

Abstract
We investigated the contribution of the intracellular calcium (Ca2+i) transient to acetylcholine (ACh)-mediated reduction of pacemaker frequency and cAMP content in rabbit sinoatrial nodal (SAN) cells. Action potentials (whole cell perforated patch clamp) and Ca2+i transients (Indo-1 fluorescence) were recorded from single isolated rabbit SAN cells, whereas intracellular cAMP content was measured in SAN cell suspensions using a cAMP assay (LANCE®). Our data show that the Ca2+i transient, like the hyperpolarization-activated “funny current” (I f) and the ACh-sensitive potassium current (I K,ACh), is an important determinant of ACh-mediated pacemaker slowing. When I f and I K,ACh were both inhibited, by cesium (2 mM) and tertiapin (100 nM), respectively, 1 μM ACh was still able to reduce pacemaker frequency by 72%. In these I f and I K,ACh-inhibited SAN cells, good correlations were found between the ACh-mediated change in interbeat interval and the ACh-mediated change in Ca2+i transient decay (r 2 = 0.98) and slow diastolic Ca2+i rise (r 2 = 0.73). Inhibition of the Ca2+i transient by ryanodine (3 μM) or BAPTA-AM (5 μM) facilitated ACh-mediated pacemaker slowing. Furthermore, ACh depressed the Ca2+i transient and reduced the sarcoplasmic reticulum (SR) Ca2+ content, all in a concentration-dependent fashion. At 1 μM ACh, the spontaneous activity and Ca2+i transient were abolished, but completely recovered when cAMP production was stimulated by forskolin (10 μM) and I K,ACh was inhibited by tertiapin (100 nM). Also, inhibition of the Ca2+i transient by ryanodine (3 μM) or BAPTA-AM (25 μM) exaggerated the ACh-mediated inhibition of cAMP content, indicating that Ca2+i affects cAMP production in SAN cells. In conclusion, muscarinic receptor stimulation inhibits the Ca2+i transient via a cAMP-dependent signaling pathway. Inhibition of the Ca2+i transient contributes to pacemaker slowing and inhibits Ca2+i-stimulated cAMP production. Thus, we provide functional evidence for the contribution of the Ca2+i transient to ACh-induced inhibition of pacemaker activity and cAMP content in rabbit SAN cells.
Keywords: Acetylcholine, Ca2+ transient, cAMP, Pacemaker cells, Sinoatrial node
Introduction
The mechanisms underlying pacemaker activity of sinoatrial node (SAN) cells are still a matter of debate [14, 17, 19, 20, 24]. Classically, β-adrenergic and muscarinic modulation of SAN pacemaker activity is explained by cAMP and PKA-dependent acceleration or deceleration of the “membrane clock”, which consists of time- and voltage-dependent ion channels located in the plasma membrane of SAN cells [26]. Noradrenaline (NA) increases the inward currents that are active during diastolic depolarization [4], including the hyperpolarization-activated cation current (I f) [7], L-type Ca2+ current (I Ca,L) [12] and sustained inward current (I st) [11, 32]. In addition, outward currents are reduced by hastening deactivation of the total delayed rectifier current (I K) [22]. On the other hand, acetylcholine (ACh) slows the diastolic depolarization [4, 52] by decreasing the inward currents, I f [8], I Ca,L [34] and I st [10], and activating the outward current I K,ACh [37].
Only during the last decade, spontaneous Ca2+ releases from the sarcoplasmic reticulum (SR) have been widely recognized as an additional pacemaker mechanism: the “calcium clock” [26]. It was demonstrated that inhibition of the “calcium clock” largely prevented β-adrenergic speeding of the pacemaker rate [25, 36]. Although pacemaker cells lack T-tubules, have sparsely developed SR, and contain little contractile proteins [29], they are endowed with significant intracellular Ca2+ (Ca2+i) transients [23]. β-adrenergic stimulation augments the amplitude of the Ca2+i transients and the local Ca2+ releases (LCRs) from the SR late during the diastolic depolarization [26]. Besides the amplitude, also the frequency at which the LCRs occur increases [26]. The accelerated “calcium clock” dynamically interacts with the “membrane clock” through the LCR-driven activation of an inwardly directed Na+/Ca2+ exchange current (I NCX) [28], which substantially contributes to the diastolic depolarization rate and to the ignition of the action potential (AP) [3]. The augmented Ca2+i transients and the accelerated “calcium clock” are the consequence of β-adrenergic-mediated cAMP production and increased PKA-dependent phosphorylation of Ca2+ handling proteins [46]. The Ca2+i transients and “calcium clock” are interdependent [26]. At every beat, the “calcium clock” is reset by I Ca,L that subsequently triggers a Ca2+i transient. Only after the Ca2+i transient has sufficiently declined and enough Ca2+ is restored in the SR, LCRs can again occur. This makes the Ca2+i transient an integral part of the “calcium clock” [25–27].
It has been hypothesized that Ca2+i also indirectly couples to the “membrane clock” via a route involving cAMP, based on the observation that Ca2+i transient inhibition abolished I f modulation by β-adrenergic agonists, while a membrane-permeable cAMP analog restored it [5]. Indeed, Mattick et al. [30] recently identified Ca2+-stimulated adenylyl cyclase in guinea pig SAN cells that could explain this interesting observation.
In summary, the afore-mentioned data from literature show that β-adrenergic stimulation of SAN cells accelerates both the “membrane clock” and the “calcium clock” and augments the Ca2+i transient via cAMP–PKA-dependent signaling pathways. Moreover, evidence has been provided that Ca2+i stimulates cAMP production. We hypothesized that muscarinic receptor stimulation would lead to the opposite effects. In the present study, we carried out experiments on isolated rabbit SAN cells to determine (1) whether Ca2+i transients, such as I f and I K,ACh, are important determinants of ACh-mediated pacemaker slowing, and (2) whether inhibition of Ca2+i transients by muscarinic agonists lowers cAMP production. Our results demonstrate the importance of Ca2+i transients in ACh-induced inhibition of pacemaker frequency and cAMP content in rabbit SAN cells.
Methods
Ethical approval and cell preparation
All experiments were carried out in accordance with guidelines of the local institutional animal care and use committee. In addition, the investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). Single SAN cells were isolated from 42 rabbit hearts by an enzymatic dissociation procedure as previously described [42]. The hearts were obtained from New Zealand White rabbits (body weight: 3.0–3.5 kg) were first sedated with a 1 ml kg−1 intramuscular injection of Hypnorm® (10 mg ml−1 fluanisone and 0.315 mg ml−1 fentanyl citrate; Janssen Pharmaceutics, Tilburg, The Netherlands) and 15 min later anaesthetized by an injection of 3.0–3.5 ml Nembutal® (60 mg ml−1 pentobarbital sodium; Sanofi, Maassluis, The Netherlands) in the marginal ear vein together with 0.3 ml heparin (5,000 IU ml−1 thromboliquine; Organon, Oss, The Netherlands). For all measurements, except cAMP determination, spindle and elongated spindle-like cells displaying regular contractions in normal Tyrode’s solution were selected.
Electrophysiology
APs were recorded at 36 ± 0.2°C with the amphotericin B perforated patch-clamp technique using an Axopatch 200B patch-clamp amplifier (Molecular Devices Corporation, Sunnyvale, CA, USA). Patch pipettes (2–5 MΩ) were pulled from borosilicate glass, and data acquisition and analysis were accomplished using pCLAMP 8 software (Molecular Devices). Signals were low-pass filtered with a 1 kHz cutoff frequency, and digitized at 2 kHz. Potentials were corrected for the calculated liquid junction potential (13 mV). Cell membrane capacitance was estimated by dividing the decay time constant of the capacitive transient in response to 5-mV hyperpolarizing voltage clamp steps from 0 mV by the series resistance, and amounted to 34 ± 3 pF (mean ± SEM; n = 12).
Ca2+i measurements
[Ca2+]i was measured in Indo-1 loaded cells. In brief, cells were loaded with 5 μM Indo-1-AM for 10 min at room temperature, and subsequently superfused with normal Tyrode’s solution for 15 min at 36 ± 0.2°C to remove excess indicator and allow full de-esterification. A rectangular adjustable slit was used to select a single cell and to reduce background fluorescence. Dye-loaded cells were excited with 340 nm light (75 W Xenon arc lamp) and fluorescent light intensities were measured at 405 (I405) and 505 (I505) nm, using photomultiplier tubes. Signals were digitized at 1 kHz, filtered at 100 Hz, and corrected for background fluorescence before the I405/I505 ratio values were calculated.
