

Abstract
A sensitive and specific fluorimetric assay for the determination of pyruvate is reported. This assay is based on the oxidation of pyruvate in the presence of pyruvate oxidase. Hydrogen peroxide generated by pyruvate oxidase reacts with non-fluorescent amplex red at a 1:1 stoichiometry to form the fluorescent product, resorufin. The assay is optimized with respect to pH of reaction buffer, enzyme concentration, dye concentration, and the time course. The usefulness of the assay is demonstrated by the accurate measurement of intracellular and extracellular pyruvate concentrations. The limit of detection (LOD) of the assay is 5 nM.
Keywords: metabolism, pyruvate, pyruvate oxidase, L-phenyalanine, enzymatic assay, amplex red
Introduction
Pyruvate (pyruvic acid), which is produced by the final step of glycolysis, lies at the intersection of multiple metabolic pathways. Most notably, pyruvate links glycolysis to the tricarboxylic acid cycle. It is produced by pyruvate kinase (EC2.7.1.40; 2-O-phoshotransferase), which catalyzes the conversion of phosphoenolpyruvate and ADP into ATP and enzyme-bound enolpyruvate, which undergoes ketonization to form pyruvate. Aberrant serum pyruvate levels correlate with several diseases, especially those exhibiting metabolic acidosis. Examples include: sepsis, motor neuron disease, necrotizing encephalomyelopathy, MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes), Kearns Sayre syndrome, uremia, and others [e.g., 1]. As intracellular pyruvate concentrations regulate the kinetics of Icrac channels [2], calcium signal transduction events are likely to be regulated by this metabolite. Thus, accurate assessment of pyruvate levels can provide valuable clinical and cellular information.
Various methods for the quantitation of pyruvate have been reported, such as enzyme-based amperometric biosensors [3-5], HPLC coupled with colorimetric and fluorimetric detection [6-8], and enzymatic assays with fluorescence [9, 10] or absorbance measurements [11-14]. Chromatographic methods require expensive equipment and, as pyruvate has no chromophore, pre-column or post-column derivatization is usually required. The LOD of HPLC methods is at the μM level [7]. Enzymatic assays are specific, rapid and convenient to perform. The major drawbacks of enzyme-based amperometric methods are the relatively low sensitivity (LOD is 5 μM, ref 3), the costs of electronic signal detection, and the tedious immobilization of enzymes on the electrodes. The most widely used method for pyruvate detection is the lactate dehydrogenase (LDH)-based spectrophotometric assay [11-14]. In this assay, pyruvate is converted to lactate by the catalytic action of LDH with NADH as a cofactor; the decrease in NADH concentration, measured by the change in absorbance at 339 nm, is proportional to the pyruvate concentration. The LOD of the LDH assay is ΔA=0.002 or 0.3 μM pyruvate [14]. To provide accurate pyruvate measurements in small biological samples, we developed a more sensitive fluorescent assay. The chemical reactions of this method of pyruvate detection are depicted in Figure 1. Pyruvate is oxidized by pyruvate oxidase via enzyme reactions to generate acetate, carbon dioxide and hydrogen peroxide (H2O2). H2O2 then reacts with amplex red in a 1:1 stoichiometry to form the red fluorescent product, resorufin [15, 16]. The fluorescence of resorufin is proportional to the initial pyruvate concentration in the solution. The LOD of the assay is 5 nM.
Fig. 1.
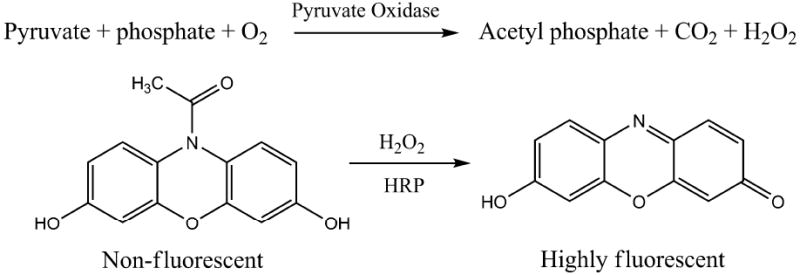
The chemical reactions of the pyruvate assay are shown. (HRP: horseradish peroxidase)
Materials and methods
Chemicals
Pyruvate, flavin adenine dinucleotide (FAD), cocarboxylase (thiamine pyrophosphate, TPP), horseradish peroxidase (HRP), bacterial pyruvate oxidase (cat. # P4591; POX-1) and pyruvate oxidase from Aerococcus sp. (cat. # P4105; POX-2) were purchased from Sigma Chem. Co. (St. Louis, MO). Amplex red was obtained from Invitrogen (Carlsbad, CA).
