

Abstract
Loss of VHR phosphatase causes cell cycle arrest in HeLa carcinoma cells, suggesting that VHR inhibition may be a useful approach to halt the growth of cancer cells. We recently reported that VHR is upregulated in several cervix cancer cell lines as well as in carcinomas of the uterine cervix. Here we report the development of multidentate small-molecule inhibitors of VHR that inhibit its enzymatic activity at nanomolar concentrations and exhibit antiproliferative effects on cervix cancer cells. Chemical library screening was used to identify hit compounds, which were further prioritized in profiling and kinetic experiments. SAR analysis was applied in the search for analogs with improved potency and selectivity, resulting in the discovery of novel inhibitors that are able to interact with both the phosphate-binding pocket and several distinct hydrophobic regions within VHR’s active site. This multidentate binding mode was confirmed by Xray crystallography. The inhibitors decreased the proliferation of cervix cancer cells, while growth of primary normal keratinocytes was not affected. These compounds may be a starting point to develop drugs for the treatment of cervical cancer.
Introduction
Protein tyrosine phosphatases (PTPs) have recently been implicated in various human diseases, including cancer, and have been suggested as potential drug targets (i,ii,iii,iv,v). Vaccinia H1-related (VHR) phosphatase is a relatively small member of the sub-class of dual-specificity phosphatases (vi) with only 185 amino acids (Mr 21 kDa) and no apparent targeting domain or docking site (vii). Compared to the phospho-tyrosine (pTyr)-specific classical PTPs, the crystal structure of VHR revealed a much shallower active site, which allows VHR to act on both pTyr and phospho-threonine (pThr) in its substrates (viii). VHR has been reported to dephosphorylate extracellular signal-regulated kinases Erk1/2 and c-Jun N-terminal kinases Jnk1/2, but not p38 (ix,x,xi). These mitogen-activated protein kinases (MAP kinases) mediate major signaling pathways triggered by extracellular growth factor, stress, or cytokines (xii), and regulate cellular processes such as differentiation, proliferation, and apoptosis (xiii,xiv). MAP kinases are activated within the kinase cascade by phosphorylation at a Thr-X-Tyr motif in their activation loop, whereas dephosphorylation of this motif by VHR or other MAP kinase phosphatases (MKPs) leads to a conformational change, leaving the kinase domain in an inactive, closed conformation (xv).
Unlike many MKPs, VHR expression is not induced in response to activation of MAP kinases (ix), but is instead regulated during cell cycle progression (xvi). Using RNA interference to knock down endogenous VHR, we have previously shown that HeLa cervix carcinoma cells that are lacking VHR arrested at the G1-S and G2-M transitions of the cell cycle and showed initial signs of senescence. Loss of VHR increased the expression of the cyclin-dependent kinase inhibitor p21Cip-waf1, whereas genes of cell cycle regulators, DNA replication, transcription, and mRNA processing were downregulated (xvi). In addition, there have been several reports showing that prolonged activation of MAP kinase pathways results in cell cycle arrest and cell senescence (xvii,xviii,xix,xx,xxi).
In our recent study (xxii), we established a link between VHR and cervical cancer. We found VHR protein levels upregulated in several cervix cancer cell lines compared to normal keratinocytes, including human papillomavirus (HPV) positive cell lines CaSki, HeLa, and SiHa, as well as HPV negative cell lines HT3 and C33. Moreover, we also found higher expression levels of VHR in primary cervix cancer biopsies, including squamous intraepithelial lesions and squamous cell carcinomas of the uterine cervix (xxii). This suggested to us that VHR might be a novel and promising drug target for the treatment of cervical cancer, and that small-molecule inhibitors of VHR should be valuable tools to validate this new target. Combining high-throughput chemical library screening and structure-activity relationship (SAR) analysis, we developed compounds that specifically bind to VHR’s active site in a multidentate fashion. This binding mode, which makes use of unique features at the protein surface surrounding the catalytic pocket, was confirmed by X-ray crystallography. Several of the inhibitors were active in cell-based assays at low micromolar concentrations and decreased the proliferation of cervix cancer cell lines significantly.