[Ca2+]i was computed from [Ca2+]i = β × K d × (R − R min)/(R max − R), where β represented the ratio of maximal I505 and minimal I505 (2.2), K d was 250 nM (data sheet from Molecular Probes, Eugene, OR, USA), and R min and R max were the minimal and maximal ratio, respectively. To determine R min and R max, the cells were made Ca2+ permeable with a high potassium solution containing 1.7 μM A23187, 5 μM gramicidin and 10 μM nigericin, while Ca2+ re-uptake was inhibited by 5 μM thapsigargin. In the absence of Ca2+, we obtained an R min value of 0.31 ± 0.04 (n = 24), whereas in the presence of 10 mM extracellular Ca2+ the R max value was 2.21 ± 0.24 (n = 24).
The SR Ca2+ content of cells was studied by a brief exposure, via a nearby pipette, to 20 mM caffeine and 5 mM nickel to release Ca2+ from the SR and to prevent Ca2+ efflux by Na+/Ca2+ exchange, respectively. The fractional SR Ca2+ release was determined by normalizing the preceding Ca2+i transient amplitude (average of 6) to the caffeine-evoked Ca2+i transient amplitude.
cAMP measurements
The LANCE cAMP kit (PerkinElmer, Zaventem, Belgium) was used to determine cAMP concentrations (according to the manufacturer’s instruction) using a Victor plate reader (Wallac, PerkinElmer, Zaventem, Belgium). Cell suspension was added to a 384-well OptiPlate (PerkinElmer, Waltham, MA, USA; 5 μl per well) and incubated for 1 h at room temperature with metacholine (MCh) at concentrations ranging from 10−10 to 10−5 M. All experiments were performed in the presence of 0.05% bovine serum albumin (BSA) and 500 μM of the phosphodiesterase inhibitor isobutyl-methyl-xanthine (IBMX) to allow accumulation of cAMP.
For every experiment, the yields of two SAN cell isolations were pooled and divided over different wells. All MCh–cAMP response curve measurements were performed in triplo and averaged. For each intervention (500 nM NA, 3 μM ryanodine, 25 μM BAPTA-AM), a control MCh–cAMP response curve was measured in the presence of the vehicle used. The obtained cAMP values were normalized to the control cAMP value measured in the absence of MCh. To determine the effect of Ca2+i on cAMP content, SAN cells were pre-incubated with BAPTA-AM or ryanodine for 10 min.
Solutions and drugs
Solutions
Tyrode’s solution contained (mM): 140 NaCl, 5.4 KCl, 1.8 CaCl2, 1.0 MgCl2, 5.5 glucose, 5.0 HEPES; pH 7.4 (2.6 NaOH). The pipette solution contained (mM): 120 K-gluconate, 20 KCl, 5 NaCl, 0.22 amphothericin B, 10 NMDG-Cl (N-methyl-d-glucammonium chloride), 10 HEPES; pH 7.2 (5.5 KOH). The high potassium solution contained (mM): 130 KCl, 10 NaCl, 1 MgCl2, 10 HEPES; pH 7.2 (5.5 KOH).
Drugs
All drugs were obtained from Sigma (Zwijndrecht, The Netherlands), except ryanodine (Calbiochem-Novabiochem, Darmstadt, Germany; 10 mM), noradrenaline (Centrafarm, Etten-Leur, The Netherlands; 6 mM), Indo-1, BAPTA-AM, and thapsigargin (Molecular Probes). Except for noradrenaline, all drugs were prepared as a stock solution in DMSO (amphothericin B (65 mM), Indo-1 (5 mM), A23187 (1.7 mM), thapsigargin (10 mM), BAPTA-AM (25 mM), forskolin, (10 mM)), in ethanol (gramicidin (50 mM), nigericin (10 mM)), or in H2O (ACh (1 mM), ryanodine (3 mM), MCh (10 mM)). The values in parentheses indicate the concentration of the stock. The ryanodine and BAPTA-AM concentrations were such that Ca2+i transients were largely, but not fully, inhibited, whereas the cesium and tertiapin concentrations were selected to maximally inhibit I f and I K,ACh, respectively, without exerting major effects on other membrane currents. We chose 3 μM ryanodine since it has often been used, e.g., by Bogdanov et al. [3], Bucchi et al. [5] and Vinogradova et al. [46], and higher concentrations are known to block I Ca,L [38]. A sub-maximal NA concentration (500 nM) was selected to study the anti-adrenergic effects of ACh on Ca2+i transients and pacemaker frequency. Ryanodine, BAPTA-AM, MCh, ACh and NA were prepared freshly on the day of the experiment. The effect of ACh, NA, cesium and tertiapin on pacemaker frequency was determined after 2 min exposure time, whereas the effect of ryanodine and BAPTA-AM was determined after 10 min.
Statistics
Results are expressed as mean ± SEM, with the number of cells or cell suspensions denoted by n. Statistical analysis was performed using SPSS 10 software (SPSS Inc., Chicago, IL, USA). Two-way ANOVA with repeated measurements was used to compare concentration effect curves, whereas the EC50 values (derived from a Hill equation fit) and the pacemaker frequencies of Fig. 2b were compared with a Student’s t test. One-way ANOVA with repeated measurements, followed by a Student–Newman–Keuls test for pairwise comparsions, was used to compare the pacemaker frequencies of Fig. 2f. P values of <0.05 were considered to be significant.
Fig. 2.
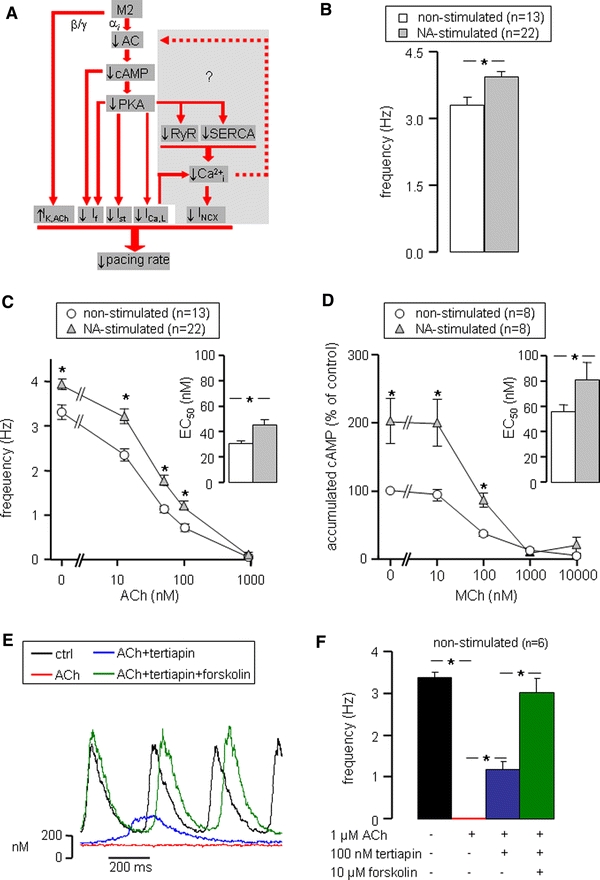
Muscarinic agonist-induced pacemaker slowing. a Muscarinic receptor (M2) stimulation reduces pacemaker rate by down-regulation of inward membrane currents (I f, I st, I Ca,L) through inhibition of cAMP production and up-regulation of the outward I K,ACh. The main objective of the present study is to elucidate the possible role of Ca2+i transients in muscarinic agonist-induced pacemaker slowing (gray area). b Intrinsic pacemaker frequency of non-stimulated (white bars) and 500 nM NA-stimulated (gray bars) SAN cells in the absence of muscarinic agonists. c Frequency–ACh relationship for non-stimulated and NA-stimulated SAN cells. Inset shows average EC50 values of the frequency–ACh relationships of individual cells. d cAMP–MCh relationship for non-stimulated and NA-stimulated SAN cells. Inset shows average EC50 values of the cAMP–MCh relationships of individual cells. e Typical Ca2+i transients of a SAN cell successively exposed to normal Tyrode’s solution (black trace), 1 μM ACh (red trace), 1 μM ACh + 100 nM tertiapin (blue trace) and 1 μM ACh + 100 nM tertiapin + 10 μM forskolin (green trace). f Mean frequency of six non-stimulated SAN cells successively exposed to the solutions of panel E. Quiescent SAN cells (ACh) regained spontaneous activity on inhibition of I K,ACh (ACh + tertiapin), whereas pacemaker frequency was fully restored to control values on additional stimulation of cAMP production (ACh + tertiapin + forskolin). Asterisks indicate significant differences (P < 0.05)
Results
Determination of pacemaker frequency by Ca2+i transients
Consistent with previous reports [20], spontaneous APs in single SAN cells are accompanied by Ca2+i transients in a 1:1 fashion, both under control conditions (Fig. 1a, black traces) and in the presence of NA (Fig. 1a, red traces) or both NA and ACh (Fig. 1a, blue traces). Consequently, a good correlation between the cycle length of the spontaneous Ca2+i transients and that of the associated APs was observed (r 2 = 0.98) (Fig. 1b). Therefore, the average cycle length of ten consecutive spontaneous Ca2+i transients was used to determine pacemaker frequency. While every AP is accompanied by a Ca2+i transient, APs and Ca2+i transients are not exactly synchronized. In the absence of ACh and NA, maximal upstroke velocity of the Ca2+i transient occurred 21.3 ± 0.6 ms (n = 8) later than the maximum upstroke velocity of the action potential.