Cell culture
Jurkat cells (ATCC, Manassas, VA) were cultivated in RPMI-1640 medium (Invitrogen) containing 10% FCS and 1% antibiotics. For pyruvate experiments, the cells were centrifuged at 400 × g for 5 min.; medium was aspirated and then the cell pellet was washed with 15 ml of PBS (1×, pH7.4) followed by 10 ml of fresh RPMI-1640 medium. Then cells were seeded evenly into two new 250 ml culture flasks. Control flasks contained Jurkat cells in 15 ml of medium. Experimental flasks contained Jurkat cells in 15 ml of medium with 6 mM L-phenylalanine (L-Phe). Each flask was seeded at a density of roughly 5×105 cells/ml. Cells were cultured in a humidified atmosphere of 95% air, 5% CO2 at 37°C for 30 hrs. The cell density at this time was roughly 2×106 cells/ml. Cells or culture supernatants were collected then treated as described below.
Extracellular pyruvate extraction
The pyruvate extraction procedure was adapted from O’Donell-Tormey et al. [9] and Lamprecht et al. [14] with some modifications. In brief, a 12 ml aliquot was withdrawn from the culture flask (Jurkat cells only or cells plus L-Phe) and centrifuged at 600 × g for 1 min. The cell pellet was used for intracellular pyruvate determination. One ml of supernatant was transferred to a 15 ml tube containing 1 ml of ice-cold 0.5 M HClO4 and then incubated for 5 min on ice. Afterwards, the acid was neutralized with 50 μl of 5 M K2CO3, and then the precipitated KClO4 and proteins were removed by filtration with a 0.2 μM PTFE syringe filter. The filtrate was centrifuged at 10,000 × g for 5 min. For the fluorescence enzyme assay, the supernatant was diluted to 1/3 of its original concentration with assay buffer (100 mM potassium phosphate with 1.0 mM EDTA, pH 6.7) to stabilize the pH.
Intracellular pyruvate extraction
The above cell pellet was washed with 12 ml of PBS; cell numbers were counted in a hemocytometer. To the cell pellet, 0.5 ml of ice-cold 0.25 M HClO4 was added, vortex-mixed and then incubated on ice for 5 min. The mixture was neutralized with 12 μl of 5 M K2CO3 (~pH6.5). The supernatant was collected for pyruvate measurement after centrifugation at 10,000 × g for 5 min.
LDH assay
The assay was performed using the procedure of Lamprecht and Heinz [14], with minor modifications. Briefly, 0.5 ml of pyruvate extract, 0.25 ml of TEA/EDTA buffer (TEA: 0.5 M, pH 7.6; EDTA: 5 mM) and 20 μl of 7 mM NADH was added to a 1 ml cuvette. The sample was mixed thoroughly and then the absorbance (A1) was recorded at 339 nm. 20 μl of 225 U/ml of LDH was added and then the sample was incubated at room temperature in the dark for 30 min. The absorbance (A2) at 339 nm was read again. The change of absorbance at 339 nm (ΔA = A1 − A2) was used to calculate pyruvate concentration (ε339 = 6.22 × 10-3 μM-1 cm-1).
Fluorescence assay
Fluorescence was measured with a FlexStation II plate reader (Molecular Devices, Sunnnyvale, CA) using a 96-well black assay plate (Nunc, Nalge Nunc International). Calibration experiments were performed with 20 μl volumes of pyruvate standards (0, 200, 400, 600, 800, 1000, 1500, 2000 pmol/well). Extracellular or intracellular pyruvate extracts were pipetted into a 96-well plate, 180 μl of an assay cocktail was added into each well, and then incubated for 30 min. at room temperature. The final reaction solution contained 100 mM potassium phosphate with 1.0 mM EDTA, pH 6.7, 1.0 mM MgCl2, 10 μM FAD, 0.2 mM thiamine pyrophosphate, 0.2 U/ml pyruvate oxidase, 50 μM amplex red and 0.2 U/ml HRP. Fluorescence at 590 nm was measured with excitation at 535 nm. Background was corrected by subtracting the value of the no pyruvate control from all sample readings.
