

Summary
Accurate in silico models for the quantitative prediction of the activity of G protein-coupled receptor (GPCR) ligands would greatly facilitate the process of drug discovery and development. Several methodologies have been developed based on the properties of the ligands, the direct study of the receptor-ligand interactions, or a combination of both approaches. Ligand-based three-dimensional quantitative structure-activity relationships (3D-QSAR) techniques, not requiring knowledge of the receptor structure, have been historically the first to be applied to the prediction of the activity of GPCR ligands. They are generally endowed with robustness and good ranking ability; however they are highly dependent on training sets. Structure-based techniques generally do not provide the level of accuracy necessary to yield meaningful rankings when applied to GPCR homology models. However, they are essentially independent from training sets and have a sufficient level of accuracy to allow an effective discrimination between binders and nonbinders, thus qualifying as viable lead discovery tools. The combination of ligand and structure-based methodologies in the form of receptor-based 3D-QSAR and ligand and structure-based consensus models results in robust and accurate quantitative predictions. The contribution of the structure-based component to these combined approaches is expected to become more substantial and effective in the future, as more sophisticated scoring functions are developed and more detailed structural information on GPCRs is gathered.
Keywords: 3D-QSAR, G protein-coupled receptors (GPCR), homology modeling, molecular docking, virtual screening (VS)
Introduction
GPCRs are a large superfamily of membrane-bound proteins that, upon recognition of specific extracellular stimuli, give rise to a complex signaling cascade generally starting with the activation of heterotrimeric G proteins. Typically, the stimuli are provided by extracellular endogenous ligands, such as neurotransmitters and hormones, exogenous ligands, such as odorants and tastes, or light photons, as it applies to the case of rhodopsin [1]. Due to the plethora of physiological implications of their signaling, GPCR constitute the single protein family most targeted by the currently marketed drugs [2].
All GPCRs share a common structure, consisting of a single polypeptide chain that transverses seven times the cell membrane with seven alpha-helical transmembrane domains (TMs). Despite the large number of GPCRs, for many years direct structural information was available only for bovine rhodopsin, mainly through the 2D projection map disclosed by Schertler and coworkers in 1993 and the three-dimensional (3D) crystal structure solved for the first time by Palczewski and coworkers in 2000 [3,4]. Thus, structure-function studies of GPCRs relied heavily on sequence and phylogenetic analyses, rhodopsin-based homology modeling, and docking studies supported by site-directed mutagenesis and ligand structure-activity relationships (SAR) data [5,6]. In this context, experimentally validated GPCR homology models have proven to be valuable tools for lead identification, and the scientific literature flourished with successful rational drug design and virtual screening examples [7–11]. In 2007 Kobilka and coworkers unveiled the 3D crystal structure of the human β2-adrenergic receptor (β2-AR), finally providing the long awaited proof that the structure of GPCRs generally resembles that of rhodopsin [12–15]. Comparison between rhodopsin-based models of the β2-AR in complex with the inverse agonist carazolol and the corresponding crystal structure supports and encourages the applicability of GPCR homology modeling and molecular docking to computer-aided drug discovery, at least for qualitative purposes [16].
However, computer-assisted optimization of lead compounds toward the design of molecules with improved affinity is not a straightforward matter, as it requires models capable of quantitatively calculating and ranking the activity of series of analogs. Predicting with accuracy free binding energy and/or potencies of ligands is in general a daunting task. Due to the paucity of direct structural information, the quantitative prediction of the biological activity of GPCR ligands presents even greater difficulties. A number of approaches have been proposed to address this challenging issue: most of them rely on ligand-based QSAR, while others attempt a direct calculation of receptor-ligand interactions.
Ligand-based 3D-QSAR
Under the label of ligand-based 3D-QSAR are enlisted several QSAR methodologies that attempt to correlate the biological activities of molecules with a number of location-dependent measures that describe their molecular properties, without any explicit calculation of their interactions with the targets [17,18].
Among the various 3D-QSAR approaches, Comparative Molecular Field Analysis (CoMFA) is one of the earliest examples and perhaps still the most widespread. Introduced in 1988 by Cramer and coworkers at Tripos, CoMFA relies on a 3D description of molecular properties based on the analysis of molecular fields detected by a probe atom and mapped on a grid [19]. Fundamental to the development and success of CoMFA has been the introduction of the Partial Least Square (PLS) statistical approach applied to chemistry by Wold in 1984, which allows meaningful correlations in situations in which the variables outnumber the observations [20]. A CoMFA study typically analyzes electrostatic fields, in the form of a Coulombic function, and steric fields, in the form of a Lennard-Jones function [17]. In 1994, Klebe and coworkers introduced the Comparative Molecular Similarity Indices Analysis (CoMSIA) approach, in which molecular fields are expressed in the form of Gaussian-type functions. To attempt an account of the entropic contribution to receptor-ligand binding, alongside steric and electrostatic fields CoMSIA considers also hydrophobic fields [21].
