

Abstract
An accumulation of research over the years has demonstrated the utility of nanoparticles as antigen carriers with adjuvant activity. Herein we defined the adjuvanticity of a novel lecithin-based nanoparticle engineered from emulsions. The nanoparticles were spheres of around 200 nm. Model protein antigens, bovine serum albumin (BSA) or Bacillus anthracis protective antigen (PA) protein, were covalently conjugated onto the nanoparticles. Mice immunized with the BSA-conjugated nanoparticles developed strong anti-BSA antibody responses comparable to that induced by BSA adjuvanted with incomplete Freund's adjuvant and 6.5-fold stronger than that induced by BSA adsorbed onto aluminum hydroxide. Immunization of mice with the PA-conjugated nanoparticles elicited a quick, strong, and durable anti-PA antibody response that afforded protection of the mice against a lethal dose of anthrax lethal toxin challenge. The potent adjuvanticity of the nanoparticles was likely due to their ability to move the antigens into local draining lymph nodes, to enhance the uptake of the antigens by antigen-presenting cells (APCs), and to activate APCs. This novel nanoparticle system has the potential to serve as a universal protein-based vaccine carrier capable of inducing strong immune responses.
Keywords: Emulsions, adjuvanticity, antigen uptake, lymph nodes, anthrax
1. Introduction
Recombinant protein antigens may help overcome the toxicity concerns associated with many of the traditional vaccines prepared with live, attenuated or killed pathogens. However, recombinant proteins are often weakly immunogenic or non-immunogenic on their own, and a vaccine adjuvant is usually needed to enhance the resultant immune responses. Aluminum adjuvant, such as the aluminum hydroxide (Alum), remains to be the only adjuvant approved for human use in the U.S. It forms a precipitate when combined with soluble antigen, and the slow release of the antigen from the precipitate at the injection site causes prolonged, strong antibody responses [1-2]. However, Alum is a relatively weak adjuvant and has various limitations [2]. Therefore, there is a critical need to search for or to devise alternative vaccine adjuvants to improve the immune responses induced by recombinant protein antigens.
In recent years, particles of nanometer or micrometer scales are increasingly used as antigen carriers, and it is generally accepted that microparticles and nanoparticles have adjuvant activity [3-5], which is likely due to the easiness for antigen-presenting cells (APCs) to take up antigens associated with the particles, as compared to free antigens in solution. In theory, one expects that nanoparticles have a more potent adjuvant activity than microparticles. It was reported that particles with a diameter of 500 nm or less were optimal for uptake by APCs such as dendritic cells (DCs) and macrophages [6-7]. In addition, data from a recent study showed that small nanoparticles (20-200 nm) can freely drain to the lymph nodes (LNs) for antigen presentation, whereas DCs were required for the transport of large microparticles (0.5-2 μm) from the injection site to the LNs [8]. Therefore, it is likely that antigens associated with nanoparticles can reach the draining LNs for antigen presentation not only by the trafficking of APCs, but also by direct draining, which is expected to lead to a strong immune response. However, recent data from studies aimed at correlating the particle size of the antigen carriers and the resultant immune responses are rather controversial. For example, Wendorf et al. (2008) reported that comparable immune responses were induced in mice by protein antigens (Env from HIV-1 and MenB from Neisseria meningitides) adsorbed onto anionic microparticles (∼1 μm) and nanoparticles (110 nm) prepared with poly (lactic-co-glycolic acid) (PLGA) polymers [9]. However, data from a study by Gutierro et al. (2002) showed that when BSA was entrapped in PLGA particles of different sizes (200 nm, 500 nm, and 1 μm), the 1 μm microparticles generally induced a stronger serum anti-BSA IgG response than the 200 nm or 500 nm nanoparticles [10]. Similarly, Kanchan and Panda (2007) reported that the HBsAg entrapped in PLGA particles of 200-600 nm induced a weaker anti-HBsAg antibody response than HBsAg entrapped in PLGA particles of 2-8 μm, when injected intramuscularly into rats [7]. On the contrary, data from many other studies showed that the adjuvanticity of smaller nanoparticles was more potent than that of the larger microparticles using protein antigens or plasmid DNA [11-15]. More interestingly, Fifis et al. (2004) reported that the optimal carrier particle size was in the range of 40-50 nm when they compared the immune responses, both antibody and cellular, induced by polystyrene particles ranging from 20 nm to 2 μm with antigens conjugated on their surface [12]. However, the 40-50 nm was the size of the polystyrene particles before the protein antigen was conjugated onto their surface. When the OVA as an antigen was conjugated onto the 49 nm particles, the size of the resultant particles was reported to be 232 nm [13], which was more relevant. It is unclear why the conjugation of the OVA onto the 49 nm polystyrene particles quadrupled the diameter of the resultant particles. Nevertheless, it demonstrated that particles with a diameter of around 200 nm are ideal antigen carriers when the antigens are on the surface of the particles. Finally, it is becoming evident that the size of the particles may have different effects on the antigen-specific antibody or cellular immune responses[7, 16-17]. It seemed that microparticles promote humoral response, whereas nanoparticles (< 1 μm) promote cellular response [7]. For example, Caputo et al. (2009) showed that HIV TAT protein adsorbed on cationic polymeric nanoparticles (220 nm or 630 nm) tended to induce a stronger cellular immune response and a weaker antibody response than the same protein adsorbed on large microparticles (∼ 2 μm) made of the same materials [16]. To make it more interesting, data from Mann et al.'s recent study using oral bilosomes (bile salt in lipid vesicles) with influenza A antigens showed that the larger bilosomes (400-2000 nm) generated an immune response that had a significantly greater Th1 bias than the smaller bilosomes (10-100 nm) [17].
Previously, we reported the engineering of 100-200 nm nanoparticles using lecithin as the matrix material [18]. Lecithins are components of cell membranes and are regularly consumed as part of a normal diet. They are used extensively in pharmaceutical applications as emulsifying, dispersing, and stabilizing agents and are included in intramuscular and intravenous injectables and parenteral nutrition formulations [19]. In the present study, we modified the lecithin nanoparticles by including glyceryl monostearate (GMS) in the matrix and replacing the previous cationic surfactant with a non-ionic surfactant. We then evaluated the adjuvanticity of the modified nanoparticles in a mouse model using two model antigens, BSA or the Bacillus anthracis protective antigen protein. The antigens were chemically conjugated onto the surface of the nanoparticles because it was shown that when an antigen was covalently coupled to nanoparticles, it induced a stronger immune response than when it was simply adsorbed to the nanoparticles [12]. The anthrax PA protein was the functional antigen used in this study. Anthrax is a toxin-caused disease. There is no evidence that a cellular immune response is needed to control the anthrax toxins [20]. Therefore, we only focused on the evaluation of the antibody responses.