Statistical Analysis
Unless otherwise mentioned, data are presented as means ± standard deviations of triplicate measurements. P values were calculated from paired two-tailed t-tests and differences are considered significant for P < 0.05. Inter and intra-run precision and accuracy of assays were evaluated by one-way analysis of variance (ANOVA). All calculations were performed with microsoft Excel analysis ToolPak.
Results and discussion
Optimization of the assay
In the present study we have developed a highly sensitive amplex red-based fluorescence assay for pyruvate, as illustrated in Fig. 1. An H2O2-producing pyruvate oxidase was necessarily employed to provide substrate for the coupled reaction with amplex red. However, the pH optima of H2O2-producing pyruvate oxidases vary significantly among bacterial species [17]. Moreover, the absorption maximum and fluorescence quantum yield of resorufin is markedly reduced at pH<6.0 (Invitrogen product information for A22188). Therefore, we tested a wide spectrum of experimental conditions on the assay’s performance. As Fig. 2A shows, the standard curve loses linearity at pH 5.7. Amplex red’s recommended pH range (pH7.0-8.0) is substantially higher than pyruvate oxidase’s pH optimum. Optimal pH conditions appear to be near neutral pH. This result is consistent with the conclusion of an amperometric study that found the highest signal to background ratio at pH 7.0 [3]. We perform the pyruvate assay at pH 6.7.
Fig. 2.
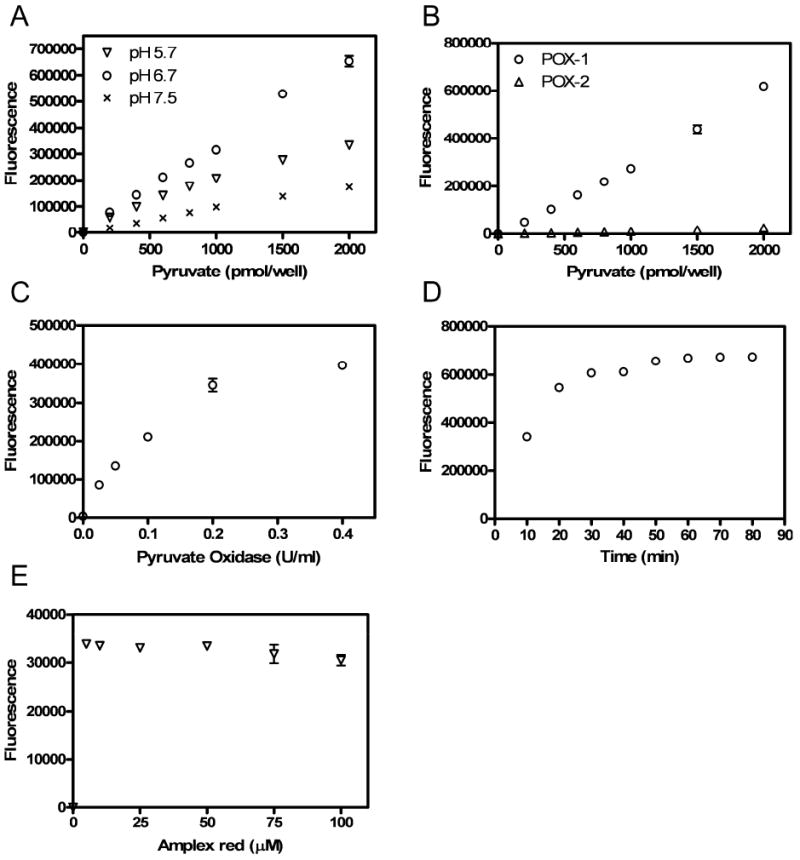
Experiments designed to optimize the assay are shown. A. pH effect on the fluorescence signals. The reaction mixtures contained 100 mM potassium phosphate with 1.0 mM EDTA, 1.0 mM MgCl2, 10 μM FAD, 0.2 mM thiamine pyrophosphate, 0.2 U/ml pyruvate oxidase from bacterial, 50 μM amplex red and 0.2U/ml HRP. B. Effect of pyruvate oxidase type on the fluorescence signals (POX-1: bacterial pyruvate oxidase and POX-2: pyruvate oxidase from Aerococcus sp.) Assays were performed at pH 6.7. C. Relationship of fluorescence intensity and final concentrations of pyruvate oxidase in the reaction mixture (pyruvate: 1 nmol/well). D. Time course of pyruvate assay (pyruvate: 2 nmol/well). E. Dye concentration effect on the fluorescent signals (100 pmol/well). Each datum point is a mean of triplicate measurements; error bars are standard deviations of three separate wells. Error bars are not shown when their sizes are less than those of the symbols.