Before the disclosure of the 3D structure of rhodopsin, the receptor-based design of GPCR ligands was not even feasible through homology modeling. Thus, many researchers turned to 3D-QSAR in an attempt to shed light on the still vague receptor-ligand interactions. An article published by Greco and coworkers in 1991 is probably the first account of the applicability of CoMFA to GPCRs [22]. In their paper, the authors described the application of CoMFA to the study of a set of non-congeneric agonists of the muscarinic receptors, which led to a model consistent with the postulated mechanism of interaction. In the following years, CoMFA and CoMSIA continued to be successfully applied to the prediction of the activities of GPCR ligands. The publication of the crystal structure of rhodopsin gave a new impetus to 3D-QSAR studies, as receptor docking became a practical way of superimposing ligands in the 3D space – a special section of this review is dedicated to combined ligand and structure-based modeling. Among GPCRs, the adenosine receptors have been extensively studied by means of CoMFA and CoMSIA. The first paper featuring the description of a CoMFA study applied to adenosine receptors was published by Jacobson and coworkers in 1995 and dealt with the 3D-QSAR of a series of N6-benzyladenosine-5’-uronamide analogs as agonists of the A3 subtype [23]. Many similar studies followed in subsequent years, conducted by the same group as well as various other laboratories [24–37]. In this context, in 2007 Kim and Jacobson developed CoMFA and CoMSIA models based on structurally diverse adenosine analogues and successfully elucidated not only the molecular basis for their affinity but also those for their relative efficacy at the human A3 receptor [35].
As an alternative to CoMFA, the GRID/GOLPE combination has also been successfully used to generate predictive 3D-QSAR models. The program GRID, published by Goodford in 1985, allows a thorough exploration of the molecular interaction fields with a large number of different molecular probes [38]. The descriptors produced with GRID can then be subjected to chemometric analysis with the program GOLPE (General Optimal Linear PLS Estimation), developed by Clementi and coworkers, which offers the advantage of an advanced variable selection procedure [39]. The GRID/GOLPE analysis has been successfully applied to the development of 3D-QSAR models for the prediction of the activity of ligands of a number of GPCRs, including adrenergic, dopamine, serotonin and opioid receptors [40–44].
Another viable and convenient way for the derivation of descriptors for 3D-QSAR models was identified by Gasteiger and coworkers in the calculation of autocorrelation vectors of molecular electrostatic potentials (MEPs) mapped over the molecular surfaces of the ligands [45,46]. It is well accepted that MEPs considerably affect and govern molecular recognition. Furthermore, spatial autocorrelation vectors offer the advantage of being independent of the 3D orientation of the molecules. On these bases, Moro and coworkers recently introduced the autocorrelation of molecular electrostatic surface properties combined with partial least square analysis (autoMEP/PLS) as an attractive alternative to CoMFA [36,47–49]. The authors applied the novel strategy to the study of the activity of antagonists of the adenosine A3 receptor. Their results demonstrate that autoMEP/PLS has a predictive power comparable to that of CoMFA, with the advantage of not requiring 3D alignment of ligands. This property makes autoMEP/PLS a particularly attractive tool to generate 3D-QSAR models in cases in which the receptor-ligand interactions have not been clearly established, thus preventing the use of structure-based methodologies to generate the 3D alignment of the ligands.
Aside from 3D-QSAR, other approaches to QSAR have also been successfully applied to the prediction of the activity of GPCR ligands, as extensively reviewed by Tropsha and coworkers [50].
Structure-based methodologies
The most direct method for the quantitative prediction of ligand-protein affinity is the calculation of the free energy of binding from the 3D structure of the complexes while employing first principle techniques, such as free energy perturbations (FEP) or thermodynamic integration (TI). Especially when dealing with high-resolution protein crystal structures, these methodologies have proven the ability to reproduce with accuracy experimentally measured binding affinities [51–54]. In the field of GPCRs, a successful attempt to calculate the free energy of binding between the cholecystokinin-1 receptor (CCK1R) and a cholecystokinin peptide composed of nine amino acids (CCK9) was made by performing “alchemical” transformation of the peptide into a mutated one with known affinity for the receptor using FEP in a water-lipid environment [55]. The initial protein-ligand complex for this study was generated by means of a number of optimizations of a rhodopsin-based homology model of CCK1R conducted in an iterative manner with site-directed mutagenesis experiments. The very encouraging reproduction of relative free binding energies supported the validity of the model and the experimentally derived assumptions on the basis of which it was constructed.
However, first principle calculations are time-consuming and require a great deal of effort for the preparation of the system and the optimization of the sampling. Therefore, they are not suitable for the study of many ligand-protein complexes. To overcome these limitations, in 1994 Aqvist and coworkers introduced the linear-interaction energy (LIE) method, also known as the linear response method (LRM), which offers a more simplified and rapid approach to the calculation of the free energy of binding [56,57]. By means of molecular dynamics or Monte Carlo simulations, average electrostatic and van der Waals energies are calculated for the ligand in the bound state and free in solution. In the earliest formulation of the LIE method, the free energy of binding is represented as a linear combination of the differences between the energies in the two states according to the formula:
where the scaling factor α is empirically determined, while β is set to 0.5. Subsequently, the methodology has been extensively developed and refined giving rise to a number of alternative variants. Notably, the most recent versions feature an empirically determined β factor and a third term representing the solvent accessible surface area (SASA), as proposed by Jorgensen and coworkers [58]. Furthermore, the methodology has been adapted in order to function with Poisson-Boltzmann (PB) and Generalized Born (GB) continuum models of solvation with levels of accuracy comparable to those obtained with explicit solvent [59].