Fig. 1.

Ca2+i transients and action potentials in a rabbit SAN cell. a A typical simultaneous recording of action potentials (upper panel) and Ca2+i transients (lower panel) in normal Tyrode’s solution (black traces), in the presence of 500 nM NA alone (red traces) or combined with 50 nM ACh (blue traces). b Cycle length of action potentials versus cycle length of the associated Ca2+i transients (r 2 = 0.98)
Inhibition of cAMP production and activation of IK,ACh underlie pacemaker slowing
Classically, reduction of pacemaker frequency by ACh is explained by slowing of the “membrane clock” through a muscarinic receptor-regulated signaling pathway [4]. Our main objective in this study is to elucidate the possible role of Ca2+i transients in muscarinic agonist-induced pacemaker slowing (Fig. 2a, gray area). We hypothesize that muscarinic agonists slow pacemaking partly by reducing Ca2+i transients through inhibiting cAMP–PKA-dependent phosphorylation of Ca2+i handling proteins. This subsequently affects Ca2+-dependent currents and reduces pacemaker frequency by decreasing I NCX, Ca2+-stimulated cAMP production, and cAMP and PKA-dependent inward currents (Fig. 2a, gray area). Before we study the role of Ca2+i transients in muscarinic agonist-induced pacemaker slowing, we will first examine some classical pathways in our rabbit SAN cell model. As illustrated in Fig. 2a, muscarinic receptor stimulation directly activates I K,ACh and indirectly decreases I f, I Ca,L and I st via reduced cAMP or PKA-dependent modulation. Because the effect of muscarinic agonists largely depends on the absolute cAMP levels, studies were performed in the absence (non-stimulated) and presence of 500 nM NA (NA stimulated).
In NA-stimulated SAN cells, pacemaker frequency, as derived from Ca2+i transients, was significantly higher compared to non-stimulated SAN cells (3.94 ± 0.11 Hz, n = 22, vs. 3.31 ± 0.17 Hz, n = 13, respectively) (Fig. 2b). In both groups, pacemaker frequency decreased with increasing ACh concentrations (12.5, 50, 100 and 1,000 nM; Fig. 2c). However, the relative decrease in pacemaker frequency was significantly larger in non-stimulated compared to NA-stimulated cells at ACh concentrations of 12.5, 50 and 100 nM (Fig. 2c, asterisks). Accordingly, the averaged EC50 value of the individual frequency–ACh relations of the 13 non-stimulated SAN cells tested (30 ± 2 nM) was significantly smaller than that for the 22 NA-stimulated cells tested (45 ± 4 nM) (Fig. 2c, inset).
Determination of cAMP accumulation in SAN cell suspensions showed that stimulation of the muscarinic receptor by metacholine (MCh), an ACh analog that cannot be hydrolyzed by acetylcholinesterase possibly present in the cell suspension, inhibits cAMP accumulation concentration dependently in both non-stimulated (n = 8) and NA-stimulated (n = 8) cells (Fig. 2d). In NA-stimulated SAN cells, the accumulated cAMP was significantly higher in basal conditions (no MCh) compared to non-stimulated cells and showed a smaller percent decrease with increasing MCh concentrations (ANOVA, P < 0.05). Accordingly, the averaged EC50 value of the individual cAMP accumulation–MCh relations (81 ± 14 vs. 55 ± 6 nM, Fig. 2d, inset) proved significantly higher in NA-stimulated compared to non-stimulated cells. Interestingly, the EC50 values of the pacemaker frequency–ACh and the cAMP accumulation–MCh relations (Fig. 2c, d, insets) are both increased by ≈50% in NA-stimulated cells compared to non-stimulated cells, suggesting a strong correlation between muscarinic agonist-induced frequency and cAMP changes.
Next, we examined whether activation of I K,ACh and inhibition of cAMP production can explain pacemaker slowing by muscarinic agonists. Therefore, we tested the effects of the I K,ACh blocker tertiapin and the adenylyl cyclase activator forskolin on 1 μM ACh effects in non-stimulated SAN cells (n = 6). Typical Ca2+i transients recorded under these conditions are depicted in Fig. 2e. Spontaneous activity recovered partially by I K,ACh blockade (tertiapin 100 nM) and fully when in addition adenylyl cyclase was activated (forskolin 10 μM) (Fig. 2f; blue and green bars, respectively). From these data, we conclude that inhibition of cAMP production and activation of I K,ACh could explain the effect of muscarinic receptor stimulation on pacemaker frequency.
Contribution of If and IK,ACh to ACh-mediated pacemaker slowing
Next, we examined the contribution of the cAMP-dependent I f and cAMP-independent I K,ACh pathways to ACh-mediated pacemaker slowing. Although other membrane ionic currents, specifically I Ca,L and I st (Fig. 2a), also contribute to ACh-mediated pacemaker slowing, we focused on I f and I K,ACh, because specific blockers of these currents are available and full blockade of these currents does not result in pacemaker arrest. In non-stimulated cells and in the absence of ACh, I f inhibition (2 mM cesium) significantly reduced pacemaker frequency (Fig. 3a, left panel), whereas I K,ACh inhibition (100 nM tertiapin) did not affect pacemaker frequency (Fig. 3a, right panel). Similar observations were made in NA-stimulated cells (not shown). The percent reduction in pacemaker frequency on inhibition of I f was similar in non-stimulated and NA-stimulated cells (≈30 and ≈33%, respectively; Fig. 3b, c, data at 0 nM ACh).
Fig. 3.

Contribution of I f and I K,ACh to muscarinic agonist-induced pacemaker slowing. I f and I K,ACh were inhibited by 2 mM cesium and 100 nM tertiapin, respectively. a Typical Ca2+i transients of non-stimulated SAN cells in the absence (Tyrode) and presence of cesium (left panel) or tertiapin (right panel). b, c Frequency–ACh relationship under control conditions (ctrl), in the presence of cesium, and in the presence of tertiapin in non-stimulated (b) and NA-stimulated (c) cells. Asterisks indicate significant difference from control at the associated ACh concentration
In non-stimulated SAN cells, the If blocker cesium (2 mM; n = 10) slowed pacemaking only at low ACh concentrations (<50 nM), whereas the I K,ACh blocker tertiapin (100 nM; n = 9) opposed pacemaker slowing at all ACh concentrations tested (Fig. 3b) (ANOVA, P < 0.05). In NA-stimulated cells, pacemaker slowing by the I f blocker cesium (n = 8) was visible up to 100 nM ACh (ANOVA, P < 0.05), whereas the I K,ACh blocker tertiapin (n = 4) no longer significantly opposed pacemaker slowing (Fig. 3c). These results demonstrate that I f and I K,ACh contribute to ACh-mediated pacemaker slowing. I f gains importance and I K,ACh loses importance in ACh-mediated pacemaker slowing under NA-stimulated conditions. However, ACh-mediated pacemaker slowing was still clearly present during I f or I K,ACh blockade, indicating the contribution of other mechanisms (see below).
Contribution of Ca2+i transients to ACh-mediated pacemaker slowing
SAN pacemaker activity has been shown to be modulated also by SR Ca2+ loading and release [26]. Therefore, we next examined the role of Ca2+i transients in ACh-induced pacemaker slowing.
First, we tested the effect of Ca2+i transient inhibition on spontaneous activity of SAN cells in the absence of ACh. Figure 4a shows typical Ca2+i transients of non-stimulated SAN cells exposed to the SR Ca2+ release blocker ryanodine (3 μM) or the Ca2+ chelator BAPTA-AM (5 μM). Both drugs decreased the maximum systolic Ca2+i concentration, with the larger decrease in the presence of BAPTA-AM. In addition, both drugs caused an elevation of the minimum diastolic Ca2+i level, which was highest in the presence of 3 μM ryanodine. On average, 10 min exposure to ryanodine or BAPTA-AM significantly (P < 0.05) reduced pacemaker frequency in non-stimulated SAN cells by 22% (n = 8) and 37% (n = 7), respectively, and in NA-stimulated SAN cells by 16% (n = 10) and 30% (n = 4) (Fig. 4b, c, at 0 nM ACh). This indicates that Ca2+i transients contribute to pacemaker activity of both non-stimulated and NA-stimulated SAN cells.