Additional assay parameters were checked to further optimize the experimental protocol. Fig. 2B shows the measured resorufin fluorescence intensities as a function of pyruvate concentration for two pyruvate oxidases (referred to as POX-1 and POX-2) obtained from Sigma (see Materials and Methods). Although Aerococcus sp. pyruvate oxidase produces H2O2, it was not effective in our assay conditions. Therefore, we use bacterial pyruvate oxidase (POX-1; Sigma cat. no. P4591) in subsequent pyruvate assays.
We next assessed the assay’s performance at different concentrations of POX-1. As Fig. 2C shows, the fluorescence signals increase with pyruvate oxidase concentration, especially in the low dose range (0 - 0.1U/ml). Therefore, in subsequent experiments we employ 0.2 U/ml of pyruvate oxidase in the reaction mixture.
The generation of fluorescence was also measured as a function to time. As shown in Fig. 2D, the fluorescence signal rapidly increases from 0-20 min. After 20 min. of incubation, the reaction rate substantially declines. Simultaneously, the background noise increases with time (data not shown). The optimal S/N ratio is observed at an incubation time of 30 min. Unless otherwise mentioned, we incubate the reactions for 30 min. at room temperature in all assays.
In this assay, H2O2 detection is based on the oxidation of nonfluorescent amplex red to highly fluorescent resorufin. However, high H2O2 levels can further oxidize resorufin into non-fluorescent resazurin [16, 18]. To avoid this secondary reaction, the amplex red concentration must be at least a 5-fold higher greater than the amount of H2O2 [16]. However, too high a concentration may cause inner-filter effects. Therefore, an appropriate dye concentration is important for the accuracy of the assay. As indicated in Fig. 2E, amplex red at 10 μM to 100 μM produced consistent fluorescence signals for pyruvate at 1 nmol/well and 100 pmol/well. We observed a reduction in signal at high amplex red levels. We use 50 μM of amplex red in subsequent assays.
To further assess the quality of the pyruvate assay, we performed the assay using optimized conditions to calculate the Z’ factor. The Z’ factor is defined as [19]:
Where μc+ and μc- are the means of the positive control and negative control signals, respectively, and σc+ and σc- are the signals’ standard deviations. The means and standard deviations were calculated from nine wells with buffer alone and nine wells with 2 nmol/well of pyruvate. The Z’ factor was found out to be 0.89, indicating that this is an excellent assay.
Validation of the assay
Reaction specificity was first tested using dropout experiments in which each reagent was independently omitted from the reaction mixture. The results show that the complete reaction mixture demonstrates high levels of fluorescence (Fig. 3A). Although FAD and TPP are coenzymes in the reaction, the assay is minimally affected by their omission. However, the assay does require key factors such as pyruvate, pyruvate oxidase, amplex red, and HRP. To assess substrate specificity, the assay was performed with pyruvate replaced by structurally similar compounds. As compounds such as lactate and α-ketoacids might interfere the measurement of pyruvate, we evaluated fluorescence signals generated using pyruvate and other α-ketoacids as substrates under otherwise identical conditions. The results show that L-lactate and phosphoenolpyruvate do not interfere with the assay, whereas 2-oxobutyrate and oxaloacetate only show 6.8% and 8.7% signal intensities, respectively.
Fig. 3.
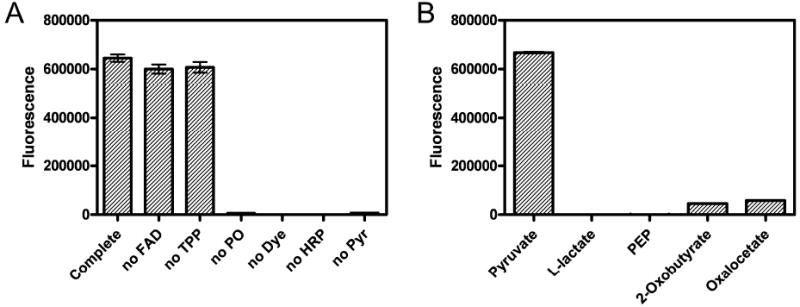
The specificity of the pyruvate assay is illustrated. A. Dropout experiments are shown (pyruvate 2 nmol/well except the no pyruvate experiments). B. Interference experiment are shown (compound content: 2 nmol/well). Data are the mean ± SD of triplicate wells. Error bars are not shown when their sizes are less than those of the symbols.