Regarding GPCRs, there have been a few attempts of calculating ligand binding energies through the LIE method. In particular, Gutiérrez-de-Terán and coworkers applied it with the aim of discriminating among two different binding modes of agonists of the adenosine A1 receptor generated by automatic docking [60]. More recently, we applied the LIE method to the calculation of the activity of a series of antagonists of the P2Y1 receptor [7]. The method did not show the ability to rank the activity of a test set of adenosine-3’-5’-bisphosphate analogs. However, it yielded relatively low root-mean-square errors of predictions and demonstrated substantial independence from the training set, supporting the applicability of structure-based methodologies to the discrimination between binders and non-binders. The combination of this specific model with ligand-based 3D-QSAR toward the prediction of the activity of P2Y1 antagonists is further discussed in the next section.
A more simplified and rapid quantitative description of ligand-protein interactions can be obtained through the application of scoring functions, which are intended for a rough high-throughput estimation of binding affinities. Scoring functions can be classified into three groups: force field-based, empirical, and knowledge-based [18,61–66]. The first are based on the evaluation of the affinity on the basis of molecular mechanics energy terms for non-bonded interactions, such as van der Waals and electrostatic energies. Knowledge-based scoring functions are based on the decomposition of ligand-protein binding into a list of interactions between atom pairs, which are evaluated with respect to a database of experimentally determined protein-ligand complexes [63–66]. The interaction between a particular atom type of a protein and a particular atom type of a ligand located at a specific distance is considered more or less favorable proportionally to the number of times it occurs in the database. Lastly, empirical scoring functions are based on a number of descriptors intended to reflect different contributions to binding affinity, such as steric and electrostatic interactions, hydrogen bonds, lipophilic and entropic effects, and solvation, all calibrated to fit experimental data on the basis of given training sets.
The empirical scoring function of the internal coordinate mechanics (ICM) program includes terms accounting for van der Waals interactions, ligand conformational entropy loss, hydrogen bond formation and disruption, solvation electrostatic energy change, hydrophobic free energy gain. In 2003, Cavasotto and coworkers demonstrated the applicability of this scoring function to the identification of the binding site of retinal in bovine rhodopsin and to the discrimination between binders and nonbinders of rhodopsin [67]. Similarly, using molecular databases spiked with known binders, a number of studies based on combinations of different scoring functions (such as Gold [68], Dock [69], CScore (Tripos), Fresno [70], Score [71], FlexX [72], and PMF [66]) have demonstrated the applicability of molecular docking at GPCR models to virtual screening [9,10,73]. In agreement with the expectations set by these studies, a variety of docking programs and scoring functions have been successfully applied to the discovery of novel ligands for a plethora of GPCRs, including neurorokinin [74,75], adrenergic [76], chemokine [75,77], dopamine [75], serotonin [75,78], cannabinoid [79], and free fatty acid receptors [80], among others.
Combining ligand and structure-based methodologies
A crucial element in the construction of 3D-QSAR models is the structural alignment of the ligands, which may prove particularly challenging in the case of structurally diverse or highly flexible compounds. To overcome these difficulties, in 1993 Marshall and coworkers proposed the utilization of alignment techniques based on binding site geometries and minimizations within binding sites as effective alternatives to conformational searches and superimpositions solely based on the molecular properties of the ligands [81,82]. In the following years, with the development of sophisticated automatic docking algorithms and programs, this combined approach became increasingly more feasible and appealing. Much work needs to be done to improve the scoring functions towards a direct estimation of free energies of binding (see structure-based methodologies section). However, docking programs have demonstrated a substantial reliability in the prediction of ligand binding modes and bioactive conformations [83]. Dealing with crystal structures of nuclear hormone receptors and enzymes, Sippl amply demonstrated the advantages of combining the accuracy of ligand alignments obtained through docking with the computational efficiency of 3D-QSAR methodologies [84–87]. Receptor-based 3D-QSAR – in which receptor-ligand complexes are not used to directly calculate binding interactions, but only to generate the 3D alignment of the ligands – has also been applied successfully to GPCRs, by means of docking studies conducted at homology models derived using rhodopsin as a structural template [7,37,44,48,88–91]. Notably, Moro and coworkers adopted this approach to obtain a quantitative model for the prediction of the activities of antagonists of the human A3 receptor. On the basis of this model, the authors designed, synthesized, and tested a set of novel compounds. The experimentally determined affinities of the novel antagonists were remarkably close to their predicted values, with an r2 of 0.873 [37,48].
Recently, we combined receptor-based QSAR techniques and purely structure-based techniques to generate a model for the prediction of the activity of antagonists of the P2Y1 receptor [7]. We docked a set of antagonists to a rhodopsin-based model of the receptor and, as mentioned earlier in the paper, calculated docking scores and free binding energies via the LIE method, as implemented in the Schrödinger software package. In parallel, we used the alignment resulting from docking for the construction of receptor-based CoMFA and CoMSIA models. Additionally, in order to benefit from the strength of each technique and to compensate for their inherent limitations, we generated a consensus PLS model named LIST-CM, featuring as descriptors the activities independently predicted through receptor-based 3D-QSAR and through the direct calculation of receptor-ligand interactions. A schematic representation of this combined approach is provided in Figure 1. To test the predictive power of this model, we designed, synthesized, and tested a challenging test set of novel antagonists, each of which featured at least one substituent not present in the training set. With the training set, receptor-based 3D-QSAR methodologies demonstrated a predictivity far superior to that of purely receptor-based techniques. These results were expected, considering that homology models cannot provide a sufficiently high resolution picture of the binding site to rank with accuracy the activities of a series of analogues. Although not endowed with the ability to rank analogs, as mentioned, purely receptor-based methodologies showed relatively low root-mean-square errors of prediction together with a substantial independence from the training set, thus confirming their applicability to lead identification.
