment than complete virus. Virology. 2002;294(2):296–304. doi: 10.1006/viro.2001.1340. [DOI] [PubMed] [Google Scholar]
- 68.Teng MN, Collins PL. Identification of the respiratory syncytial virus proteins required for formation and passage of helper-dependent infectious particles. J Virol. 1998;72(7):5707–5716. doi: 10.1128/jvi.72.7.5707-5716.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Takimoto T, Taylor GL, Connaris HC, Crennell SJ, Portner A. Role of hemagglutinin-neuraminidase protein in the mechanism of paramyxovirus-cell membrane fusion. J. Virol. 2002;76:13028–13033. doi: 10.1128/JVI.76.24.13028-13033.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Walsh EE, Hruska J. Monoclonal antibodies to respiratory syncytial virus proteins: identification of the fusion protein. J. Virol. 1983;47:171–177. doi: 10.1128/jvi.47.1.171-177.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang E, Sun X, Qian Y, Zhao L, Tien P, Gao GF. Both heptad repeats of human respiratory syncytial virus fusion protein are potent inhibitors of viral fusion. Biochem. Biophysical. Res. Communications. 2003;302:469–475. doi: 10.1016/s0006-291x(03)00197-9. [DOI] [PubMed] [Google Scholar]
- 72.Wharton SA, Skehel JJ, Wiley DC. Studies of influenza haemagglutination-mediated membrane fusion. Virology. 1986;149(1):27–35. doi: 10.1016/0042-6822(86)90083-8. [DOI] [PubMed] [Google Scholar]
- 73.Wharton SA, Skehel JJ, Wiley DC. Temperature dependence of fusion by sendai virus. Virology. 2000;271:71–78. doi: 10.1006/viro.2000.0280. [DOI] [PubMed] [Google Scholar]
- 74.Wild C, Oas T, McDanal C, Bolognesi D, Matthews T. A synthetic peptide inhibitor of human immunodeficiency virus replication: correlation between solution structure and viral inhibition. Proc. Natl. Acad. Sci. U S A. 1992;89(21):10537–10541. doi: 10.1073/pnas.89.21.10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wild CT, Shugars DC, Greenwell TK, McDanal CB, Matthews TJ. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. U S A. 1994;91(21):9770–9774. doi: 10.1073/pnas.91.21.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yin HS, Paterson RG, Wen X, Lamb RA, Jardetzky TS. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc. Natl. Acad. Sci. U S A. 2005;102(26):9288–9293. doi: 10.1073/pnas.0503989102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yin HS, Wen X, Paterson RG, Lamb RA, Jardetzky TS. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature. 2006;439(7072):38–44. doi: 10.1038/nature04322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yoshimoto J, Kakui M, Iwasaki H, Fujiwara T, Sugimoto H, Hattori N. Identification of a novel HA conformational change inhibitor of human influenza virus. Arch. Virol. 1999;144:865–878. doi: 10.1007/s007050050552. [DOI] [PubMed] [Google Scholar]
- 79.Zelus BD, Schickli JH, Blau DM, Weiss SR, Holmes KV. Conformational changes in the spike glycoprotein of murine coronavirus are induced at 37 degrees C either by soluble murine CEACAM1 receptors or by pH 8. J Virol. 2003;77(2):830–840. doi: 10.1128/JVI.77.2.830-840.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhao X, Singh M, Malashkevich VN, Kim PS. Structural Characterization of the human respiratory syncytial virus fusion protein core. Proc. Natl. Acad. Sci. USA. 2000;97(26):14172–14177. doi: 10.1073/pnas.260499197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zimmer G, Trotz I, Herrler G. N-glycans of F protein differentially affect fusion activity of human respiratory syncytial virus. J. Virol. 2001;75(10):4744–51. doi: 10.1128/JVI.75.10.4744-4751.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]