2. Materials and Methods
2.1. Engineering of nanoparticles from emulsions
Nanoparticles were prepared from emulsions. Briefly, soy lecithin (3.5 mg, Alfa Aesar, Ward Hill, MA) and GMS (0.5 mg, Gattefosse Corp., Paramus, NJ) were weighed into a 7-ml glass scintillation vial. One ml of de-ionized and filtered (0.2 μm) water was added into the vial, followed by heating on a hot plate to 70-75°C with stirring and brief intermittent periods of sonication (Ultrasonic Cleaner Model 150T, VWR International, West Chester, PA). Upon formation of a homogenous milky slurry, Tween 20 was added in a step-wise manner to a final concentration of 0-1.2% (v/v). The resultant emulsions were cooled to room temperature while stirring to form nanoparticles. The particle size was determined using a Coulter N4 Plus Submicron Particle Sizer (Beckman Coulter Inc., Fullerton, CA).
The conjugation of proteins (BSA from Sigma-Aldrich or PA from BEI resources, Manassas, VA) to the nanoparticles was completed as previously described [21]. A reactive maleimide group was incorporated into the nanoparticles by adding 5% (w/w) of 1, 2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[4-(p-maleimidophyl)butyramide] (Avanti Polar Lipids, Alabaster, AL) to form m-nanoparticles (m-NPs). The proteins were thiolated using 2-iminothiolane (Traut's reagent, Sigma-Aldrich). Briefly, the proteins were diluted into PBS (0.1 M with 3.0 mM EDTA, pH 8), followed by the addition of Traut's reagent (20 × molar excess). After 60 min of incubation at room temperature, the proteins were purified using a PD10 column (Amersham Biosciences, Bellefonte, PA). To react the thiolated proteins with the m-NP, 1 ml of freshly prepared m-NPs were mixed with the thiolated proteins (1 mg BSA or 0.25 mg PA) in PBS (0.1 M, pH 7.4). The pH was adjusted to 6.5, and the reaction mixture was stirred under N2 gas for 12-14 h at room temperature. Unconjugated proteins were separated from the nanoparticles by gel permeation chromatography (GPC, Sepharose 4B, 6 × 150 mm) or ultracentrifugation (1.0 × 105 g). In the GPC, the eluted fractions were analyzed for nanoparticles by measuring the absorption at 269 nm, and the protein content in every fraction was determined using a Bradford's reagent (BioRad, Hercules, CA). As an additional method to confirm the conjugation of the proteins to the nanoparticles, the fraction containing the BSA-conjugated nanoparticles (BSA-NPs) was diluted 100-fold in carbonate buffer and used as the capture antigen to coat the plates in an enzyme-linked immunosorbent assay (ELISA). The primary antibody was mouse anti-BSA anti-serum.
2.2. Transmission electron micrography (TEM)
The size and morphology of the nanoparticles were examined using transmission electron microscopy (TEM, Philips CM12 TEM/STEM) in the Oregon State University Electron Microscope Facility [22].
2.3. Stability of nanoparticles in simulated biological media
To evaluate the stability of the nanoparticles in simulated biological media, the nanoparticle suspension was diluted in normal saline (0.9% NaCl) or 10% FBS (v/v) in normal saline and incubated at 37°C for 30 min. The particle sizes at 0 and 30 min were measured.
2.4. In vitro uptake of nanoparticles by DC2.4 cells
Nanoparticles were conjugated with BSA that was pre-labeled with fluorescein-5(6)-isothiocyanate (FITC, Sigma-Aldrich). Thiolated BSA was labeled with FITC following the manufacturer's instruction (Promega). DC2.4 cells (1.0 × 106 cells/well), a mouse dendritic cell line [23], were seeded into 24-well plates and allowed to grow overnight. The cells were then incubated with 50 μl of the FITC-BSA-NPs for 6 h at 37°C under 5% CO2 or 4°C. The cells were then washed three times with PBS (10 mM, pH 7.4), lysed with 0.5% SDS (w/v), and analyzed for fluorescence intensity using a BioTek Synergy HT Multi-Detection Microplate Reader (BioTek Instruments, Inc. Winooski, VT). Fluorescence intensity was normalized using FITC-BSA as a control. Data were presented as the fold of fluorescence intensity increase (FITC-BSA-NP over FITC-BSA). Alternatively, the nanoparticles were labeled with FITC directly by incorporating 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-carboxyfluorescein (Avanti Polar Lipids) (5% w/w) into the nanoparticles prior to the conjugation of the BSA to generate BSA-NPs-FITC, which were used in the following fluorescence microscopic study and in vivo distribution experiments.
To microscopically evaluate the uptake of the nanoparticles, DC2.4 cells (2 × 104) were plated on poly-D-lysine-coated glass coverslips for 24 h. Cells were incubated with BSA-NPs-FITC and maintained at 4°C or 37°C for 6 h. Cells were then washed with PBS (10 mM, pH 7.4), fixed in 3% paraformaldehyde for 20 min, and washed three additional times prior to mounting on slides with Fluoromount G® (SouthernBiotech, Birmingham, AL). Bright-field and fluorescent images were obtained using a Zeiss AutoImager Z1 microscope (Carl Zeiss, Thornwood, NY) with a Zeiss 20× objective.
2.5. In vivo distribution and uptake of the nanoparticles in LNs
All mouse studies were carried out following NIH guidelines for animal use and care. BALB/c mice (female, 6 -8 weeks) were from Simonsen Laboratories Inc. (Gilroy, CA). Twenty-five μl of BSA-NPs-FITC or FITC-labeled nanoparticles without BSA (FITC-NPs) in suspension were subcutaneously (s.c.) injected into the footpads of the hind legs of the mice (n = 3). The lipopolysaccharide (LPS) from E. coli (Sigma-Aldrich, 100 ng per footpad) was used as a positive control. Mice in the negative control group were injected with sterile PBS (10 mM, pH 7.4) or left untreated. Mice were euthanized 23-24 h later, and their popliteal (and inguinal) LNs were removed and pooled to prepare single cell suspension [24]. To examine the activation of DCs, the cells from the popliteal LNs were stained with antibodies against CD11c and CD86 (BD Pharmingen, San Diego, CA) [24] and analyzed using a flow cytometer (FC500 Beckman Coulter EPICS V Dual Laser Flow Cytometer, Fullerton, CA).