Fig. 4.

Contribution of Ca2+i transients to muscarinic agonist-induced pacemaker slowing. Ca2+i transients were inhibited by 3 μM ryanodine or 5 μM BAPTA-AM. a Typical Ca2+i transients of non-stimulated SAN cells before (Tyrode) and after treatment with ryanodine (left panel) or BAPTA (right panel). b, c Frequency–ACh relationship under control conditions (ctrl), in the presence of ryanodine, and in the presence of BAPTA in non-stimulated (b) and NA-stimulated (c) cells. Asterisks indicate significant difference from control at the associated ACh concentration
Second, we tested the effects of ryanodine and BAPTA-AM on ACh-induced pacemaker slowing. In non-stimulated SAN cells, BAPTA-AM, but not ryanodine, caused a clear slowing at the low ACh concentration of 12.5 nM (Fig. 4b), whereas in NA-stimulated cells both agents exaggerated pacemaker slowing at ACh concentrations of 12.5, 50 and 100 nM (Fig. 4c) (ANOVA, P < 0.05). Taken together, these data demonstrate that Ca2+i transients can contribute to ACh-induced pacemaker slowing.
Effect of ACh on Ca2+i transients
Ca2+i transients of SAN cells
According to the important role of Ca2+i transients in ACh-induced pacemaker slowing (Fig. 4), we analyzed the effects of ACh on Ca2+i transient characteristics in detail (Fig. 5). We discerned five distinct phases (numbered I–V) in Ca2+i transients of SAN cells. Figure 5a shows a typical example of the various Ca2+i transient phases in a non-stimulated cell in the absence of ACh. The Ca2+i transient was quantified by (1) the maximum rate of the fast systolic [Ca2+]i rise during phase I following the fast AP upstroke, (2) the maximum systolic [Ca2+]i level during phase II, (3) the time constant of the monoexponential [Ca2+]i decay during phase III, (4) the minimum diastolic [Ca2+]i level during phase IV reached after completing about two-thirds of diastolic depolarization, and (5) the mean rate of the slow diastolic [Ca2+]i rise during phase V following the final third of diastolic depolarization until the next AP upstroke. As well known [26], NA stimulation increased d[Ca2+]i/dt during the upstroke (phase I (Fig. 5b)), the systolic Ca2+ concentration (phase II (Fig. 5c)) and diastolic d[Ca2+]i/dt (phases III (Fig. 5d) and V (Fig. 5f)).
Fig. 5.

ACh inhibits Ca2+i transients. a Superimposed action potentials (red trace) and associated Ca2+i transients (blue trace) recorded in a non-stimulated rabbit SAN cell in the absence of ACh. Numbers I–V indicate different phases of the Ca2+i transient. b–f Effects of ACh on rate of fast systolic [Ca2+]i rise (b), maximum systolic [Ca2+]i (c), time constant of diastolic [Ca2+]i decay (d), minimum diastolic [Ca2+]i (e) and rate of diastolic [Ca2+]i rise (f) in non-stimulated (circles) and NA-stimulated (triangles) rabbit SAN cells. Asterisks indicate significant difference (P < 0.05) between non-stimulated and NA-stimulated SAN cells at the associated ACh concentration
Effect of ACh on Ca2+i transients
We then analyzed the effects of ACh on the Ca2+i transient characteristics in both non-stimulated (n = 13) and NA-stimulated (n = 22) SAN cells in detail. Figure 5b, c, d, e, f shows that all five phases were affected concentration dependently in both non-stimulated and NA-stimulated cells. The fast systolic [Ca2+]i rise slowed (Fig. 5b), the maximum systolic [Ca2+]i fell (Fig. 5c), the diastolic [Ca2+]i decay slowed (Fig. 5d), the minimum diastolic [Ca2+]i fell (Fig. 5e) and the diastolic [Ca2+]i rise slowed (Fig. 5f). Of note, phase V of the Ca2+i transient proved most sensitive to ACh and was inhibited by 94 and 87% at 100 nM ACh in non-stimulated and NA-stimulated SAN cells, respectively. Moreover, the diastolic [Ca2+]i rise of phase V proved also most sensitive to NA stimulation and increased by 70% at 0 nM ACh (Fig. 5f).
From these data, we conclude that ACh inhibited the Ca2+i transients in both non-stimulated and NA-stimulated SAN cells.
Effect of ACh on SR Ca2+ content
To test whether the changed Ca2+i transients were associated with reduced SR Ca2+ loading and/or reduced SR Ca2+ release, we studied SR Ca2+ content and fractional SR Ca2+ release. Therefore, we measured SR Ca2+ release in response to 20 mM caffeine, in the presence of 5 mM nickel to block the Na+/Ca2+ exchanger. Figure 6a shows an example of three consecutive caffeine-evoked Ca2+i transients measured in one cell in the absence and presence of ACh (12.5 and 50 nM). In all conditions, emptying of the SR abolished spontaneous Ca2+i transients until the SR Ca2+ content was restored. Figure 6b summarizes the caffeine-induced Ca2+ release in non-stimulated and NA-stimulated cells in the absence and presence of ACh. In both non-stimulated and NA-stimulated cells, the SR Ca2+ release fell concentration dependently with increasing ACh concentrations. In NA-stimulated SAN cells, the SR Ca2+ release proved significantly higher (ANOVA, P < 0.05). As shown in Fig. 6c, the fractional SR Ca2+ release was not significantly affected by ACh and did also not differ between both non-stimulated and NA-stimulated SAN cells.
Fig. 6.
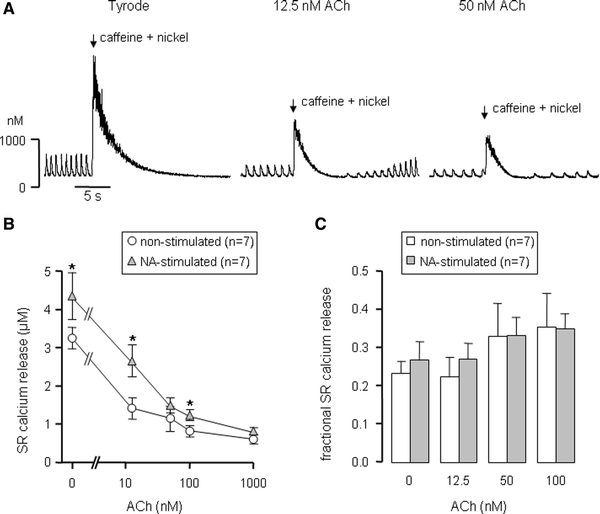
ACh lowers SR Ca2+ content. a Typical effects of caffeine in a non-stimulated SAN cell. Instantaneous release of Ca2+ from the SR was evoked by application of a brief exposure of the cell to 20 mM caffeine plus 5 mM nickel to prevent Ca2+i efflux through the Na+/Ca2+ exchanger (arrows). b ACh dependence of SR Ca2+ release on exposure to caffeine in non-stimulated (circles) and NA-stimulated (triangles) cells. c ACh dependence of the fractional SR Ca2+ release in both non-stimulated (white bars) and NA-stimulated (gray bars) cells. Asterisks indicate significant difference between non-stimulated and NA-stimulated SAN cells at the associated ACh concentration
From these data we conclude that ACh lowers SR Ca2+ content, which could explain the inhibitory effect of ACh on Ca2+i transients (Fig. 5).
Phases III and V of the Ca2+i transient correlate with pacemaker slowing
ACh-mediated slowing of the diastolic Ca2+i transient phases III and V could contribute to the ACh-mediated slowing of the diastolic depolarization rate and pacemaker frequency. Phase III reflects SERCA activity, whereas phase V likely reflects the LCRs observed by confocal microscopy [25–27]. Both SERCA activity (phospholamban phosphorylation) and LCRs have been shown to exhibit a strong correlation with pacemaker frequency of rabbit SAN cells [47]. Here, we examined the correlation between the ACh-mediated change in the diastolic Ca2+i phases III and V and the ACh-mediated change in interbeat interval (IBI) in non-stimulated SAN cells (n = 14), when the other important contributors to ACh-mediated pacemaker slowing I f and I K,ACh were blocked by 2 mM cesium and 100 nM tertiapin, respectively. Figure 7a shows typical examples of Ca2+i transients recorded from a non-stimulated SAN cell with inhibited I f and I K,ACh that was successively exposed to increasing ACh concentrations (0, 12.5, 50, 100 and 1,000 nM). Under these conditions, ACh inhibited Ca2+i transients (Fig. 7a) and slowed pacemaker frequency (Fig. 7b) in a concentration-dependent manner. Both the ACh-mediated changes in the diastolic Ca2+i transient decay time (Fig. 7c, phase III) and the diastolic Ca2+i rise (Fig. 7d, phase V) revealed close correlations with the ACh-mediated changes in IBI. These data support our hypothesis that ACh-mediated inhibition of the diastolic Ca2+i transient phases III and V contributes to ACh-mediated pacemaker slowing.