Results
Chemical Library Screening
A 96-well format in vitro assay was used to screen a set of 50,000 drug-like molecules of the DIVERSet™ library from ChemBridge (ChemBridge, Inc.). At the used concentration of 0.02 mg/mL, 221 compounds inhibited VHR’s enzymatic activity >60% (average of n=2) compared to a no-inhibitor control. For further evaluation, a total of 56 compounds were picked that inhibited VHR >90% (average of n=2). Michaelis-Menten kinetic studies revealed 21 hits that inhibited the enzyme with Ki values <20 μM (Table 1). Clustering the 21 compounds by a Tanimoto distance (xxiii) of 0.5 resulted in 12 different clusters and singletons, respectively, indicating a quite diverse chemical space covered by these molecules. The most active hit, 2-((Z)-4-oxo-5-((E)-3-phenylallylidene)-2-thioxothiazolidin-3-yl)ethane-sulfonic acid 1, inhibited VHR with a Ki value of 0.81 μM. In a counter screen against the protein tyrosine phosphatases PTP1B, CD45, HePTP, Shp2, and YopH, only 1 and 1,4-dimethoxyanthracene-9,10-dione 8 were found to be selective for VHR (Figure 1). Of these two compounds, only 1 showed a clear competitive inhibition pattern as shown in the Lineweaver-Burk plot in Figure 2A.
Table 1.
21 most potent VHR screening hits (Ki value <20 μM).
| # | Structure* | Mol. Weight | Ki, μM |
|---|---|---|---|
| 1 | 355.441 | 0.809 | |
| 2 | 451.4374 | 1.76 | |
| 3 | 332.3482 | 1.86 | |
| 4 | 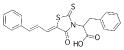 |
395.4902 | 2.01 |
| 5 | 261.338 | 2.09 | |
| 6 | 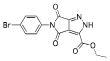 |
364.1547 | 2.64 |
| 7 | 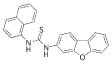 |
368.4522 | 2.94 |
| 8 | 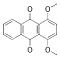 |
268.2684 | 3.07 |
| 9 | 429.9075 | 3.13 | |
| 10 | 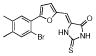 |
377.2549 | 3.19 |
| 11 | 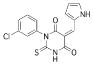 |
331.7759 | 3.49 |
| 12 | 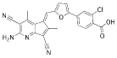 |
442.8605 | 3.87 |
| 13 | 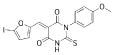 |
454.2384 | 4.44 |
| 14 | 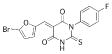 |
395.2022 | 12.1 |
| 15 | 231.2506 | 12.8 | |
| 16 | 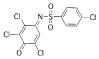 |
385.0484 | 13.4 |
| 17 | 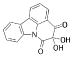 |
267.2404 | 13.8 |
| 18 | 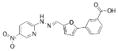 |
352.3056 | 13.8 |
| 19 | 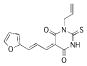 |
288.3204 | 15.8 |
| 20 | 359.208 | 15.9 | |
| 21 | 481.2585 | 16.2 |
Compounds 11, 13, 19, and 21 are mixtures of E/Z-isomers.
Figure 1.

Profiling of the 21 most active VHR screening hits at 20 μM against VHR, PTP1B, CD45, HePTP, Shp2, and YopH. Black arrows indicate compounds 1 and 8, which were found to be selectively inhibiting VHR.
Figure 2.

Characterization of compound 1. A. Michaelis-Menten kinetic measurements, Lineweaver-Burk plot for 1 with VHR. B. FlexX docking of 1 into the active site of VHR crystal structure (PDB code 1J4X). The color code of the MOLCAD surface represents the lipophilic potential (brown: most hydrophobic, blue: most hydrophilic).
In silico docking
To study the molecular basis for inhibition of VHR, we first used in silico docking to dock 1 and 8 into the active site of the VHR crystal structure (PDB code 1J4X, ref. xxiv). Compound 8 failed to dock into the catalytic pocket, further supporting an inhibition mechanism other than competitive. Since quinones like 8 are known to deactivate PTPs by oxidizing the catalytic cysteine residue (xxv), we discarded 8 from further investigation. As for compound 1, the docking suggested that the sulfonic acid moiety functions as a phosphate mimic and binds into the catalytic pocket, forming a network of hydrogen bond interactions with the phosphate binding loop, also called P-loop (Figure 2B). In support of this, similar compounds lacking the sulfonic acid moiety did not inhibit VHR. Furthermore, the docking suggested that the oxo-thioxothiazolidine ring interacts with the rim of the catalytic pocket, whereas the diene linker and the phenyl ring make van der Waals interactions with a hydrophobic region, mainly formed by Leu25 and Tyr128 (Figure 2B).