2.6. Immunization Studies
The vaccine formulations were administered to mice by s.c. injection. To quantify the amount of antigen proteins on the nanoparticles, the antigens (BSA or PA) were labeled with FITC before being conjugated onto the nanoparticles, and the fluorescence intensity was measured using a fluorescence spectrometer. We have repeated this method multiple times to make sure that the same amount of protein antigens were consistently conjugated to a fixed amount of nanoparticles. In the control groups, the dose of the aluminum hydroxide (Alum) was 50 μg/mouse, and the dose of the incomplete Freund's adjuvant (IFA) was 100 μl/mouse (mixed with BSA in PBS at a ratio of 1:2). Except where mentioned, mice were dosed on days 0, 14, and 28 and euthanized on day 42. To measure the durability of the antibody response, mice were bled via the craniofacial vein. ELISA, splenocyte proliferation assay, and the in vitro and in vivo lethal toxin neutralization assays were all completed as previously described [25]. For the ELISA, EIA/RIA flat bottom, medium binding, polystyrene, 96-well plate (Corning Costar, Corning, NY) were coated with 100 ng of PA in 100 μl of carbonate buffer (0.1 M, pH 9.6) or BSA in PBS (10 mM, pH 7.4) overnight at 4 °C. For anti-PA Ab measurement, plates were washed with PBS/Tween 20 (10 mM, pH 7.4, 0.05% Tween 20, Sigma–Aldrich) and blocked with 4% (w/v) BSA in PBS/Tween 20 for 1 h at 37 °C. Samples were diluted two-fold serially in 4% BSA/PBS/Tween 20, added to the plates following the removal of the blocking solution, and incubated for an additional 3 h at 37 °C. The serum samples were removed, and the plates were washed five times with PBS/Tween 20. Horse radish peroxidase (HRP)-labeled goat anti-mouse immunoglobulin (IgG, 5000-fold dilution in 1% BSA/PBS/Tween 20, Southern Biotechnology Associates Inc., Birmingham, AL) was added into the plates, followed by another hour of incubation at 37 °C. Plates were again washed five times with PBS/Tween 20. The presence of bound Ab was detected following a 30 min incubation at room temperature in the presence of 3,3′,5,5′-tetramethylbenzidine solution (TMB, Sigma–Aldrich), followed by the addition of 0.2 M sulfuric acid as the stop solution. The absorbance was read at 450 nm. Anti-BSA Abs were determined similarly; except that the 4% BSA in PBS/Tween 20 was replaced by 5% (v/v) horse serum (Sigma–Aldrich) in PBS/Tween 20 in the plate blocking and sample dilution steps. In addition, the secondary Abs were diluted in 1.25% (v/v) horse serum in PBS/Tween 20. The antibody titers were determined by considering any OD value higher than the mean plus two times of the standard deviation (mean + 2 × S.D.) of the negative control group as positive. In the splenocyte proliferation assay, the splenocytes were stimulated with 12.5 μg/ml of PA protein[25]. In the in vivo lethal toxin neutralization assay, the dose of the anthrax lethal toxin was 90 μg of PA83 and 45 μg of LF per mouse (i.e., 7.5 × LD50). Finally, mice used in the in vivo challenge experiment were different from those used to collect serum samples for the in vitro neutralization assay.
2.7. Statistics
Statistical analyses were completed using ANOVA followed by the Fischer's protected least significant difference procedure. Standard linear regression analysis was used to analyze the PA dose response data. A p-value of ≤ 0.05 (two-tail) was considered statistically significant.
3. Results
3.1. The lecithin-based nanoparticles were spherical and stable in simulated biological media
The nanoparticles were engineered from emulsions, consisting of 3.5 mg/ml lecithin and 0.5 mg/ml GMS. As shown in Fig. 1A, at least 0.4% of Tween 20 was required to emulsify the lecithin and GMS. Increasing the concentration of the Tween 20 resulted in smaller nanoparticles and a decrease in the turbidity of the nanoparticle suspensions. When the Tween 20 was increased to 1%, nanoparticles of 178 ± 20 nm were generated, and further increasing the concentration of the Tween 20 did not lead to a further decrease in the resultant particle size and the turbidity of the particle suspension. Thus, nanoparticles prepared with a final Tween 20 concentration of 1% were used for further studies. Shown in Fig. 1B is a typical TEM micrograph of the unpurified nanoparticles. The nanoparticles were spherical (Fig. 1B and inset). Finally, the nanoparticles did not aggregate during a 30 min incubation period at 37°C in normal saline or 10% FBS in normal saline (Fig. 1C).
Fig. 1. Lecithin-based nanoparticles were spherical and stable in simulated biological media.
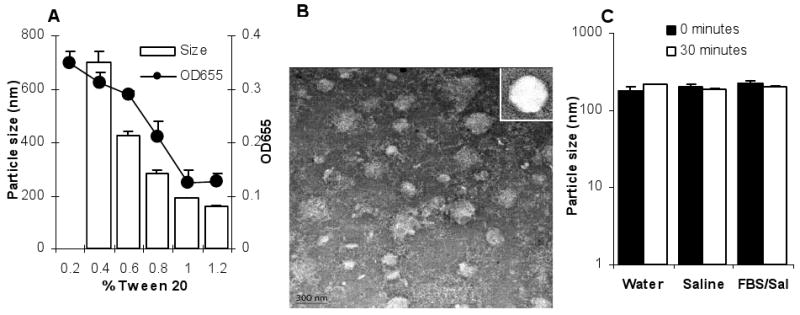
A. The effect of the concentration of the Tween 20 on the size of the resultant nanoparticles (white bars) and the turbidity (OD655) of nanoparticle suspension (filled circles).
B. TEM of the nanoparticles. Inset is to show the spherical nature of the nanoparticles.
C. The nanoparticles did not aggregate in normal saline or 10% FBS in normal saline. Particle sizes were measured immediately and following a 30-min incubation period at 37°C. Data reported are mean ± S.D. (n = 3).
3.2. BSA was conjugated onto the surface of the nanoparticles
Two approaches were used to confirm that the BSA was conjugated onto the surface of the nanoparticles, GPC and ELISA. Data in Fig. 2A showed that the GPC column can be used to separate the nanoparticles from free BSA protein. Data in Figs. 2B and C demonstrated the conjugation of BSA onto the nanoparticles. After the BSA was reacted with the nanoparticles, the particle peak and the BSA peak overlapped at fraction 7. However, when the nanoparticles and BSA were simply physically mixed for the same amount of time as the conjugation reaction, the particle peak and the BSA peak did not overlap. In the ELISA, BSA was only detected when the fraction 7 from the reaction product of the nanoparticles and BSA, not when the fraction 7 from the nanoparticles alone or from the physical mixture of the nanoparticles and BSA was used to coat the plates (Fig. 2D), demonstrating the conjugation of the BSA onto the nanoparticles. The size of the resultant BSA-conjugated nanoparticles was 201 ± 5 nm.