Fig. 7.

Diastolic Ca2+i transient parameters correlate with pacemaker frequency. a Typical Ca2+i transients of a non-stimulated SAN cell with inhibited I f (2 mM cesium) and I K,ACh (100 nM tertiapin) that was successively exposed to normal Tyrode’s solution (black trace), 12.5 nM ACh (red trace), 50 nM ACh (blue trace), 100 nM ACh (green trace) and 1,000 nM ACh (purple trace). b Average frequency–ACh relationship of non-stimulated SAN cells (n = 14) with inhibited I f and I K,ACh. c Correlation between fold change in interbeat interval (IBI) and fold change in time constant of Ca2+i transient decay. Solid line is the linear best fit to the data (r 2 = 0.98). d Correlation between fold change in IBI and fold change in the slow diastolic Ca2+i rise. Solid line is the linear best fit to the data (r 2 = 0.73)
Effect of ryanodine and BAPTA-AM on intracellular cAMP content
We found that ACh reduced pacemaker frequency, reduced intracellular cAMP content and inhibited the Ca2+i transients. Recently, Ca2+-stimulated adenylyl cyclases have been identified in guinea pig SAN, suggesting that Ca2+i could influence cAMP production [30]. In a final series of experiments, we therefore examined whether inhibition of the Ca2+i transients by ryanodine or BAPTA-AM affected the intracellular cAMP content. Figure 8 shows the effects of muscarinic receptor stimulation by MCh on cAMP content in both non-stimulated and NA-stimulated cells in the presence of ryanodine or BAPTA-AM. Inhibition of Ca2+i transients by 3 μM ryanodine significantly (ANOVA, P < 0.05) reduced intracellular cAMP content in both non-stimulated and NA-stimulated cells (Fig. 8a, b), with larger effects in the NA-stimulated cells. Comparable significant (ANOVA, P < 0.05) effects were found in the presence of BAPTA-AM (Fig. 8c, d). Taken together, these data demonstrate that inhibition of Ca2+i transients, e.g., by ryanodine, BAPTA-AM or muscarinic receptor stimulation, depresses cAMP production in rabbit SAN cells. This is consistent with the presence of Ca2+-stimulated adenylyl cyclases, as identified in guinea pig SAN cells [30].
Fig. 8.

Intracellular cAMP production depends on Ca2+i transients. cAMP was measured in non-stimulated (a and c) and NA-stimulated (b and d) SAN cell samples, either pre-treated with vehicle (open symbols) or Ca2+i transient inhibiting drugs (filled symbols). a, b cAMP–MCh relationship in the absence (vehicle) and presence of 3 μM ryanodine in non-stimulated (a) and NA-stimulated (b) cells. c, d cAmp–MCh relationship in the absence (vehicle) and presence of 25 μM BAPTA-AM in non-stimulated (c) and NA-stimulated (d) cells. All cAMP values were normalized to the control cAMP levels of non-stimulated SAN cells in the absence of ACh. Asterisks indicate significant difference between vehicle and Ca2+i transient inhibiting drug at the associated MCh concentration
Discussion
In this study, we demonstrate for the first time that muscarinic agonists inhibit Ca2+i transients in SAN cells, and that the pacemaker slowing effects of muscarinic agonists are augmented by Ca2+i transient inhibition. Moreover, we provide evidence that, like muscarinic agonists, Ca2+i transient inhibition reduces cAMP levels in SAN cells. Both inhibition of Ca2+i transients and reduction of cAMP levels will inhibit Ca2+i and cAMP-dependent ionic currents. These data suggest that the negative chronotropic effect of muscarinic agonists is in part obtained via Ca2+i transient inhibition and the subsequent reduction in cAMP.
Classical pathways in muscarinic agonist-induced pacemaker slowing
In our rabbit single SAN cell preparation, we demonstrate that muscarinic agonists inhibit pacemaker frequency (Figs. 1, 2, 3, 4, 7) and cAMP production (Figs. 2d, 8) in a concentration-dependent manner. Our data thus support the classical view that cAMP is an important determinant of pacemaker frequency. The classical view that activation of I K,ACh is a cAMP-independent pathway by which muscarinic agonists exert their negative chronotropic effect [4] (cf. Fig. 2a) is also supported by our data (Figs. 2e, f, 3). Moreover, we demonstrated that NA stimulation, which is known to increase cAMP production, reduced the contribution of the cAMP-independent pathway I K,ACh to ACh-mediated pacemaker slowing. This explains the limited effect of ACh on the maximum diastolic potential seen in Fig. 1a. In contrast, NA stimulation increased the contribution of the cAMP-dependent pathway I f to ACh-mediated pacemaker slowing. This can be explained by the NA-mediated increase in cAMP production.
Muscarinic agonists slow pacemaker rate by inhibition of Ca2+i transients
The importance of Ca2+i in pacemaking has been demonstrated in non-stimulated and NA-stimulated conditions. It has been shown that blunting Ca2+i transients (by ryanodine) or chelating Ca2+i (by BAPTA) slowed or arrested pacemaking and prevented NA-induced pacemaker acceleration [25]. Although ryanodine and BAPTA both inhibit Ca2+i transients, it should be kept in mind that there are several differences in actions between ryanodine and BAPTA. For instance, BAPTA increases the intracellular Ca2+ buffering power. Consequently, the free Ca2+i concentration will fall and any increase in [Ca2+]i through Ca2+ release (from the SR) or Ca2+ influx (through I Ca,L) is blunted and slowed. This will affect the LCRs, Ca2+i transients and all Ca2+i-dependent currents including I Ca,L. Ryanodine, on the other hand, reduces the open probability of the RyR channels. Consequently, less ryanodine channels are available for SR Ca2+ release. This will reduce fractional SR Ca2+ release and the amplitude of LCRs and Ca2+i transients. However, ryanodine will not blunt any Ca2+ influx and will affect Ca2+i-dependent modulation of I Ca,L to a lesser extent. These differences in actions between ryanodine and BAPTA may explain their quantitatively different effects on pacemaker frequency.
In the present study, we confirm that Ca2+i transients contribute to the intrinsic pacemaker activity of non-stimulated SAN cells (Fig. 4b) and to pacemaker acceleration of NA-stimulated cells (Fig. 4c). In addition, we provide evidence for the involvement of Ca2+i transients in muscarinic agonist-induced pacemaker slowing.
First, both ryanodine and BAPTA-AM facilitated pacemaker slowing at all ACh concentrations in NA-stimulated SAN cells. In non-stimulated SAN cells, the effects of ryanodine and BAPTA on ACh-mediated pacemaker slowing were less abundant and restricted to low ACh concentrations. The latter is likely explained by the concomitant increase in minimum diastolic Ca2+i concentration (Fig. 4a) and the subsequent activation of I NCX. A ryanodine-induced increase in diastolic [Ca2+]i has been previously reported in rabbit SAN cells [38]. The diastolic Ca2+i increase was larger in the presence of ryanodine than in the presence of BAPTA-AM. This could underlie the quantitative differences between the effects of both agents on ACh-mediated pacemaker slowing. Moreover, Bucchi et al. [6] recently reported that ryanodine (3 μM) depolarizes the take-off potential of the AP, which by itself slows pacemaker frequency.
Second, we show that ACh depresses all five (I–V) distinct phases of the Ca2+i transients (Fig. 5b, c, d, e, f) and lowers the SR Ca2+ content (Fig. 6b) of rabbit SAN cells in a concentration-dependent manner, all of which is compatible with reduced cAMP–PKA-dependent phosphorylation of the Ca2+i handling proteins. In accordance, in NA-stimulated cells, where cAMP–PKA signaling is stimulated, higher ACh concentrations were required to obtain a similar level of the maximum systolic [Ca2+]i (Fig. 5c), slow diastolic [Ca2+]i rise (Fig. 5f) and SR Ca2+ content (Fig. 6b) as compared to non-stimulated SAN cells. Moreover, higher ACh concentrations were required to increase the time constant of Ca2+i transient decay in NA-stimulated cells (Fig. 5d). Of interest, the slow diastolic [Ca2+]i rise that probably reflects the LCRs proved the Ca2+i transient parameter with the highest ACh-sensitivity (Fig. 5f).