Figure 1.

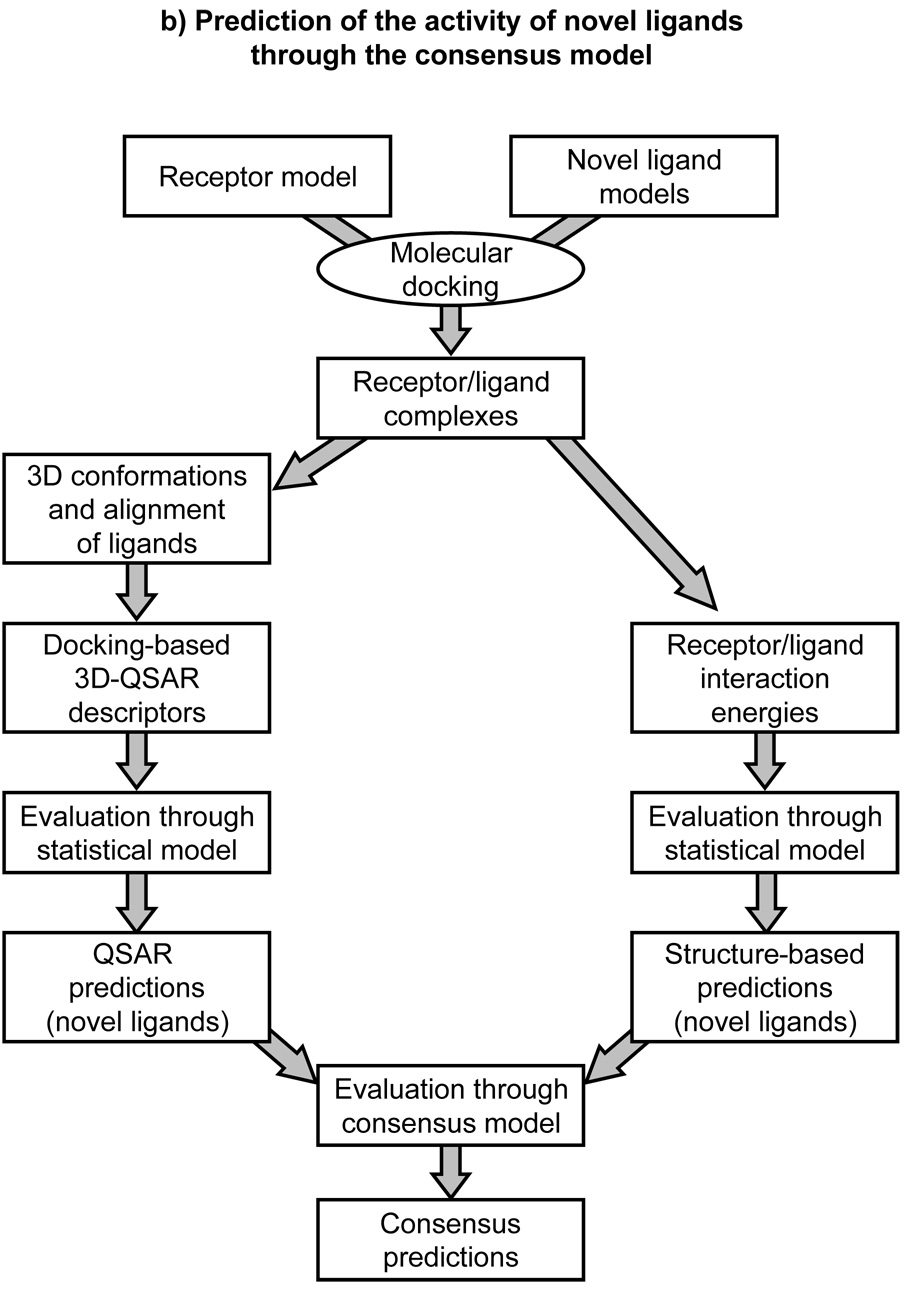
Schematic representation of LIST-CM, a combined ligand and structure-based approach to the quantitative prediction of ligand activities [7]. Scheme (a) depicts the construction of a predictive consensus model on the basis of a training set of ligands with known activities. Geometries of receptor-ligand complexes are obtained trough molecular docking of the training set. Subsequently, ligand-based 3D-QSAR descriptors are calculated and used to generate statistical models for the prediction of the activities of the training set (left side of the scheme). In parallel, receptor-ligand interaction energies are also calculated and used to generate statistical models for the prediction of the activities of the training set (right side of the scheme). Ligand and structure-based predictions are then combined together to yield a consensus model. Scheme (b) depicts the prediction of the activity of novel ligands through the consensus model. Geometries of receptor-ligand complexes are obtained through molecular docking of the novel ligands and used to calculate 3D-QSAR descriptors (left side of the scheme) and receptor-ligand interactions (right side of the scheme), which are in turn used to generate predictions on the basis of the statistical models previously obtained with the training set. Ligand and structure-based predictions are then used to generate consensus predictions through the consensus model also previously obtained with the training set.
With a challenging test set, our consensus model that combines receptor-based 3D-QSAR and purely receptor-based techniques proved to be more robust than any individual model, and resulted in the accurate prediction of the activity of six out of the eight new compounds [7]. These results suggest that the combination of a number of orthogonal methodologies might work synergistically toward obtaining improved, more robust models.