Fig. 2. BSA was conjugated onto the nanoparticles.
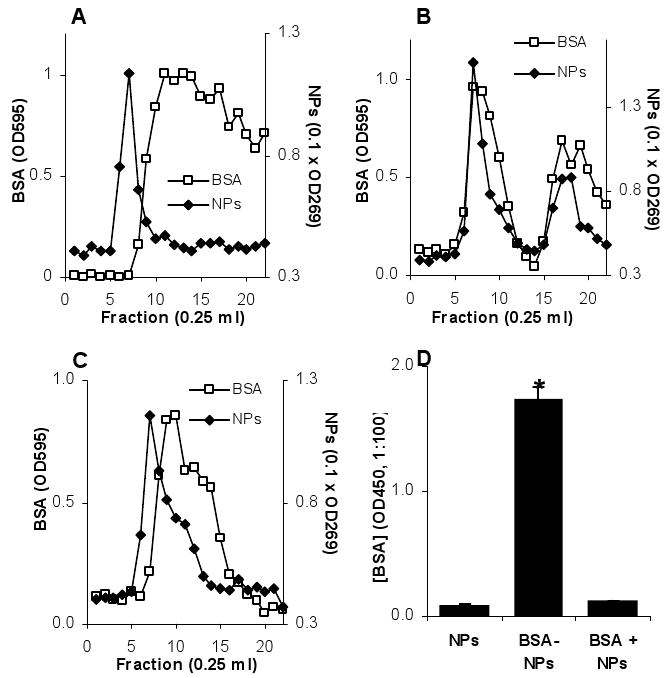
A. The Sepharose 4B column can separate the nanoparticles from free BSA protein. Nanoparticles and the BSA were applied to the column separately. Nanoparticles were detected by measuring the absorption at 269 nm. BSA was detected at 595 nm after staining with Bradford's reagent.
B, C. The chromatogram of the reaction product of BSA and nanoparticles or the physical mixture of BSA and nanoparticles. In A, B, and C, data reported were the mean values from three independent determinations. The S.D. was not shown for clarity. In all cases, fraction 7 contained nanoparticles that could be reasonably separated from free BSA.
D. ELISA confirmed the conjugation of the BSA onto the nanoparticles. Fraction 7 from the nanoparticles alone (A), the reaction product of the BSA and nanoparticles (B), and the physical mixture of the BSA and nanoparticles (C) were used to coat the ELISA plate. The primary Ab was anti-BSA antiserum. * The value of the BSA-NPs was significantly different from that of the other two (p < 0.05). Data presented are the mean ± S.D. (n = 3).
3.3. The nanoparticles facilitated the uptake of the antigen by APCs
To identify the extent to which the nanoparticles can facilitate the uptake of antigen by APCs, FITC-BSA-NPs were incubated with DC2.4 cells. As shown in Fig. 3A, the DC2.4 cells took up significantly more BSA when the BSA was conjugated onto the nanoparticles than free BSA (p < 0.001). Moreover, the fluorescence intensity ratio was significantly stronger at 37°C than at 4°C (Fig. 3A), indicating that the nanoparticles were internalized by the DC2.4 cells, not simply bound to their surface, which was further confirmed by the micrographs shown in Fig. 3B. Strong fluorescence signal was detected only in DC2.4 cells incubated with the FITC-labeled nanoparticles at 37°C, but not in the cells incubated at 4°C.
Fig. 3. The uptake of BSA-NPs by DC2.4 cells in culture.

A. The uptake of FITC-labeled BSA conjugated onto the nanoparticles by DC2.4 cells. Cells (1.0 × 106) were incubated with FITC-BSA-NPs or FITC-BSA for 6 h at 37°C or 4°C. Data reported are the ratio of the fluorescence intensity of cells treated with FITC-BSA-NPs over that with FITC-BSA.
B. Microscopic graphs showing the uptake of the FITC-labeled, BSA-conjugated nanoparticles. Cells were incubated with BSA-NPs-FITC for 6 h at 37°C or 4°C and observed under a bright-field microscope (top panel) or a fluorescence microscope (bottom panel).
3.4. The trafficking of BSA-NPs into the LNs and the activation of APCs
Twenty-four h after the injection of the BSA-NPs-FITC in the hind footpads, the FITC was detectable in the popliteal and inguinal LNs (Fig. 4A), more in the local popliteal LNs than in the distal inguinal LNs (∼3.7% vs. 1.9%). Close to 15% of the CD11c+ cells in the popliteal LNs were found to be FITC positive or have taken up the FITC-labeled BSA-NPs (Fig. 4B, 100 × 0.43/(0.43+2.52)). Similarly, roughly 15% of the popliteal LN cells that took up the BSA-NPs (FITC+) were CD11c+ (Fig. 4B, 100 × 0.43/(0.43+2.43)). Finally, the nanoparticles increased the percent of CD11c and CD86 double positive cells in the popliteal LNs. Over 1.67% of the popliteal LN cells were CD11c+ and CD86+ 23 h after the injection of the nanoparticles, as compared to 2.04% in mice injected with the E. coli LPS (100 ng/footpad) (Fig. 4C).
Fig. 4. Trafficking of BSA-NPs into the LNs.