Third, ACh was still able to slow pacemaker frequency of non-stimulated SAN cells in a concentration-dependent manner when the important contributors to ACh-mediated pacemaker slowing, I f and I K,ACh, were both inhibited (Fig. 7b). Thus, other mechanisms, such as Ca2+i releases [19], I Ca,L [34, 43] and I st [10], involved in ACh-mediated pacemaker slowing must be active. Both the Ca2+i transient decay and the slow diastolic Ca2+i rise of SAN cells with inhibited I f and I K,ACh showed close correlations with the ACh-induced changes in interbeat interval (Fig. 7c, d), suggesting that the ACh-mediated modulation of SERCA activity and of LCRs, and thus the rate of SR Ca2+ refilling and release, are important determinants of ACh-mediated pacemaker slowing. This is in accordance with previous publications by Lakatta and co-workers who found strong correlations between beating frequency and phospholamban phosphorylation status, and beating frequency and the periodicity of LCRs in rabbit SAN cells [27, 47].
Fourth, Ca2+i transient inhibition by ryanodine and BAPTA-AM facilitated MCh-mediated inhibition of cAMP, an important second messenger that is positively correlated with pacemaker frequency (Figs. 2, 8).
The effect of ACh on Ca2+i transients can be explained by reduced cAMP–PKA-dependent phosphorylation of ryanodine receptors and phospholamban. These phosphorylation changes lead to reduced open probability of the ryanodine receptor and to slower SERCA-mediated Ca2+i re-uptake into the SR, respectively. Moreover, in voltage clamp experiments, ACh inhibits the I Ca,L of rabbit SAN cells [51], which may result in Ca2+i transient inhibition. Further experiments are required to elucidate the exact role of I Ca,L in ACh-mediated pacemaker slowing.
Membrane clock and calcium clock
The main mechanism by which Ca2+i is thought to interact with the “membrane clock” is through electrogenic Ca2+ extrusion via the Na+/Ca2+ exchanger [3]. For instance, NA stimulates cAMP–PKA-dependent phosphorylation of Ca2+i handling proteins, thereby increasing the amplitude and frequency of the LCRs and subsequently the contribution of I NCX to diastolic depolarization [3, 45]. It should be taken into account, however, that various ion currents can also be modulated by Ca2+i (e.g., I Ca,L [39], I f [35], the T-type calcium current (I Ca,T) [18], chloride currents [1, 44] and the slow delayed rectifier current (I Ks) [41]) and by calmodulin or Ca2+/calmodulin-dependent protein kinase II (CaMKII) (e.g., I Ca,L [9], I Ca,T [21] and I Ks [33]). In the present study, we did not assess each of the ionic currents that are affected by the muscarinic agonist-induced Ca2+i transient changes, nor did we study the effect of muscarinic agonists on LCRs as part of the “calcium clock”. Nevertheless, our Ca2+i transient data are still relevant for understanding the “calcium clock”, since the Ca2+i transient and the “calcium clock” are interdependent [25]. LCRs ignite the AP by activating I Ca,L, which resets the “calcium clock” and triggers a Ca2+i transient. Only after the Ca2+i transient has sufficiently declined and enough Ca2+ is restored in the SR, another LCR can occur.
Thus, muscarinic agonists slow SAN cell pacemaker activity by inhibition of Ca2+i transients. However, the relative contribution of Ca2+i-dependent mechanisms and “membrane clock” mechanisms, including inhibition of I f and activation of I K,ACh, to ACh-mediated pacemaker slowing is difficult to assess because they are so strongly interdependent (see above). Only if we assume that ACh-mediated pacemaker slowing is fully explained by the action of ACh on I f, I K,ACh and a Ca2+-dependent mechanism, we can estimate their relative contributions. Pacemaker frequency of uninhibited SAN cells was slowed by 66, 79 and 100% when exposed to 50, 100 and 1,000 nM ACh, respectively (Fig. 3b), whereas pacemaker frequency of SAN cells with inhibited I f and I K,ACh was slowed by 51, 58 and 72% when exposed to 50, 100 and 1,000 nM ACh, respectively (Fig. 7b). Thus, one could say that at 50–1,000 nM ACh, the Ca2+-dependent mechanism is responsible for ≈75% pacemaker slowing and the combined inhibition of I f and opening of I K,ACh for ≈25% pacemaker slowing. Further experiments are required to elucidate the ionic currents involved in the Ca2+-dependent mechanism of ACh-mediated pacemaker slowing.
Muscarinic agonists slow pacemaker rate by inhibition of Ca2+i-stimulated cAMP production
It was recently demonstrated that basal cAMP levels are almost tenfold higher in SAN cells as compared to atrial and ventricular myocytes [46]. The presence of Ca2+-stimulated adenylyl cyclases in SAN cells may account for the relatively high basal cAMP level [30] and PKA activity [46]. The latter proved essential for the spontaneous rhythmic LCRs and “calcium clock” in SAN cells. The presence of these Ca2+-stimulated adenylyl cyclases in SAN cells [30] provides a potential indirect mechanism by which Ca2+i influences cAMP-dependent ionic currents and thus pacemaker frequency [5, 30].
Our data add functional evidence for the presence of a Ca2+i-dependent cAMP production in rabbit SAN cells. While the present paper was in preparation, Younes et al. [49] published their work on Ca2+-stimulated basal adenylyl cyclase activity of rabbit SAN pacemaker cells. In this study, they showed that rabbit SAN cells express two Ca2+-stimulated adenylyl cyclases (AC1 and AC8), located in the lipid rafts, which are largely responsible for the basal adenylyl cyclase activity and account for ≈30% of the high basal cAMP levels in SAN cells. In our non-stimulated SAN cells, Ca2+i-dependent cAMP production contributes to 33% to the basal cAMP levels, as demonstrated by the reduction in cAMP production on inhibition by BAPTA (Fig. 8c). From our results and the similar observations by Younes et al. [49], we conclude that in rabbit SAN cells, Ca2+-stimulated adenylyl cyclases (AC1 and AC8) in part contribute to the basal cAMP levels and that the Ca2+–independent adenylyl cyclases (AC5 and AC6) may provide the remaining cAMP [48, 49].
In addition, from our data we conclude that the Ca2+i-dependent cAMP production can provide the increase in cAMP in NA-stimulated SAN cells. Thus, the NA-augmented Ca2+i transients may have stimulated the extra cAMP production required for pacemaker acceleration (Figs. 2d, 8d). This finding contrasts with the observation by Younes et al. [49] that BAPTA did not affect the cAMP production in SAN cells stimulated with a β-adrenergic agonist, but it is in line with the earlier report of Bucchi et al. [5] who showed that a membrane-permeable cAMP analog restored I f modulation by β-adrenergic agonists in ryanodine treated SAN cells.
Furthermore, we demonstrate that Ca2+i transient inhibition facilitates the ACh-mediated inhibition of cAMP production in non-stimulated SAN cells (Fig. 8a, c). Thus, muscarinic agonist-mediated inhibition of the Ca2+i transient may in part underlie its inhibitory effect on cAMP production and pacemaker slowing (Fig. 2c). The remaining muscarinic agonist-induced cAMP reduction is likely obtained via the classical Gi-mediated inhibition of adenylyl cyclases [13].
Limitations
The present study has several limitations. First, it is difficult to draw firm conclusions with respect to the role of Ca2+i transients in muscarinic agonist-induced pacemaker slowing due to the interdependence of Ca2+i transients, cAMP content and pacemaker frequency. Ideally, one should vary one parameter while keeping other parameters constant. Unfortunately, this is impossible in SAN cells, even under AP clamp conditions, where pacemaker frequency, but not ion flow, is controlled. Nevertheless, our observations that muscarinic agonists reduce pacemaker frequency, Ca2+i transients and cAMP content dose dependently, and that Ca2+i transient inhibition (by ryanodine or BAPTA) in turn facilitates muscarinic agonist-mediated pacemaker slowing and cAMP inhibition provides valuable insight into the processes underlying pacemaker frequency modulation. Second, the exact spatial and temporal relation between Ca2+i and cAMP may be highly complex, since local concentration differences of both exist within a cell [48]. Phosphodiesterases, which in our cAMP assays were inhibited to allow cAMP accumulation, underlie these spatial differences and have been recently shown to be constitutively active in SAN cells [47]. Nevertheless, also in PDE-inhibited SAN cells, MCh, NA, ryanodine and BAPTA changed cAMP levels, indicating that cAMP production was still subject to modulation. Third, it should be noted that 2 mM cesium not only largely inhibits I f [15], but also slightly reduces outward potassium current [50], which may have led to an underestimation of the contribution of I f to pacemaker slowing in our experiments. However, “more specific” I f channel blockers also affect other ion currents present in SAN cells [2]. In addition, I f channel blockers may also directly influence Ca2+i transients, since the I f channel has been recently shown to be permeable to calcium ions [31]. A further limitation is that I K,ACh may not have been completely blocked by 100 nM tertiapin, as suggested by the experiments of Kitamura et al. [16], who found a ≈95% block of I K,ACh by 100 nM tertiapin in rabbit atrial myocytes. Therefore, the actual contribution of I K,ACh to ACh-mediated pacemaker slowing may have been underestimated in the present study. Moreover, the potential contribution of I Ca,L to ACh-mediated pacemaker slowing has not been investigated because of the lack of appropriate pharmacological tools to assess the role of I Ca,L without stopping pacemaking or affecting the action potential waveform. Finally, MCh is less potent compared to ACh in stimulating muscarinic receptor type 2 [40]. Therefore, a direct quantitative comparison between the cAMP–MCh curves and the frequency–ACh response curves is not allowed.