To demonstrate the assay’s linear range, experiments were performed over a broad range of pyruvate concentrations. Data from two experiments (0-200 pmol/well and 0-2 nmol/well) are plotted together in Fig. 4. When analyzed using linear regression, a high correlation coefficient of 0.995 was obtained. Hence, excellent linearity was achieved throughout the range from 3 pmol/well to 2 nmol/well.
Fig. 4.
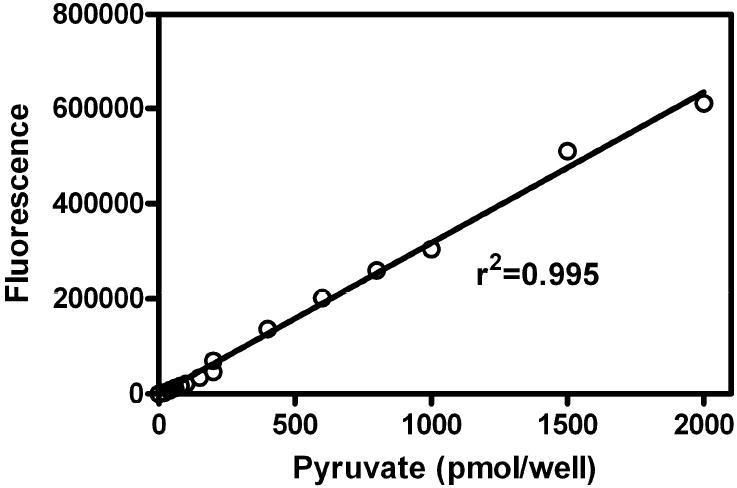
The calibration curve for this novel fluorimetric pyruvate assay is shown (each datum point is a mean of triplicate measurements). (r2 = 0.995) (y = 317 x)
The LOD and limit of quantitation (LOQ) were calculated [20] as: LOD = 3σ/S and LOQ = 10σ/S, where σ = standard deviation of the background and S = the slope of the calibration curve. The value of σ was obtained by measuring the fluorescence intensity of blank samples in which all reagents except pyruvate were added. The LOD and LOQ of this assay were calculated to be: LOD = 1.0 pmol/well (5 nM) and LOQ = 3.0 pmol/well (15 nM), (n=9) respectively. As the amount of amplex red in each well is 10 nmol, the pyruvate concentration must not be more than 2 nmol to obtain a linear calibration curve [16]. Thus, upper limit of quantitation (ULOQ) of the assay is 2 nmol/well.
Further statistical analyses were performed. Sets of control samples containing LOQ (3 pmol/well), midpoint (1 nmol) and ULOQ (2 nmol) were assayed on six consecutive days with four replicates for each sample. Data were evaluated by a one-way analysis of variance (ANOVA) to calculate the inter- and intra-run precision and accuracy of the assay [21]. The accuracy of assay is expressed as a percentage of relative error (% RE): %RE = 100% × (mean value - nominal value)/nominal value. Intra-run precisions and inter-run precisions of the assay were determined by percent coefficient of variation (%CV) in which %CV = [(mean square variance)0.5/mean value] × 100. The intra-run and inter-run mean square variances were calculated with Excel analysis ToolPak. The accuracy and precision of the measurements at the LOQ (3pmol/well) were 10.7% and 18.9%, respectively. At the highest dose tested of 2 nmol/well, the precision and accuracy were 3.8% and 5.5%, respectively. These results indicated that the new assay is accurate, and that the results are reproducible. The statistical results are summarized in Table 1.
Table 1.
The precision and accuracy of the assay for measurement of pyruvate.a
| Added amount (pmol/well) | Found amount (pmol/well) | Accuracy (% RE) | Precision (%CV) |
|
|---|---|---|---|---|
| Intra-run | Inter-run | |||
| 3 | 3.3 | 10.7 | 18.0 | 18.9 |
| 1000 | 980 | -2.1 | 2.0 | 5.4 |
| 2000 | 2076 | 3.8 | 1.8 | 5.5 |
Six runs with four replicates per run were performed.