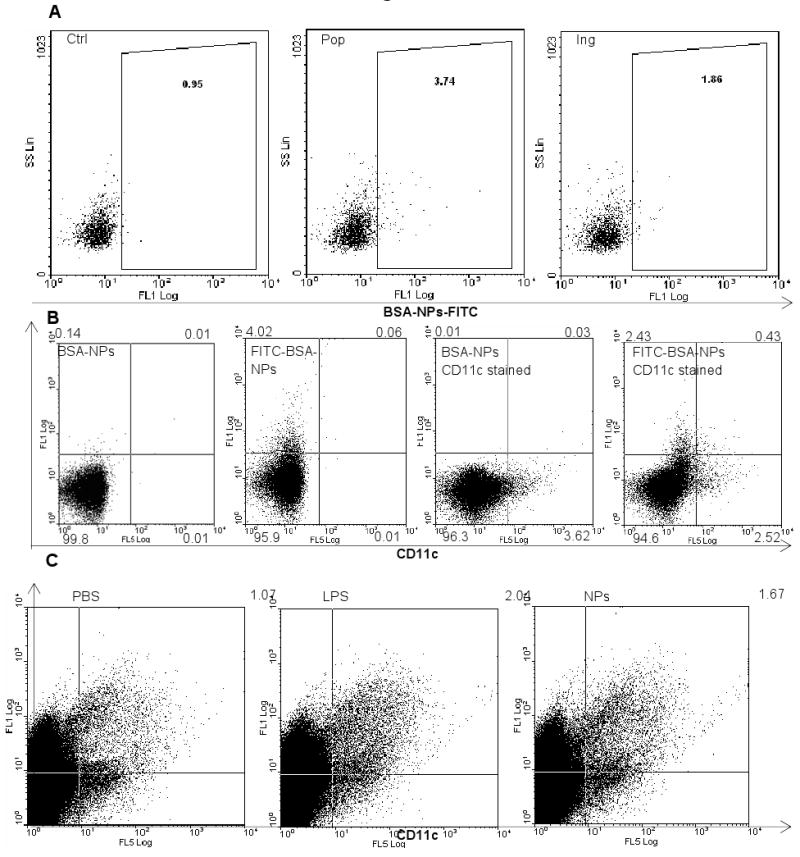
A. BSA-NPs were detectable in local and distal draining LNs. Mice (n = 3) were injected in the hind footpads with BSA-NPs-FITC. Popliteal and inguinal LN cells were collected 24 h after the injection. Ctrl indicates cells from the popliteal LNs of untreated mice. Pop, popliteal LNs; Ing, Inguinal LNs. Numbers are the % of cells that were FITC positive.
B. The uptake of BSA-NPs by DCs in the popliteal LNs. Mice were injected in the hind footpads with BSA-NPs or BSA-NPs-FITC. Popliteal LNs were collected 24 h later. Cells were stained with PE-Cy7-labeled anti-mouse CD11c.
C. Nanoparticles increased the percent of CD86 and CD11c double positive cells in the popliteal LNs. Mice (n = 3) were injected in the hind footpads with PBS, LPS from E. coli, or the NPs without BSA. Popliteal LNs were collected 23 h later and pooled. Cells were stained with PE-Cy7-anti-CD11c and FITC-anti-CD86.
3.5. Immunization of mice with the BSA-NPs induced strong anti-BSA Ab responses
Data in Table 1 showed that the BSA-specific IgG and IgM titers induced by the BSA-NPs were comparable to that induced when the IFA was used as an adjuvant (p = 0.88), but significantly higher than when the Alum was used as an adjuvant (p < 0.001). The BSA-NPs, BSA/IFA, and BSA/Alum induced comparable levels of anti-BSA IgG1, but the anti-BSA IgG2a and IgG3 induced by the BSA/Alum was significantly weaker than that induced by the BSA-NPs and the BSA/IFA (Table 1).
Table 1. Anti-BSA antibody titers in mouse serum samples.
| Treatment (titers in anti-log2) | |||
|---|---|---|---|
| BSA-NPs | BSA/IFA | BSA/Alum | |
| Anti-BSA IgG | 13.5 ± 1.5 | 14.3 ± 0.5 | 10.8 ± 1.0* |
| Anti-BSA IgG1 | 16.0 ± 1.9 | 17.5 ± 1.7 | 16.0 ± 0 |
| Anti-BSA IgG2a | 9.9 ± 1.1 | 11.1 ± 0.6 | 6.6 ± 0* |
| Anti-BSA IgG3 | 8.4 ± 1.1 | 8.4 ± 1.5 | 6.6 ± 0* |
| Anti-BSA IgM | 8.5 ± 0.9 | 7.9 ± 1.0 | 6.1 ± 0.6* |
Mice (n = 5) were dosed with BSA-NPs, BSA/Alum, BSA/IFA, or left untreated on days 0 and 14 (5 μg BSA/mouse) and bled on day 28. Data shown are mean ± S.D. The data of the BSA-NPs were the average from two independent experiments.
The anti-BSA Ab titers induced by the BSA/Alum were lower than the other treatments (p < 0.01).
3.6. Immunization of mice with PA conjugated onto the nanoparticles induced functional anti-PA antibody responses
The diameter of the PA-conjugated nanoparticles (PA-NPs) was 197 ± 6 nm. Shown in Fig. 5A is the effect of the dose of the PA protein on the resultant anti-PA IgG titers in the serum samples of mice immunized with the PA-NPs. Within the dose range of 1 to 10 μg/mouse, increasing the dose of the PA led to increased anti-PA IgG (p < 0.001 from linear regression) (Fig. 5A). Moreover, the anti-PA IgG titers induced by the PA-NPs at the dose of 5 μg PA were comparable to that induced by 5 μg PA adjuvanted with Alum. Seven days after the first dose at 5 μg of PA per mouse, anti-PA IgG was detectable in mice immunized with the PA-NPs, but not in mice dosed with the PA adjuvanted with Alum (Fig. 5B) (p < 0.001). However, after the second dose, the anti-PA antibody titers in both groups became comparable (Fig. 5B). The splenocytes isolated from mice immunized with the PA-NPs proliferated when re-stimulated with the PA protein (Fig. 5C). Finally, the anti-PA antibodies induced by the PA-NPs were able to neutralize anthrax lethal toxin and protect mouse macrophages (J774A.1) from a lethal toxin challenge in vitro (Fig. 5D). Mice immunized with the PA-NPs survived a lethal dose (7.5 × LD50) of anthrax LeTx challenge, and the lethal toxin challenge itself significantly enhanced the anti-PA IgG titers in the surviving mice (Table 2).
Fig. 5. Immunization of mice with PA-NPs induced functional anti-PA Ab responses.