Conclusions
We provide evidence that muscarinic agonists not only slow pacemaker activity by inhibition of cAMP-dependent ionic currents and activation of I K,ACh, but also by inhibition of Ca2+i transients, which reduces cAMP content through inhibition of Ca2+i-stimulated cAMP production.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
Marcel M. G. J. van Borren and Arie O. Verkerk contributed equally to this manuscript.
References
- 1.Arai A, Kodama I, Toyama J. Roles of Cl− channels and Ca2+ mobilization in stretch-induced increase of SA node pacemaker activity. Am J Physiol. 1996;270:H1726–H1735. doi: 10.1152/ajpheart.1996.270.5.H1726. [DOI] [PubMed] [Google Scholar]
- 2.Baruscotti M, Bucchi A, DiFrancesco D. Physiology and pharmacology of the cardiac pacemaker (“funny”) current. Pharmacol Ther. 2005;107:59–79. doi: 10.1016/j.pharmthera.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 3.Bogdanov KY, Maltsev VA, Vinogradova TM, Lyashkov AE, Spurgeon HA, Stern MD, Lakatta EG. Membrane potential fluctuations resulting from submembrane Ca2+ releases in rabbit sinoatrial nodal cells impart an exponential phase to the late diastolic depolarization that controls their chronotropic state. Circ Res. 2006;99:979–987. doi: 10.1161/01.RES.0000247933.66532.0b. [DOI] [PubMed] [Google Scholar]
- 4.Boyett MR, Honjo H, Kodama I. The sinoatrial node, a heterogeneous pacemaker structure. Cardiovasc Res. 2000;47:658–687. doi: 10.1016/S0008-6363(00)00135-8. [DOI] [PubMed] [Google Scholar]
- 5.Bucchi A, Baruscotti M, Robinson RB, DiFrancesco D. If-dependent modulation of pacemaker rate mediated by cAMP in the presence of ryanodine in rabbit sino-atrial node cells. J Mol Cell Cardiol. 2003;35:905–913. doi: 10.1016/S0022-2828(03)00150-0. [DOI] [PubMed] [Google Scholar]
- 6.Bucchi A, Baruscotti M, Robinson RB, DiFrancesco D. Modulation of rate by autonomic agonists in SAN cells involves changes in diastolic depolarization and the pacemaker current. J Mol Cell Cardiol. 2007;43:39–48. doi: 10.1016/j.yjmcc.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 7.DiFrancesco D. The onset and autonomic regulation of cardiac pacemaker activity: relevance of the f current. Cardiovasc Res. 1995;29:449–456. [PubMed] [Google Scholar]
- 8.DiFrancesco D, Tromba C. Muscarinic control of the hyperpolarization-activated current (If) in rabbit sino-atrial node myocytes. J Physiol. 1988;405:493–510. doi: 10.1113/jphysiol.1988.sp017344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dzhura I, Wu Y, Colbran RJ, Balser JR, Anderson ME. Calmodulin kinase determines calcium-dependent facilitation of L-type calcium channels. Nat Cell Biol. 2000;2:173–177. doi: 10.1038/35004052. [DOI] [PubMed] [Google Scholar]
- 10.Guo J, Mitsuiye T, Noma A. The sustained inward current in sino-atrial node cells of guinea pig heart. Pflügers Arch. 1997;433:390–396. doi: 10.1007/s004240050293. [DOI] [PubMed] [Google Scholar]
- 11.Guo J, Ono K, Noma A. A sustained inward current activated at the diastolic potential range in rabbit sino-atrial node cells. J Physiol. 1995;483:1–13. doi: 10.1113/jphysiol.1995.sp020563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hagiwara N, Irisawa H, Kameyama M. Contribution of two types of calcium currents to the pacemaker potentials of rabbit sino-atrial node cells. J Physiol. 1988;395:233–253. doi: 10.1113/jphysiol.1988.sp016916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hescheler J, Kameyama M, Trautwein W. On the mechanism of muscarinic inhibition of the cardiac Ca current. Pflügers Arch. 1986;407:182–189. doi: 10.1007/BF00580674. [DOI] [PubMed] [Google Scholar]
- 14.Honjo H, Inada S, Lancaster MK, Yamamoto M, Niwa R, Jones SA, Shibata N, Mitsui K, Horiuchi T, Kamiya K, Kodama I, Boyett MR. Sarcoplasmic reticulum Ca2+ release is not a dominating factor in sinoatrial node pacemaker activity. Circ Res. 2003;92:e41–e44. doi: 10.1161/01.RES.0000055904.21974.BE. [DOI] [PubMed] [Google Scholar]
- 15.Huang X, Yang P, Du Y, Zhang J, Ma A. Age-related down-regulation of HCN channels in rat sinoatrial node. Basic Res Cardiol. 2007;102:429–435. doi: 10.1007/s00395-007-0660-5. [DOI] [PubMed] [Google Scholar]
- 16.Kitamura H, Yokoyama M, Akita H, Matsushita K, Kurachi Y, Yamada M. Tertiapin potently and selectively blocks muscarinic K+ channels in rabbit cardiac myocytes. J Pharmacol Exp Ther. 2000;293:196–205. [PubMed] [Google Scholar]
- 17.Kodama I, Honjo H, Boyett MR. Are we lost in the labyrinth of the sinoatrial node pacemaker mechanism? J Cardiovasc Electrophysiol. 2002;13:1303–1305. doi: 10.1046/j.1540-8167.2002.01303.x. [DOI] [PubMed] [Google Scholar]
- 18.Lacinová L, Kurejová M, Klugbauer N, Hofmann F. Gating of the expressed T-type Cav3.1 calcium channels is modulated by Ca2+ Acta Physiol (Oxf) 2006;186(24):9–260. doi: 10.1111/j.1748-1716.2006.01539.x. [DOI] [PubMed] [Google Scholar]
- 19.Lakatta EG, DiFrancesco D. What keeps us ticking: a funny current, a calcium clock, or both? J Mol Cell Cardiol. 2009;47(2):157–170. doi: 10.1016/j.yjmcc.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lakatta EG, Maltsev VA, Bogdanov KY, Stern MD, Vinogradova TM. Cyclic variation of intracellular calcium: a critical factor for cardiac pacemaker cell dominance. Circ Res. 2003;92:e45–e50. doi: 10.1161/01.RES.0000055920.64384.FB. [DOI] [PubMed] [Google Scholar]
- 21.Lambert RC, Bessaih T, Leresche N. Modulation of neuronal T-type calcium channels. CNS Neurol Disord Drug Targets. 2006;5:611–627. doi: 10.2174/187152706779025544. [DOI] [PubMed] [Google Scholar]
- 22.Lei M, Brown HF, Terrar DA. Modulation of delayed rectifier potassium current, IK, by isoprenaline in rabbit isolated pacemaker cells. Exp Physiol. 2000;85:27–35. doi: 10.1017/S0958067000019151. [DOI] [PubMed] [Google Scholar]
- 23.Li J, Qu J, Nathan RD. Ionic basis of ryanodine’s negative chronotropic effect on pacemaker cells isolated from the sinoatrial node. Am J Physiol. 1997;273:H2481–H2489. doi: 10.1152/ajpheart.1997.273.5.H2481. [DOI] [PubMed] [Google Scholar]
- 24.Lipsius SL, Bers DM. Cardiac pacemaking: If vs. Ca2+, is it really that simple? J Mol Cell Cardiol. 2003;35:891–893. doi: 10.1016/S0022-2828(03)00184-6. [DOI] [PubMed] [Google Scholar]
- 25.Maltsev VA, Lakatta EG. Normal heart rhythm is initiated and regulated by an intracellular calcium clock within pacemaker cells. Heart Lung Circ. 2007;16:335–348. doi: 10.1016/j.hlc.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maltsev VA, Lakatta EG. Dynamic interactions of an intracellular Ca2+ clock and membrane ion channel clock underlie robust initiation and regulation of cardiac pacemaker function. Cardiovasc Res. 2008;77:274–284. doi: 10.1093/cvr/cvm058. [DOI] [PubMed] [Google Scholar]