Application of the assay
To further test this pyruvate assay, we compared the performance of our new assay with the established LDH assay for the measurement the pyruvate concentrations. In the first series of experiments, we measured the pyruvate levels in FCS. Samples were deproteinized with 0.5M HClO4, as described in the Materials and Methods. The pyruvate concentration determined with the LDH assay was 27.3±1.2 μM (n=3) whereas that determined by the new fluorescence assay was 21.3±1.6 μM (n=9). The reliability of these values was confirmed using the standard addition method wherein 5, 25, 50, and 100 μM exogenous pyruvate in FCS were deproteinized then measured (data not shown). The pyruvate recovery rates were calculated as 94.0±7.9 % (n=12) in the spiking range of 5-100 μM. Extrapolation of these data yielded a value of 21.0 μM for this assay (Fig. 5), which is in excellent agreement with the value reported above.
Fig. 5.
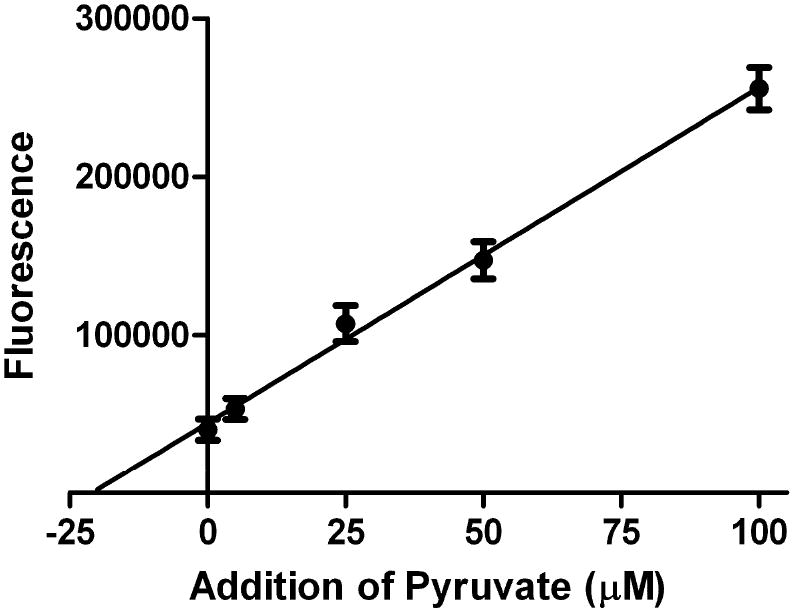
A standard addition calibration curve is shown that measures the concentration of pyruvate in FCS (each datum point is a mean of three measurements). A value of 21.0 μM was obtained. (r2 = 0.996) (y = 2120 x + 44540)
The pyruvate levels were also assessed in the tissue culture media and within Jurkat cells. In the cell system, pyruvate was measured during normal medium conditions and in the presence of 6 mM L-phenylalanine (L-Phe). L-Phe inhibits the activity of pyruvate kinase by 50% at 6 mM [22]. The results for control and L-Phe-treated samples were measured with these two assays. As shown in Table 2, the LDH assay and our fluorescence assay give very similar results for extracellular pyruvate, which is also in agreement with the substrate specificity noted above. Due to a lack of sensitivity, the intracellular pyruvate measurement is difficult to perform with the LDH assay, while the new assay can measure intracellular pyruvate levels. Assuming a Jurkat cell diameter of 7 μm, the intracellular pyruvate concentration can be calculated. After treatment of Jurkat cells with L-Phe (P<0.01), the intracellular pyruvate concentration decreased, as expected due to the reduction in pyruvate kinase activity.
Table 2.
Comparison of pyruvate measured using LDH assay and the new assay.a
| Test | Extracellular pyruvate (μM) |
Intracellular pyruvate (μM) |
|
|---|---|---|---|
| LDH assay | New assay | New assay | |
| Cells (untreated) | 126 ± 5.6 | 125 ± 8.1 | 201 ± 20.7 |
| L-Phe treated cells | 93.7 ± 2.0 | 91.4 ± 3.2 | 134 ± 26.2 |
| P valueb | <0.001 | <0.001 | <0.01 |
The means and standard deviations were calculated from five measurements performed in triplicate.