Summary and conclusions
In silico methodologies for the quantitative prediction of the affinity of small molecules of pharmaceutical interest would greatly facilitate the process of drug discovery and development. However, due to the lack of direct 3D structural information, the application of these methodologies to the study of the activity of GPCR ligands is particularly challenging.
Ligand-based 3D-QSAR techniques have been applied for years in order to rationalize the structure-activity relationships of GPCR agonists and antagonists with the aim of producing models capable of quantitatively predicting the activity of new analogs. Being based exclusively on the properties of known ligands, these techniques offer the advantage of not requiring knowledge of the receptor structure. This aspect made possible their application to the discovery of GPCR ligands even before the publication of the experimentally elucidated rhodopsin structures that opened the way to GPCR homology modeling. Ligand-based 3D-QSAR techniques are capable of generating accurate and robust models leading to precise ranking of the activity of series of analog compounds. However, these models are highly dependent on the training sets. Thus, they are not applicable to situations in which activity data for a set of ligands of a specific target are not available. Furthermore, they often fail in the prediction of the activity of molecules with a structure significantly diverging from those included in the training set.
Conversely, structure-based methodologies rely on the direct estimation of the free energy of binding from the 3D structure of receptor-ligand complexes. Although essentially independent from a priori knowledge of the activity of ligands at the studied target, direct methods for the evaluation of the receptor-ligand affinity are either too computationally expensive to be considered for high-throughput drug discovery purposes or too approximate to yield meaningful affinity rankings. Furthermore, in most cases homology models do not provide the level of accuracy necessary to rank the affinity of a series of analogues to be exploited for lead-optimization purposes. However, structure-based methodologies often have a sufficient level of accuracy to allow an effective discrimination between binders and nonbinders. This, together with their substantial independence from training sets, allows their successful application in the quest for novel and structurally diverse lead compounds by means of virtual screening techniques.
Despite the inaccuracy and vagueness of scoring functions, docking programs have demonstrated the ability of producing sensible conformations of receptor-ligand complexes. In the case of GPCRs, this is especially true if the models incorporate experimental data usually obtained through site-directed mutagenesis and chemical modification of the ligands. On this basis, molecular docking appears to be a very good alternative to techniques based exclusively on the electrostatic and steric properties of the small molecules to yield the bioactive conformations and 3D superimpositions of the ligands necessary to generate 3D-QSAR models. The receptor-based 3D-QSAR models obtained in this fashion, besides being generally characterized by accuracy and robustness, also allow the docking of the generated QSAR contours into the model of the receptor, thus generating hybrid models intended to explain the structure-activity relationships in terms of receptor-ligand interactions. Additionally, although not endowed with ranking ability when considered alone, structure-based predictions might contribute to the construction of robust consensus models when combined with 3D-QSAR predictions [7]. The contribution of structure-based methodologies to these combined approaches is expected to increase significantly in the future, with the advent of additional direct 3D structural information for GPCRs and the introduction of more sophisticated and accurate scoring functions.
Acknowledgements
The authors thank Dr. Santiago Vilar Varela for the helpful discussions. This research was supported by the Intramural Research Program of the NIH, NIDDK.
Abbreviations
- 3D
Three-dimensional
- GB
generalized Born
- GPCR
G protein coupled receptor
- LIE
linear interaction energy
- LRM
linear response method
- MEP
molecular electrostatic potentials
- QSAR
quantitative structure-activity relationships
- PB
Poisson-Boltzman
- PLS
partial least square
- SAR
structure-activity relationships
- TM
transmembrane domain
References