SAR Analysis and Analogs of Lead Compound 1
By further analyzing the lipophilic potential of VHR’s active site surface area, we found a total of three distinct hydrophobic regions surrounding the catalytic pocket (Figure 2B). Compared to active site properties of solved PTP structures and homology models (xxvi), this feature seems to be rather unique to VHR, and therefore could be exploited for designing selective inhibitors. The exposed hydrophobic patches are formed by Leu25/Tyr128 and Leu16/Tyr23, which make up large portions of the substrate binding site (xxiv), as well as by Phe68/Met69, which is part of a loop opposite of the catalytic pocket (Figure 2B). In an attempt to find analogs of 1 that could target more than one of these hydrophobic regions, we kept the oxo-thioxothiazolidinyl-ethanesulfonic acid moiety as hydrophilic pharmacophore, and subjected it to a substructure search among commercially available compounds. Nine structures were identified that fulfilled the requirement of having additional multiple hydrophobic entities to interact with multiple hydrophobic regions in VHR’s active site. All compounds contained a 1-phenyl-substituted pyrazole ring, which at its 3-position linked to another, more variable entity (Table 2). We obtained all nine analogs and subjected them to direct measurements of VHR inhibition. Indeed, compounds SA1, SA2, SA3, SA4, and SA5 showed excellent IC50 values of 18, 71, 74, 78, and 268 nM, respectively (Table 2). An aromatic group in position ‘R’, such as the benzoxy group in compounds SA1-4, seemed to be very well accommodated by the protein, whereas sterically more demanding structures, such as the piperidinsulfonyl group in SA5 or the morpholinosulfonyl group in SA8, were less favorable. On the other hand, groups such as methoxy in SA6 or methyl in SA9 seemed to be too small to provide additional binding energy through van der Waals interactions. Interestingly, compounds SA1-4 only differ in their substituents at the benzoxy group. Chlorine in para-position was most favorable (SA1), whereas substituents in ortho-position, such as chlorine (SA2) or fluorine (SA3), had no effect on inhibitory activity against VHR in vitro. Testing the five best inhibitors (SA1–5) against a panel of related PTPs demonstrated that they were at least one order of magnitude less potent for MKP-1, CD45, Cdc25A, PTP1B, and HePTP (Table 2). Of these phosphatases, MKP-1 and HePTP share the same physiological substrate with VHR, namely the MAP kinase Erk, and MKP-1 is structurally closely related to VHR.
Table 2.
In vitro efficacy and selectivity of 1-phenylpyrazole derivatives.
| R | IC50, μM | |||||||
|---|---|---|---|---|---|---|---|---|
| VHR | MKP-1 | Cdc25A | HePTP | CD45 | PTP1B | |||
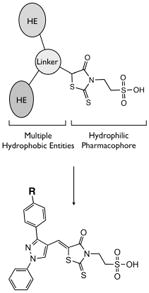 |
SA1 | 0.018 | 0.46 | 2.4 | 0.62 | 0.46 | 0.46 | |
| SA2 | 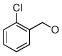 |
0.071 | 0.47 | 3.4 | 1.2 | 0.30 | 0.59 | |
| SA3 | 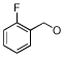 |
0.074 | 0.52 | 2.8 | 0.87 | 0.50 | 0.42 | |
| SA4 | 0.078 | 0.78 | 3.4 | 1.5 | 0.61 | 1.8 | ||
| SA5 | 0.27 | 2.8 | >10 | 2.4 | 1.8 | 2.2 | ||
| SA6 | CH3-O | 1.8 | nd | nd | nd | nd | nd | |
| SA7 | Cl | 1.8 | nd | nd | nd | nd | nd | |
| SA8 | 2.4 | nd | nd | nd | nd | nd | ||
| SA9 | CH3 | 3.1 | nd | nd | nd | nd | nd | |
Co-crystallization and Xray Structure Analysis
To investigate the nature of the multidentate binding of our inhibitors experimentally, we attempted to solve the crystal structure of SA3 bound to VHR. Using the hanging-drop vapor diffusion technique and repeated macroseeding, we obtained co-crystals of the VHR-SA3 complex. The structure was solved to 1.9 Å resolution by molecular replacement. Refinement statistics for the complex are presented in Table 3, R factor and Rfree values were 17.8 and 21.9%, respectively. After refinement, difference maps (Fo − Fc and 2Fo − Fc) were used to fit the inhibitor. Clear electron density of SA3 in the VHR active site was visible for the oxo-thioxothiazolidinyl- ethanesulfonic