The P values compare untreated cells with L-Phe-treated cells.
Implications of the assay
Although our fluorescence assay yields results comparable to the LDH assay at relatively high pyruvate concentrations (> 0.3 μM), it is able to detect pyruvate at much lower levels. An alternate fluorimetric detection method relying upon NADH autofluorescence is also available; however, the endogenous autofluorescence of biological samples may give misleading results. Moreover, its sensitivity is not suitable for intracellular pyruvate quantitation of small samples. Thus, the assay described above is especially well-suited for the assessment of low pyruvate levels, such as those encountered after extraction of cell pellets and small clinical samples.
H2O2 has been reported to cause a non-enzymatic and stoichiometric decarboxylation of pyruvate [9, 23, 24]. This competing reaction may cause a low measurement result. By comparing the concentration of pyruvate in the samples (15 nM (3 pmol/well) to 0.1 μM (2 nmol/well)) with amplex red concentration (50 μM) it seems likely that amplex red effectively suppresses decarboxylation. In addition, the side-reaction between resorufin and H2O2 was suppressed as well. We obtained a linear calibration curve in the range of 50-2000 pmol/well.
In summary, we have developed a highly sensitive, convenient and economical assay for pyruvate assay in cell extracts. In our previous paper [25], we observed that the upstream glycolytic metabolite glucose-6-phosphate increased by 20% after treatment of Jurkat cells with 6 mM of L-Phe. In this work we found the downstream product pyruvate decreases by about 20% during treatment with phenylalanine.
Acknowledgments
This work was supported, in part, by the Intramural Program of the National Institute of Child Health and Human Development, NIH, DHHS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gore DC, Jahoor F, Hibbert JM, DeMaria EJ. Lactic acidosis during sepsis is related to increased pyruvate production, not deficits in tissue oxygen availability. Ann Surg. 1996;224:97–102. doi: 10.1097/00000658-199607000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bakowski D, Parekh AB. Regulation of store-operated calcium channels by the intermediary metabolite pyruvic acid. Curr Biol. 2007;17:1076–1081. doi: 10.1016/j.cub.2007.05.041. [DOI] [PubMed] [Google Scholar]
- 3.Bergmann W, Rudolph R, Spohn U. A bienzyme modifed carbon paste electrode for amperometric detection of pyruvate. Anal Chim Acta. 1999;394:233–241. [Google Scholar]
- 4.Zapata-Bacri A, Burstein C. Enzyme electrode composed of the pyruvate oxidase from Pediucoccus species coupled to an oxygen electrode for measurements of pyruvate in biological media. Biosens. 19871988;3:227–237. doi: 10.1016/0265-928x(87)85003-4. [DOI] [PubMed] [Google Scholar]
- 5.Kulys J, Wang L, Daugvilaite N. Amperometric methylene green-mediated pyruvate electrode based on pyruvate oxidase entrapped in carbon paste. Anal Chim Acta. 1992;265:15–20. [Google Scholar]
- 6.Minniti G, Cerone R, De Toni E. Determination of lactic acid, pyruvic acid, and ketone bodies in serum and cerebrospinal fluid by HPLC. Am Clin Lab. 2001;20:21–23. [PubMed] [Google Scholar]
- 7.Tokishi H, Hironori T, Hidemi T, Hiroshi N. High-performance liquid chromatographic determination of α-keto acids in human urine and plasma. Anal Biochem. 1982;122:173–179. doi: 10.1016/0003-2697(82)90267-6. [DOI] [PubMed] [Google Scholar]