- 1.Pierce KL, Premont RT, Lefkowitz RJ. Nature Reviews Molecular Cell Biology. 2002;3:639. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 2.Overington JP, Al-Lazikani B, Hopkins AL. Nature Reviews Drug Discovery. 2006;5:993. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 3.Schertler GFX, Villa C, Henderson R. Nature. 1993;362:770. doi: 10.1038/362770a0. [DOI] [PubMed] [Google Scholar]
- 4.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Science. 2000;289:739. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 5.Moro S, Spalluto G, Jacobson KA. Trends in Pharmacological Sciences. 2005;26:44. doi: 10.1016/j.tips.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 6.Costanzi S, Ivanov AA, Tikhonova IG, Jacobson KA. In: Frontiers in Drug Design and Discovery. Caldwell GW, Rahman AU, Player MR, Chouday MI, editors. Bentham: 2007. pp. 63–79. [Google Scholar]
- 7.Costanzi S, Tikhonova IG, Ohno M, Roh EJ, Joshi BV, Colson AO, Houston D, Maddileti S, Harden TK, Jacobson KA. Journal of Medicinal Chemistry. 2007;50:3229. doi: 10.1021/jm0700971. [DOI] [PubMed] [Google Scholar]
- 8.Patny A, Desai PV, Avery MA. Current Medicinal Chemistry. 2006;13:1667. doi: 10.2174/092986706777442002. [DOI] [PubMed] [Google Scholar]
- 9.Bissantz C, Schalon C, Guba W, Stahl M. Proteins-Structure Function and Bioinformatics. 2005;61:938. doi: 10.1002/prot.20651. [DOI] [PubMed] [Google Scholar]
- 10.Bissantz C, Bernard P, Hibert M, Rognan D. Proteins-Structure Function and Genetics. 2003;50:5. doi: 10.1002/prot.10237. [DOI] [PubMed] [Google Scholar]
- 11.Evers A, Hessler G, Matter H, Klabunde T. Journal of Medicinal Chemistry. 2005;48:5448. doi: 10.1021/jm050090o. [DOI] [PubMed] [Google Scholar]
- 12.Rasmussen SGF, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VRP, Sanishvili R, Fischetti RF, Schertler GFX, Weis WI, Kobilka BK. Nature. 2007;450:383. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 13.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. Science. 2007;318:1258. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK. Science. 2007;318:1266. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 15.Day PW, Rasmussen SGF, Parnot C, Fung JJ, Masood A, Kobilka TS, Yao XJ, Choi HJ, Weis WI, Rohrer DK, Kobilka BK. Nature Methods. 2007;4:927. doi: 10.1038/nmeth1112. [DOI] [PubMed] [Google Scholar]
- 16.Costanzi S. Journal of Medicinal Chemistry. 2008 accepted. [Google Scholar]
- 17.Green SM, Marshall GR. Trends in Pharmacological Sciences. 1995;16:285. doi: 10.1016/s0165-6147(00)89052-5. [DOI] [PubMed] [Google Scholar]
- 18.Holtje HD, Sippl W, Rognan D, Folkers G. Basic Principles and Applications. Weinheim: Wiley-WCH; 2003. Molecular Modeling. [Google Scholar]
- 19.Cramer RD, Patterson DE, Bunce JD. Journal of the American Chemical Society. 1988;110:5959. doi: 10.1021/ja00226a005. [DOI] [PubMed] [Google Scholar]
- 20.Wold S, Albano C, Dunn WJ, Esbensen K, Hellberg S, Johansson E, Lindberg W, Sjostrom M. Analusis. 1984;12:477. [Google Scholar]
- 21.Klebe G, Abraham U, Mietzner T. Journal of Medicinal Chemistry. 1994;37:4130. doi: 10.1021/jm00050a010. [DOI] [PubMed] [Google Scholar]
- 22.Greco G, Novellino E, Silipo C, Vittoria A. Quantitative Structure-Activity Relationships. 1991;10:289. [Google Scholar]
- 23.Siddiqi SM, Pearlstein RA, Sanders LH, Jacobson KA. Bioorganic & Medicinal Chemistry. 1995;3:1331. doi: 10.1016/0968-0896(95)00116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moro S, van Rhee AM, Sanders LH, Jacobson KA. Journal of Medicinal Chemistry. 1998;41:46. doi: 10.1021/jm970446z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baraldi PG, Borea PA, Bergonzoni M, Cacciari B, Ongini E, Recanatini M, Spalluto G. Drug Development Research. 1999;46:126. [Google Scholar]
- 26.Li AH, Moro S, Forsyth N, Melman N, Ji XD, Jacobson KA. Journal of Medicinal Chemistry. 1999;42:706. doi: 10.1021/jm980550w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doytchinova I, Valkova I, Natcheva R. Quantitative Structure-Activity Relationships. 2001;20:124. [Google Scholar]
- 28.Song YQ, Coupar IM, Iskander MN. Quantitative Structure-Activity Relationships. 2001;20:23. [Google Scholar]
- 29.Rieger JM, Brown ML, Sullivan GW, Linden J, Macdonald TL. Journal of Medicinal Chemistry. 2001;44:531. doi: 10.1021/jm0003642. [DOI] [PubMed] [Google Scholar]
- 30.Doytchinova I. Journal of Computer-Aided Molecular Design. 2001;15:29. doi: 10.1023/a:1011150120831. [DOI] [PubMed] [Google Scholar]
- 31.Jacobson KA, Moro S, Manthey JA, West PL, Ji XD. Flavonoids in Cell Function. 2002;505:163. doi: 10.1007/978-1-4757-5235-9_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doytchinova I, Valkova I, Natcheva R. Sar and Qsar in Environmental Research. 2002;13:227. doi: 10.1080/10629360290002712. [DOI] [PubMed] [Google Scholar]