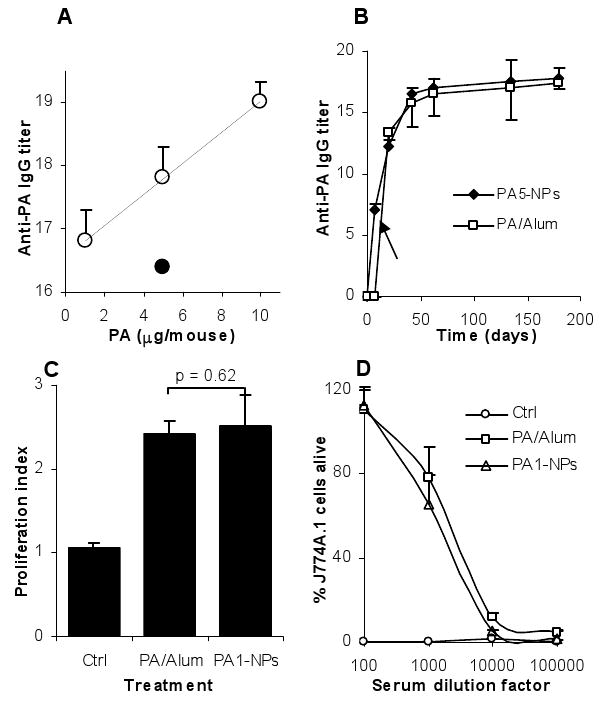
A. The effect of the dose of the PA on the resultant anti-PA IgG titer. Mice (n = 5) were dosed with PA-NPs (○) with 10, 5, or 1 μg of PA. Control mice were dosed with PA/Alum (5 μg PA) (●) or left untreated on days 0, 14, and 28 and bled on day 42. Linear regression showed that the slope of the regression line is not zero (p < 0.001). The values of the PA/Alum and the PA-NPs with 5 μg of PA were comparable (p = 0.32). Data shown are mean ± S.E.M.
B. The kinetics of the anti-PA IgG response. Mice (n = 5) were immunized on days 0, 14, and 28 with PA-NPs or PA/Alum (5 μg PA/mouse). The arrow indicates that the values of the PA5-NPs and the PA/Alum were different on day 7 (p < 0.001).
C. Splenocytes proliferated after in vitro re-stimulation with PA. Ctrl is the proliferation index from untreated mice. The values of PA1-NP (1 μg PA/mouse) and PA/Alum (5 μg PA/mouse) were comparable (p = 0.62).
D. The anti-PA antisera protected J774A.1 cells from a lethal toxin challenge in culture. Ctrl indicates serum from untreated mice. Mice in the PA1-NP group were dosed with 1 μg PA/mouse, and those in the PA/Alum group were dosed with 5 μg PA/mouse. Data reported are mean ± S.D.
Table 2. Mice immunized with the PA-NPs survived a lethal dose of anthrax lethal toxin challenge, and the lethal toxin challenge significantly boosted the anti-PA IgG titer in the surviving mice.
| Control | PA-NPs | PA/Alum | |
|---|---|---|---|
| # of mice died / # challenged a | 4/4 | 0/3 | 0/3 |
| Anti-PA IgG titer before challenge b | NA | 17.8 ± 1.1 | 16.4 ± 1.9 |
| Anti-PA IgG titer after challenge c | NA | 19.6 ± 1.1 | 20.0 ± 0 |
Mice were immunized on days 0, 14, and 28 (5 μg PA/mouse), challenged (i.v.) with anthrax lethal toxin two weeks after the last immunization, and monitored for 14 days after the challenge.
IgG titer 4 days before the challenge (n = 5).
IgG titer 14 days after the challenge (n = 3).
The values before and after the challenge were different from each other (PA-NPs, p = 0.02, PA/Alum, p = 0.03, dose of the PA was 5 μg/mouse). Titers are in anti-log2. Data shown are mean ± S.D.
4. Discussion
Our data in the present study showed that protein antigens conjugated onto the surface of nanoparticles prepared with lecithin and GMS induced quick, strong and long-lasting antibody immune responses when subcutaneously injected into mice. Depending on the antigens used, the adjuvanticity of the nanoparticles was stronger than that of the Alum and was comparable to that of the IFA. Moreover, the antibody responses developed more quickly when the antigen was adjuvanted using the nanoparticles than using Alum. The potent adjuvanticity of the nanoparticles was likely due to the nanoparticles' ability to enhance the uptake of the antigens by APCs, to move the antigens into local draining LNs, and to activate APCs.
BSA (∼66 KDa) is a serum protein with normal immunogenicity. At the beginning, we used it as an antigen to confirm the conjugation of the protein antigens onto the surface of the nanoparticles and to evaluate the adjuvanticity of the nanoparticles. Our data in Fig. 2 clearly showed that successful conjugation of the BSA onto the nanoparticles and that unconjugated BSA can be separated from the nanoparticles by GPC. Importantly, the BSA conjugated onto the nanoparticles generated anti-BSA antibody titers 5-6.5-fold higher than those induced when the BSA was adjuvanted using the Alum (Table 1, IgG and IgM). In fact, data in Table 1 showed that the nanoparticles have similar adjuvanticity as the IFA, a well known strong adjuvant [26]. The nanoparticles appeared to have adjuvant properties that favor humoral immunity, although less biased than the Alum. If a more balanced antibody response or an increased cell-mediated immune response is needed, one may have the option of including a co-adjuvant such as the low toxicity LPS [27] or the CpG oligos into the nanoparticle formulation.
In order to evaluate the functionality of the immune response induced by antigens delivered using the nanoparticles, we used the B. anthracis PA protein. The toxicity of the B. anthracis toxin is mainly from the lethal factor and the edema factor. PA is needed for the natural entrance of the LF and EF into the host cells [28]. Thus, antibodies that can neutralize the PA in serum are expected to block the transport of LF and EF into the cytosol and prevent the course of B. anthracis infection. Similar to the BSA-NPs, the PA-NPs were able to induce a strong anti-PA IgG response, and increasing the dose of the PA tended to increase the resultant anti-PA IgG titer (Fig. 5A). Slightly different from the BSA-NPs, at the dose of 5 μg/mouse, the PA-NPs and the PA admixed with Alum induced comparable anti-PA IgG titers (Fig. 5A). This may be due to that unlike the BSA, the PA protein is strongly immunogenic in inducing anti-PA antibody responses [29] and extremely foreign to the mice used in this study. In our splenocyte proliferation assay, we found a proliferation index of 2.5. The relatively small proliferation index may be due to reasons that the mice were dosed with only 1 μg of the PA protein and that the index was derived from the whole splenocytes, not the purified T cells. Shown in Fig. 5B are the kinetics of the anti-PA IgG responses induced by PA conjugated onto the nanoparticles. Interestingly, mice immunized with the PA-NPs developed detectable anti-PA IgG following a single immunization (Fig. 5B). However, mice immunized with the PA admixed with Alum failed to respond to the priming, and anti-PA IgG was detectable in those mice only after a boost immunization (Fig. 5B). The ability of an anthrax vaccine to induce an immune response immediately after the first immunization is likely critical in the case of post-exposure combination therapy of anthrax with a vaccine and an antibiotic. Finally, the anti-PA antibodies induced by PA-NPs were found to be functional. Serum from mice immunized with the PA-NPs was able to neutralize anthrax lethal toxin and protect mouse macrophages in culture (Fig. 5D). Moreover, mice immunized with the PA-NPs also survived a lethal dose of anthrax lethal toxin challenge (Table 2).