- 27.Maltsev VA, Lakatta EG. Synergism of coupled subsarcolemmal Ca2+ clocks and sarcolemmal voltage clocks confers robust and flexible pacemaker function in a novel pacemaker cell model. Am J Physiol Heart Circ Physiol. 2009;296:H594–H615. doi: 10.1152/ajpheart.01118.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maltsev VA, Vinogradova TM, Lakatta EG. The emergence of a general theory of the initiation and strength of the heartbeat. J Pharmacol Sci. 2006;100:338–369. doi: 10.1254/jphs.CR0060018. [DOI] [PubMed] [Google Scholar]
- 29.Masson-Pévet M, Bleeker WK, Gros D. The plasma membrane of leading pacemaker cells in the rabbit sinus node. A qualitative and quantitative ultrastructural analysis. Circ Res. 1979;45:621–629. doi: 10.1161/01.res.45.5.621. [DOI] [PubMed] [Google Scholar]
- 30.Mattick P, Parrington J, Odia E, Simpson A, Collins T, Terrar D. Ca2+-stimulated adenylyl cyclase isoform AC1 is preferentially expressed in guinea pig sino-atrial node cells and modulates the If pacemaker current. J Physiol. 2007;582:1195–1203. doi: 10.1113/jphysiol.2007.133439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Michels G, Brandt MC, Zagidullin N, Khan IF, Larbig R, van Aaken S, Wippermann J, Hoppe UC. Direct evidence for calcium conductance of hyperpolarization-activated cyclic nucleotide-gated channels and human native If at physiological calcium concentrations. Cardiovasc Res. 2008;78:466–475. doi: 10.1093/cvr/cvn032. [DOI] [PubMed] [Google Scholar]
- 32.Mitsuiye T, Shinagawa Y, Noma A. Sustained inward current during pacemaker depolarization in mammalian sinoatrial node cells. Circ Res. 2000;87:88–91. doi: 10.1161/01.res.87.2.88. [DOI] [PubMed] [Google Scholar]
- 33.Nitta J, Furukawa T, Marumo F, Sawanobori T, Hiraoka M. Subcellular mechanism for Ca2+-dependent enhancement of delayed rectifier K+ current in isolated membrane patches of guinea pig ventricular myocytes. Circ Res. 1994;74:96–104. doi: 10.1161/01.res.74.1.96. [DOI] [PubMed] [Google Scholar]
- 34.Petit-Jacques J, Bois P, Bescond J, Lenfant J. Mechanism of muscarinic control of the high-threshold calcium current in rabbit sino-atrial node myocytes. Pflügers Arch. 1993;423:21–27. doi: 10.1007/BF00374956. [DOI] [PubMed] [Google Scholar]
- 35.Rigg L, Mattick PA, Heath BM, Terrar DA. Modulation of the hyperpolarization-activated current (If) by calcium and calmodulin in the guinea pig sino-atrial node. Cardiovasc Res. 2003;57:497–504. doi: 10.1016/S0008-6363(02)00668-5. [DOI] [PubMed] [Google Scholar]
- 36.Rigg L, Terrar DA. Possible role of calcium release from the sarcoplasmic reticulum in pacemaking in guinea pig sino-atrial node. Exp Physiol. 1996;81:877–880. doi: 10.1113/expphysiol.1996.sp003983. [DOI] [PubMed] [Google Scholar]
- 37.Sakmann B, Noma A, Trautwein W. Acetylcholine activation of single muscarinic K+ channels in isolated pacemaker cells of the mammalian heart. Nature. 1983;303:250–253. doi: 10.1038/303250a0. [DOI] [PubMed] [Google Scholar]
- 38.Satoh H. Electrophysiological actions of ryanodine on single rabbit sinoatrial nodal cells. Gen Pharmacol. 1997;28:31–38. doi: 10.1016/s0306-3623(96)00182-6. [DOI] [PubMed] [Google Scholar]
- 39.Sipido KR, Callewaert G, Carmeliet E. Inhibition and rapid recovery of Ca2+ current during Ca2+ release from sarcoplasmic reticulum in guinea pig ventricular myocytes. Circ Res. 1995;76:102–109. doi: 10.1161/01.res.76.1.102. [DOI] [PubMed] [Google Scholar]
- 40.Sun LS, Huber F, Robinson RB, Bilezikian JP, Steinberg SF, Vulliemoz Y. Muscarinic receptor heterogeneity in neonatal rat ventricular myocytes in culture. J Cardiovasc Pharmacol. 1996;27:455–461. doi: 10.1097/00005344-199604000-00001. [DOI] [PubMed] [Google Scholar]
- 41.Tohse N. Calcium-sensitive delayed rectifier potassium current in guinea pig ventricular cells. Am J Physiol. 1990;258:H1200–H1207. doi: 10.1152/ajpheart.1990.258.4.H1200. [DOI] [PubMed] [Google Scholar]
- 42.Verheijck EE, van Ginneken ACG, Bourier J, Bouman LN. Effects of delayed rectifier current blockade by E-4031 on impulse generation in single sinoatrial nodal myocytes of the rabbit. Circ Res. 1995;76:607–615. doi: 10.1161/01.res.76.4.607. [DOI] [PubMed] [Google Scholar]
- 43.Verheijck EE, van Ginneken ACG, Wilders R, Bouman LN. Contribution of L-type Ca2+ current to electrical activity in sinoatrial nodal myocytes of rabbits. Am J Physiol. 1999;276:H1064–H1077. doi: 10.1152/ajpheart.1999.276.3.H1064. [DOI] [PubMed] [Google Scholar]
- 44.Verkerk AO, Wilders R, Zegers JG, van Borren MMGJ, Ravesloot JH, Verheijck EE. Ca2+-activated Cl− current in rabbit sinoatrial node cells. J Physiol. 2002;540:105–117. doi: 10.1113/jphysiol.2001.013184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vinogradova TM, Bogdanov KY, Lakatta EG. β-Adrenergic stimulation modulates ryanodine receptor Ca2+ release during diastolic depolarization to accelerate pacemaker activity in rabbit sinoatrial nodal cells. Circ Res. 2002;90:73–79. doi: 10.1161/hh0102.102271. [DOI] [PubMed] [Google Scholar]
- 46.Vinogradova TM, Lyashkov AE, Zhu W, Ruknudin AM, Sirenko S, Yang D, Deo S, Barlow M, Johnson S, Caffrey JL, Zhou YY, Xiao RP, Cheng H, Stern MD, Maltsev VA, Lakatta EG. High basal protein kinase A-dependent phosphorylation drives rhythmic internal Ca2+ store oscillations and spontaneous beating of cardiac pacemaker cells. Circ Res. 2006;98:505–514. doi: 10.1161/01.RES.0000204575.94040.d1. [DOI] [PubMed] [Google Scholar]
- 47.Vinogradova TM, Sirenko S, Lyashkov AE, Younes A, Li Y, Zhu W, Yang D, Ruknudin AM, Spurgeon H, Lakatta EG. Constitutive phosphodiesterase activity restricts spontaneous beating rate of cardiac pacemaker cells by suppressing local Ca2+ releases. Circ Res. 2008;102:761–769. doi: 10.1161/CIRCRESAHA.107.161679. [DOI] [PubMed] [Google Scholar]
- 48.Willoughby D, Cooper DM. Organization and Ca2+ regulation of adenylyl cyclases in cAMP microdomains. Physiol Rev. 2007;87:965–1010. doi: 10.1152/physrev.00049.2006. [DOI] [PubMed] [Google Scholar]
- 49.Younes A, Lyashkov AE, Graham D, Sheydina A, Volkova MV, Mitsak M, Vinogradova TM, Lukyanenko YO, Li Y, Ruknudin AM, Boheler KR, Eyk JV, Lakatta EG. Ca2+-stimulated basal adenylyl cyclase activity localization in membrane lipid microdomains of cardiac sinoatrial nodal pacemaker cells. J Biol Chem. 2008;283:14461–14468. doi: 10.1074/jbc.M707540200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zaza A, Micheletti M, Brioschi A, Rocchetti M. Ionic currents during sustained pacemaker activity in rabbit sino-atrial myocytes. J Physiol. 1997;505:677–688. doi: 10.1111/j.1469-7793.1997.677ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zaza A, Robinson RB, DiFrancesco D. Basal responses of the L-type Ca2+ and hyperpolarization-activated currents to autonomic agonists in the rabbit sino-atrial node. J Physiol. 1996;491:347–355. doi: 10.1113/jphysiol.1996.sp021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang H, Holden AV, Noble D, Boyett MR. Analysis of the chronotropic effect of acetylcholine on sinoatrial node cells. J Cardiovasc Electrophysiol. 2002;13:465–474. doi: 10.1046/j.1540-8167.2002.00465.x. [DOI] [PubMed] [Google Scholar]