- 33.Bhattacharya P, Leonard JT, Roy K. Journal of Molecular Modeling. 2005;11:516. doi: 10.1007/s00894-005-0273-6. [DOI] [PubMed] [Google Scholar]
- 34.Fossa P, Pestarino M, Menozzi G, Mosti L, Schenone S, Ranise A, Bondavalli F, Trincavelli ML, Lucacchini A, Martini C. Organic & Biomolecular Chemistry. 2005;3:2262. doi: 10.1039/b502831k. [DOI] [PubMed] [Google Scholar]
- 35.Kim SK, Jacobson KA. Journal of Chemical Information and Modeling. 2007;47:1225. doi: 10.1021/ci600501z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moro S, Bacilieri M, Cacciari B, Spalluto G. Journal of Medicinal Chemistry. 2005;48:5698. doi: 10.1021/jm0502440. [DOI] [PubMed] [Google Scholar]
- 37.Moro S, Braiuca P, Deflorian F, Ferrari C, Pastorin G, Cacciari B, Baraldi PG, Varani K, Borea PA, Spalluto G. Journal of Medicinal Chemistry. 2005;48:152. doi: 10.1021/jm049662f. [DOI] [PubMed] [Google Scholar]
- 38.Goodford PJ. Journal of Medicinal Chemistry. 1985;28:849. doi: 10.1021/jm00145a002. [DOI] [PubMed] [Google Scholar]
- 39.Baroni M, Costantino G, Cruciani G, Riganelli D, Valigi R, Clementi S. Quantitative Structure-Activity Relationships. 1993;12:9. [Google Scholar]
- 40.Balle T, Andersen K, Soby KK, Liljefors T. Journal of Molecular Graphics & Modelling. 2003;21:523. doi: 10.1016/S1093-3263(03)00120-7. [DOI] [PubMed] [Google Scholar]
- 41.Brea J, Rodrigo J, Carrieri A, Sanz F, Cadavid MI, Enguix MJ, Villazon M, Mengod G, Caro Y, Masaguer CF, Ravina E, Centeno NB, Carotti A, Loza MI. Journal of Medicinal Chemistry. 2002;45:54. doi: 10.1021/jm011014y. [DOI] [PubMed] [Google Scholar]
- 42.Broer BM, Gurrath M, Holtje HD. Journal of Computer-Aided Molecular Design. 2003;17:739. doi: 10.1023/b:jcam.0000017491.97244.69. [DOI] [PubMed] [Google Scholar]
- 43.Audouze K, Nielsen EO, Peters D. Journal of Medicinal Chemistry. 2004;47:3089. doi: 10.1021/jm031111m. [DOI] [PubMed] [Google Scholar]
- 44.Terzioglu N, Holtje D. Collection of Czechoslovak Chemical Communications. 2005;70:1482. [Google Scholar]
- 45.Gasteiger J, Li XZ, Rudolph C, Sadowski J, Zupan J. Journal of the American Chemical Society. 1994;116:4608. [Google Scholar]
- 46.Wagener M, Sadowski J, Gasteiger J. Journal of the American Chemical Society. 1995;117:7769. [Google Scholar]
- 47.Moro S, Bacilieri M, Ferrari C, Spalluto G. Curr.Drug Discov.Technol. 2005;2:13. doi: 10.2174/1570163053175439. [DOI] [PubMed] [Google Scholar]
- 48.Moro S, Deflorian F, Bacilieri M, Spalluto G. Current Medicinal Chemistry. 2006;13:639. doi: 10.2174/092986706776055670. [DOI] [PubMed] [Google Scholar]
- 49.Moro S, Bacilieri M, Cacciari B, Bolcato C, Cusan C, Pastorin G, Klotz KN, Spalluto G. Bioorganic & Medicinal Chemistry. 2006;14:4923. doi: 10.1016/j.bmc.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 50.Tropsha A, Wang SX. Ernst.Schering.Found.Symp.Proc. 2006;2:49. doi: 10.1007/2789_2006_003. [DOI] [PubMed] [Google Scholar]
- 51.Wang J, Dixon R, Kollman PA. Proteins-Structure Function and Genetics. 1999;34:69. [PubMed] [Google Scholar]
- 52.Chipot C, Rozanska X, Dixit SB. Journal of Computer-Aided Molecular Design. 2005;19:765. doi: 10.1007/s10822-005-9021-3. [DOI] [PubMed] [Google Scholar]
- 53.Foloppe N, Hubbard R. Current Medicinal Chemistry. 2006;13:3583. doi: 10.2174/092986706779026165. [DOI] [PubMed] [Google Scholar]
- 54.Wang JY, Deng YQ, Roux B. Biophysical Journal. 2006;91:2798. doi: 10.1529/biophysj.106.084301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Henin J, Maigret B, Tarek M, Escrieut C, Fourmy D, Chipot C. Biophysical Journal. 2006;90:1232. doi: 10.1529/biophysj.105.070599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aqvist J, Medina C, Samuelsson JE. Protein Engineering. 1994;7:385. doi: 10.1093/protein/7.3.385. [DOI] [PubMed] [Google Scholar]
- 57.Hansson T, Marelius J, Aqvist J. Journal of Computer-Aided Molecular Design. 1998;12:27. doi: 10.1023/a:1007930623000. [DOI] [PubMed] [Google Scholar]
- 58.JonesHertzog DK, Jorgensen WL. Journal of Medicinal Chemistry. 1997;40:1539. doi: 10.1021/jm960684e. [DOI] [PubMed] [Google Scholar]
- 59.Carlsson J, Ander M, Nervall M, Aqvist J. Journal of Physical Chemistry B. 2006;110:12034. doi: 10.1021/jp056929t. [DOI] [PubMed] [Google Scholar]
- 60.Gutierrez-de-Teran H, Pastor M, Centeno NB, Aqvist J, Sanz F. Chembiochem. 2004;5:841. doi: 10.1002/cbic.200300817. [DOI] [PubMed] [Google Scholar]
- 61.Coupez B, Lewis RA. Current Medicinal Chemistry. 2006;13:2995. doi: 10.2174/092986706778521797. [DOI] [PubMed] [Google Scholar]