- 8.Ewaschuk JB, Naylor JM, Barabash WA, Zello GA. High-performance liquid chromatographic assay of lactic, pyruvic and acetic acids and lactic acid stereoisomers in calf feces, rumen fluid and urine. J Chromatogr B. 2004;805:347–351. doi: 10.1016/j.jchromb.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 9.O’Donnell-Tormey J, Nathan CF, Lanks K, DeBoer CJ, De la Harpe J. Secretion of pyruvate, an antioxidant defense of mammalian cells. J Exp Med. 1987;165:500–514. doi: 10.1084/jem.165.2.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olsen C. Enzymic fluorometric micromethod for the determination of acetoacetate, β-hydroxybutyrate, pyruvate, and lactate. Clin Chim Acta. 1971;33:293–300. doi: 10.1016/0009-8981(71)90486-4. [DOI] [PubMed] [Google Scholar]
- 11.Neville JF, Jr, Gelder RL. Modified enzymatic methods for the determination of L-(+)-lactic and pyruvic acids in blood. Am J Clin Pathol. 1971;55:152–158. doi: 10.1093/ajcp/55.2.152. [DOI] [PubMed] [Google Scholar]
- 12.Artuch R, Vilaseca MA, Farre C, Ramon F. Determination of lactate, pyruvate, beta-hydroxybutyrate and acetoacetate with a centrifugal analyzer. Eur J Clin Chem Clin Biochem. 1995;33:529–533. [PubMed] [Google Scholar]
- 13.Hansen JL, Freier EF. Direct assays of lactate, pyruvate, β-hydroxybutyrate, and acetoacetate with a centrifugal analyzer. Clin Chem. 1978;24:475–479. [PubMed] [Google Scholar]
- 14.Lamprecht W, Heinz F. In: Methods of Enzymatic Analysis. 3. Bergmeyer H, editor. Chapter 3.10. Verlag Chemie; Weinheim: 1984. pp. 570–577. [Google Scholar]
- 15.Zhou M, Diwu Z, Panchuk-Voloshina N, Haugland R. A stable nonfluorescent derivative of resorufin for the fuorometric determination of trace hydrogen peroxide: applications in detecting the activity of phagocyte NADPH oxidase and other oxidases. Anal Biochem. 1997;253:162–168. doi: 10.1006/abio.1997.2391. [DOI] [PubMed] [Google Scholar]
- 16.Mohanty JG, Jaffe JS, Schulman ES, Raible DG. A highly sensitive fluorescent micro-assay of H2O2 release from activated human leukocytes using a dihydroxyphenoxazine derivative. J Immunol Methods. 1997;202:133–141. doi: 10.1016/s0022-1759(96)00244-x. [DOI] [PubMed] [Google Scholar]
- 17.Carlsson J, Edlund MB, Lundmark SK. Characteristics of a hydrogen peroxide-forming pyruvate oxidase from Streptococcus sanguis. Oral Microbiol Immunol. 1987;2:15–20. doi: 10.1111/j.1399-302x.1987.tb00264.x. [DOI] [PubMed] [Google Scholar]
- 18.Towne V, Will M, Oswald B, Zhao Q. Complexities in horseradish peroxidase-catalyzed oxidation of dihydroxyphenoxazine derivatives: appropriate ranges for pH values and hydrogen peroxide concentrations in quantitative analysis. Anal Biochem. 2004;334:290–296. doi: 10.1016/j.ab.2004.07.037. [DOI] [PubMed] [Google Scholar]
- 19.Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 20.Long GL, Winefordner JD. Limit of detection. A closer look at the IUPAC definition. Anal Chem. 1983;55:712A–724A. [Google Scholar]
- 21.Findlay JWA, Smith WC, Lee JW, Nordblom GD, Das I, DeSilva BS, Khan MN, Bowsher RR. Validation of immunoassays for bioanalysis: a pharmaceutical industry perspective. J Pharm Biomed Anal. 2000;21:1249–73. doi: 10.1016/s0731-7085(99)00244-7. [DOI] [PubMed] [Google Scholar]
- 22.Webber G. Inhibition of human brain pyruvate kinase and hexokinase by phenylalanine and phenylpyruvate: possible relevance to phenylketonuric brain damage. Proc Natl Acad Sci USA. 1969;63:1365–1369. doi: 10.1073/pnas.63.4.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Desagher S, Glowinski J, Prémont J. Pyruvate protects neurons against hydrogen peroxide-induced toxicity. J Neurosci. 1997;17:9060–9067. doi: 10.1523/JNEUROSCI.17-23-09060.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giandomenico AR, Cerniglia GE, Biaglow JE, Stevens CW, Koch CJ. The importance of sodium pyruvate in assessing damage produced by hydrogen peroxide. Free Radic Biol Med. 1997;23:426–434. doi: 10.1016/s0891-5849(97)00113-5. [DOI] [PubMed] [Google Scholar]
- 25.Zhu A, Romero R, Petty HR. An enzymatic fluorimetric assay for glucose-6-phosphate: Application in an in vitroWarburg-like effect. Anal Biochem. 2009;388:97–101. doi: 10.1016/j.ab.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]