The ability of the nanoparticles to enhance the immune responses induced by antigens conjugated on their surface was likely due to their ability to enhance the trafficking of the antigens into local draining LNs, to improve the uptake of antigens by APCs, and to activate the APCs. For example, our data in Fig. 3 clearly showed that nanoparticles facilitated the uptake of the BSA antigen conjugated on their surface, likely because the DCs were more effectively taking up particulates than the BSA protein in solution. Data from previous studies showed that particles with a diameter of 500 nm or below were optimal for uptake by APCs such as DCs and macrophages [6-7]. Our BSA-NPs was around 200 nm, which is within the optimal size range for the uptake by APCs. Moreover, our data in Fig. 4A showed that close to 4% of the cells in local draining popliteal LNs became FITC+ at 24 h after the BSA-NPs labeled with FITC were injected into the footpad of the hind legs, suggesting that the FITC-labeled nanoparticles were present in ∼4% of the LN cells. It is unclear to what extent those particles moved into the LNs by direct draining or by the trafficking of the DCs. However, only about 15% of the FITC+ cells in the popliteal LNs were CD11c+, suggesting that the direct draining of the nanoparticles into the local draining LNs may have been extensive. Nevertheless, Manolova et al. (2008) reported that particles of 20-200 nm drained freely to LNs [8]. Finally, our data indicated that the nanoparticles also activated APCs in the draining LNs because an increased percent of the cells in popliteal LNs became CD11c+ and CD86+ after the subcutaneous injection of the nanoparticles (Fig. 3C).
Finally, our nanoparticles were engineered with lecithin and GMS as the matrix materials and Tween 20 as an emulsifying agent. Lecithin is a complex mixture of phosphatides consisting chiefly of phosphatidylcholine, phosphatidylethanolamine, phosphatidylserine, phosphatidylinositol and other substances such as triglycerides and fatty acids. It is GRAS (generally regarded as safe) listed and accepted in the FDA Inactive Ingredients Guide for parenterals (e.g., 0.3-2.3% for intramuscular injection)[30]. Tween 20 is a polyoxyethylene derivative of sorbitan monolaurate. It is also GRAS listed and included in the FDA Inactive Ingredients Guide for parenterals[30]. GMS is used in a variety of food, pharmaceutical, and cosmetic applications and is also GRAS listed. Its LD50 in mouse was reported to be 0.2 g/kg by the intraperitoneal route[30]. Therefore, although more experiments will have to be carried out in the future to define the safety profile of the nanoparticles, we consider this nanoparticle-based antigen delivery system relatively safe.
5. Conclusions
We engineered a lecithin-based nanoparticle as a promising antigen delivery system. Immunization of mice with antigens conjugated on the surface of the nanoparticles induced a quick, strong, durable, and functional immune response. The adjuvant activity of the nanoparticles was likely due to their ability to facilitate the uptake of the antigens by APCs, to improve the trafficking of the antigens into local draining LNs, and to activate APCs. When measured on day 473, the anti-PA IgG level in mice immunized with the PA5-NPs in Fig. 5B remained strong (with a titer of 17 ± 0.8).
Acknowledgments
This work was supported in part by National Institute of Allergy and Infectious Diseases grants AI070538, AI078304, and AI065774 to Z.C. and AI076812 to Y.H. and Z.C. BRS was supported in part by the OSU P.F. Yerex & Nellie Buck Yerex Graduate Fellowship and the OSU Sports Lottery Scholarship. The NIAID-ATCC BEI Resources kindly provided the PA and LF. Flow cytometry analysis was completed in the Flow Cytometry and Cell Sorting Facilities in the Environmental Health Science Center at the Oregon State University, which is supported by a NIEHS grant (P30 ES000210). We would like to thank Nikki B. Marshall and Danielle King for assistant in acquiring the flow cytometry data.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lindblad EB. Aluminium compounds for use in vaccines. Immunol Cell Biol. 2004;82:497–505. doi: 10.1111/j.0818-9641.2004.01286.x. [DOI] [PubMed] [Google Scholar]
- 2.Baylor NW, Egan W, Richman P. Aluminum salts in vaccines--US perspective. Vaccine. 2002;20 3:S18–23. doi: 10.1016/s0264-410x(02)00166-4. [DOI] [PubMed] [Google Scholar]
- 3.Singh M, Chakrapani A, O'Hagan D. Nanoparticles and microparticles as vaccine-delivery systems. Expert Rev Vaccines. 2007;6:797–808. doi: 10.1586/14760584.6.5.797. [DOI] [PubMed] [Google Scholar]
- 4.Cui Z, Mumper RJ. Microparticles and nanoparticles as delivery systems for DNA vaccines. Crit Rev Ther Drug Carrier Syst. 2003;20:103–137. doi: 10.1615/critrevtherdrugcarriersyst.v20.i23.10. [DOI] [PubMed] [Google Scholar]
- 5.Peek LJ, Middaugh CR, Berkland C. Nanotechnology in vaccine delivery. Adv Drug Deliv Rev. 2008;60:915–928. doi: 10.1016/j.addr.2007.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foged C, Brodin B, Frokjaer S, Sundblad A. Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int J Pharm. 2005;298:315–322. doi: 10.1016/j.ijpharm.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 7.Kanchan V, Panda AK. Interactions of antigen-loaded polylactide particles with macrophages and their correlation with the immune response. Biomaterials. 2007;28:5344–5357. doi: 10.1016/j.biomaterials.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 8.Manolova V, Flace A, Bauer M, Schwarz K, Saudan P, Bachmann MF. Nanoparticles target distinct dendritic cell populations according to their size. Eur J Immunol. 2008;38:1404–1413. doi: 10.1002/eji.200737984. [DOI] [PubMed] [Google Scholar]