- 62.Konstantinou-Kirtay C, Mitchell JBO, Lumley JA. Bmc Bioinformatics. 2007;8 doi: 10.1186/1471-2105-8-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gohlke H, Hendlich M, Klebe G. Journal of Molecular Biology. 2000;295:337. doi: 10.1006/jmbi.1999.3371. [DOI] [PubMed] [Google Scholar]
- 64.Ishchenko AV, Shakhnovich EI. Journal of Medicinal Chemistry. 2002;45:2770. doi: 10.1021/jm0105833. [DOI] [PubMed] [Google Scholar]
- 65.Mitchell JBO, Laskowski RA, Alex A, Thornton JM. Journal of Computational Chemistry. 1999;20:1165. [Google Scholar]
- 66.Muegge I, Martin YC. Journal of Medicinal Chemistry. 1999;42:791. doi: 10.1021/jm980536j. [DOI] [PubMed] [Google Scholar]
- 67.Cavasotto CN, Orry AJW, Abagyan RA. Proteins-Structure Function and Genetics. 2003;51:423. doi: 10.1002/prot.10362. [DOI] [PubMed] [Google Scholar]
- 68.Jones G, Willett P, Glen RC. Journal of Molecular Biology. 1995;245:43. doi: 10.1016/s0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]
- 69.Kuntz ID. Science. 1992;257:1078. doi: 10.1126/science.257.5073.1078. [DOI] [PubMed] [Google Scholar]
- 70.Rognan D, Lauemoller SL, Holm A, Buus S, Tschinke V. Journal of Medicinal Chemistry. 1999;42:4650. doi: 10.1021/jm9910775. [DOI] [PubMed] [Google Scholar]
- 71.Wang RX, Liu L, Lai LH, Tang YQ. Journal of Molecular Modeling. 1998;4:379. [Google Scholar]
- 72.Rarey M, Kramer B, Lengauer T, Klebe G. Journal of Molecular Biology. 1996;261:470. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 73.Vaidehi N, Schlyer S, Trabanino RJ, Floriano WB, Abrol R, Sharma S, Kochanny M, Koovakat S, Dunning L, Liang M, Fox JM, de Mendonca FL, Pease JE, Goddard WA, Horuk R. Journal of Biological Chemistry. 2006;281:27613. doi: 10.1074/jbc.M601389200. [DOI] [PubMed] [Google Scholar]
- 74.Evers A, Klebe G. Journal of Medicinal Chemistry. 2004;47:5381. doi: 10.1021/jm0311487. [DOI] [PubMed] [Google Scholar]
- 75.Becker OM, Marantz Y, Shacham S, Inbal B, Heifetz A, Kalid O, Bar-Haim S, Warshaviak D, Fichman M, Noiman S. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:11304. doi: 10.1073/pnas.0401862101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Evers A, Klabunde T. Journal of Medicinal Chemistry. 2005;48:1088. doi: 10.1021/jm0491804. [DOI] [PubMed] [Google Scholar]
- 77.Kellenberger E, Springael JY, Parmentier M, Hachet-Haas M, Galzi JL, Rognan D. Journal of Medicinal Chemistry. 2007;50:1294. doi: 10.1021/jm061389p. [DOI] [PubMed] [Google Scholar]
- 78.Becker OM, Dhanoa DS, Marantz Y, Chen DL, Shacham S, Cheruku S, Heifetz A, Mohanty P, Fichman M, Sharadendu A, Nudelman R, Kauffman M, Noiman S. Journal of Medicinal Chemistry. 2006;49:3116. doi: 10.1021/jm0508641. [DOI] [PubMed] [Google Scholar]
- 79.Chen JZ, Wang JM, Xie XQ. Journal of Chemical Information and Modeling. 2007;47:1626. doi: 10.1021/ci7000814. [DOI] [PubMed] [Google Scholar]
- 80.Tikhonova IG, Sum CS, Neumann S, Engel S, Raaka BM, Costanzi S, Gershengorn MC. Journal of Medicinal Chemistry. 2008;51:625. doi: 10.1021/jm7012425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Waller CL, Oprea TI, Giolitti A, Marshall GR. Journal of Medicinal Chemistry. 1993;36:4152. doi: 10.1021/jm00078a003. [DOI] [PubMed] [Google Scholar]
- 82.Depriest SA, Mayer D, Naylor CB, Marshall GR. Journal of the American Chemical Society. 1993;115:5372. [Google Scholar]
- 83.Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS. Journal of Medicinal Chemistry. 2006;49:5912. doi: 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]
- 84.Sippl W. Journal of Computer-Aided Molecular Design. 2000;14:559. doi: 10.1023/a:1008115913787. [DOI] [PubMed] [Google Scholar]
- 85.Sippl W, Holtje HD. Journal of Molecular Structure-Theochem. 2000;503:31. [Google Scholar]
- 86.Sippl W. Bioorganic & Medicinal Chemistry. 2002;10:3741. doi: 10.1016/s0968-0896(02)00375-9. [DOI] [PubMed] [Google Scholar]
- 87.Sippl W. Journal of Computer-Aided Molecular Design. 2002;16:825. doi: 10.1023/a:1023888813526. [DOI] [PubMed] [Google Scholar]
- 88.Jojart B, Marki A. Journal of Molecular Graphics & Modelling. 2007;25:711. doi: 10.1016/j.jmgm.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 89.Li MY, Lu JF, Xia L. Acta Chimica Sinica. 2005;63:1875. [Google Scholar]
- 90.Xu Y, Liu H, Niu CY, Luo C, Luo XM, Shen JH, Chen KX, Jiang HL. Bioorganic & Medicinal Chemistry. 2004;12:6193. doi: 10.1016/j.bmc.2004.08.045. [DOI] [PubMed] [Google Scholar]
- 91.Iskander MN, Leung LM, Buley T, Ayad F, Di Iulio J, Tan YY, Coupar IM. European Journal of Medicinal Chemistry. 2006;41:16. doi: 10.1016/j.ejmech.2005.07.017. [DOI] [PubMed] [Google Scholar]