- 9.Wendorf J, Chesko J, Kazzaz J, Ugozzoli M, Vajdy M, O'Hagan D, Singh M. A comparison of anionic nanoparticles and microparticles as vaccine delivery systems. Hum Vaccin. 2008;4:44–49. doi: 10.4161/hv.4.1.4886. [DOI] [PubMed] [Google Scholar]
- 10.Gutierro I, Hernandez RM, Igartua M, Gascon AR, Pedraz JL. Size dependent immune response after subcutaneous, oral and intranasal administration of BSA loaded nanospheres. Vaccine. 2002;21:67–77. doi: 10.1016/s0264-410x(02)00435-8. [DOI] [PubMed] [Google Scholar]
- 11.Olbrich C, Muller RH, Tabatt K, Kayser O, Schulze C, Schade R. Stable biocompatible adjuvants--a new type of adjuvant based on solid lipid nanoparticles: a study on cytotoxicity, compatibility and efficacy in chicken. Altern Lab Anim. 2002;30:443–458. doi: 10.1177/026119290203000407. [DOI] [PubMed] [Google Scholar]
- 12.Fifis T, Gamvrellis A, Crimeen-Irwin B, Pietersz GA, Li J, Mottram PL, McKenzie IF, Plebanski M. Size-dependent immunogenicity: therapeutic and protective properties of nano-vaccines against tumors. J Immunol. 2004;173:3148–3154. doi: 10.4049/jimmunol.173.5.3148. [DOI] [PubMed] [Google Scholar]
- 13.Kalkanidis M, Pietersz GA, Xiang SD, Mottram PL, Crimeen-Irwin B, Ardipradja K, Plebanski M. Methods for nano-particle based vaccine formulation and evaluation of their immunogenicity. Methods. 2006;40:20–29. doi: 10.1016/j.ymeth.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 14.Minigo G, Scholzen A, Tang CK, Hanley JC, Kalkanidis M, Pietersz GA, Apostolopoulos V, Plebanski M. Poly-L-lysine-coated nanoparticles: a potent delivery system to enhance DNA vaccine efficacy. Vaccine. 2007;25:1316–1327. doi: 10.1016/j.vaccine.2006.09.086. [DOI] [PubMed] [Google Scholar]
- 15.Singh M, Briones M, Ott G, O'Hagan D. Cationic microparticles: A potent delivery system for DNA vaccines. Proc Natl Acad Sci U S A. 2000;97:811–816. doi: 10.1073/pnas.97.2.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caputo A, Castaldello A, Brocca-Cofano E, Voltan R, Bortolazzi F, Altavilla G, Sparnacci K, Laus M, Tondelli L, Gavioli R, Ensoli B. Induction of humoral and enhanced cellular immune responses by novel core-shell nanosphere- and microsphere-based vaccine formulations following systemic and mucosal administration. Vaccine. 2009;27:3605–3615. doi: 10.1016/j.vaccine.2009.03.047. [DOI] [PubMed] [Google Scholar]
- 17.Mann JF, Shakir E, Carter KC, Mullen AB, Alexander J, Ferro VA. Lipid vesicle size of an oral influenza vaccine delivery vehicle influences the Th1/Th2 bias in the immune response and protection against infection. Vaccine. 2009;27:3643–3649. doi: 10.1016/j.vaccine.2009.03.040. [DOI] [PubMed] [Google Scholar]
- 18.Cui Z, Qiu F, Sloat BR. Lecithin-based cationic nanoparticles as a potential DNA delivery system. Int J Pharm. 2006;313:206–213. doi: 10.1016/j.ijpharm.2006.01.042. [DOI] [PubMed] [Google Scholar]
- 19.Williams KJ, Werth VP, Wolff JA. Intravenously administered lecithin liposomes: a synthetic antiatherogenic lipid particle. Perspect Biol Med. 1984;27:417–431. doi: 10.1353/pbm.1984.0031. [DOI] [PubMed] [Google Scholar]
- 20.Wang JY, Roehrl MH. Anthrax vaccine design: strategies to achieve comprehensive protection against spore, bacillus, and toxin. Med Immunol. 2005;4:4. doi: 10.1186/1476-9433-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cui Z, Lockman PR, Atwood CS, Hsu CH, Gupte A, Allen DD, Mumper RJ. Novel D-penicillamine carrying nanoparticles for metal chelation therapy in Alzheimer's and other CNS diseases. Eur J Pharm Biopharm. 2005;59:263–272. doi: 10.1016/j.ejpb.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 22.Cui Z, Mumper RJ. Genetic immunization using nanoparticles engineered from microemulsion precursors. Pharm Res. 2002;19:939–946. doi: 10.1023/a:1016402019380. [DOI] [PubMed] [Google Scholar]
- 23.Shen Z, Reznikoff G, Dranoff G, Rock KL. Cloned dendritic cells can present exogenous antigens on both MHC class I and class II molecules. J Immunol. 1997;158:2723–2730. [PubMed] [Google Scholar]
- 24.Cui Z, Han SJ, Vangasseri DP, Huang L. Immunostimulation mechanism of LPD nanoparticle as a vaccine carrier. Mol Pharm. 2005;2:22–28. doi: 10.1021/mp049907k. [DOI] [PubMed] [Google Scholar]
- 25.Sloat BR, Cui Z. Nasal immunization with a dual antigen anthrax vaccine induced strong mucosal and systemic immune responses against toxins and bacilli. Vaccine. 2006;24:6405–6413. doi: 10.1016/j.vaccine.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 26.Chang JCC, Diveley JP, Savary JR, Jensen FC. Adjuvant activity of incomplete Freund's adjuvant. Advanced Drug Delivery Reviews. 1998;32:173–186. doi: 10.1016/s0169-409x(98)00009-x. [DOI] [PubMed] [Google Scholar]
- 27.Goldstein J, Hoffman T, Frasch C, Lizzio EF, Beining PR, Hochstein D, Lee YL, Angus RD, Golding B. Lipopolysaccharide (LPS) from Brucella abortus is less toxic than that from Escherichia coli, suggesting the possible use of B. abortus or LPS from B. abortus as a carrier in vaccines. Infect Immun. 1992;60:1385–1389. doi: 10.1128/iai.60.4.1385-1389.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA. Identification of the cellular receptor for anthrax toxin. Nature. 2001;414:225–229. doi: 10.1038/n35101999. [DOI] [PubMed] [Google Scholar]
- 29.Kenney RT, Yu J, Guebre-Xabier M, Frech SA, Lambert A, Heller BA, Ellingsworth LR, Eyles JE, Williamson ED, Glenn GM. Induction of protective immunity against lethal anthrax challenge with a patch. J Infect Dis. 2004;190:774–782. doi: 10.1086/422694. [DOI] [PubMed] [Google Scholar]
- 30.Weller PJ, Wade A, editors. Handbook of pharmaceutical excipients. 2. Washington: American Pharmaceutical Association; London: The Pharmaceutical Press; 1994. [Google Scholar]