acid moiety (Figure 3B). This moiety occupies the phosphate-binding pocket in VHR, representing the exact same position and orientation as the phospho-tyrosine group of a substrate peptide found in the 1J4X.pdb structure (ref. xxiv, Figure 3A). Thereby, the sulfonic acid group undergoes multiple hydrogen bond interactions with backbone nitrogen atoms of the P-loop, and forms a salt bridge with the guanidinium group of Arg130 (Figure 3B). Modeling of SA3 into the visible electron density clearly exhibited the multidentate binding mode of the inhibitor, including van der Waals interactions of the fluoro-benzoxy group with Leu16/Tyr23, and the phenyl-pyrazole ring with Phe68/Met69 in VHR (Figure 3C). Modeling also indicated that the SA3 hydrophobic entities might have some flexibility in interacting with these amino acid residues, which could be the reason for the not defined electron density for these parts of the molecule. In any case, however, modeling of SA3 into the visible electron density always found the fluoro-benzoxy and phenyl-pyrazole groups interacting with multiple hydrophobic patches in a bidentate fashion. Comparison of the VHR-SA3 complex with the previously reported VHR structure (1J4X.pdb) also supported our binding model. Significant differences in the side chain positions of Tyr23, Phe68, and Met69 were observed, which account for specific interactions with SA3 (Figure 3A). Moreover, parts of the substrate recognition loop immediately following the N-terminal helix α1 (residues Ser17, Asp18, Gly19, Ser20, Gly21, and Cys22) were shifted by as much as 5.7 Å, compared to 1J4X.pdb, most-likely to accommodate the fluoro-benzoxy group of SA3 and to avoid unfavorable interactions between SA3 and hydrophilic amino acid residues within this loop region.
Table 3.
Summary of crystallographic data and refinement statistics for the VHR-SA3 complex. Values for the highest resolution shell (1.95 to 1.90 Å) are given in parentheses.
| Crystallographic data | |
|---|---|
| Space group | P21 |
| Unit cell (Angstrom) | a=50.406 b=59.186 c=60.656 a=90.00 b=98.36 g=90.00 |
| Molecules/asymmetric unit | 2 monomers |
| Resolution range (Å) | 50-1.9 |
| Data completeness (%) | 99.1(99.7) |
| Unique reflections | 26330(2028) |
| Redundancy | 6.8 |
| Rsym (%) | 5.9 |
| Refinement statistics | |
| R factor (%) | 17.8 (18.4) |
| Rfree (%) | 21.9 (23.6) |
| Average B factor (Å2) | 15.5 |
| No. protein atoms | 2784 |
| No. inhibitor atoms | 28 |
| No. water atoms | 227 |
| R.m.s.d. bond length (Å) | 0.016 |
| R.m.s.d. bond angle | 1.724 |
Figure 3.

Xray structural analysis of the VHR-SA3 complex. A. Stereo ribbon diagram of the overall co-crystal structure of the VHR-SA3 complex (green), aligned with Xray coordinates of a VHR-substrate peptide complex (1J4X.pdb, VHR grey, peptide light-blue). Tyr23, Phe68, and Met69 (green and grey, respectively) as well as the oxo-thioxothiazolidinyl-ethanesulfonic acid moiety of SA3 (magenta) and pTyr from the peptide (light-blue) are highlighted in stick representation. B. Close-up of SA3 bound to VHR’s active site with visible electron density. Yellow dashed lines indicate hydrogen bond interactions of the sulfonic acid moiety with amino acid residues of the P-loop at the bottom of the catalytic pocket. C. Modeling of SA3 into visible electron density. White arrows indicate multiple van der Waals interactions with hydrophobic regions at the surface surrounding VHR’s catalytic pocket.
Proliferation of Cervix Cancer Cells
We recently found VHR protein levels upregulated in several cervix cancer cell lines, as well as in primary cervix cancer biopsies (xxii). We also reported that VHR was necessary for proliferation of HeLa cervix carcinoma cells (xvi). Therefore, we set out to determine the effects of our five best VHR inhibitors on cervix cancer cell lines HeLa and CaSki, as well as on primary normal keratinocytes. As shown in Figure 4A, proliferation of primary normal keratinocytes, as measured by cell numbers, was not affected by any of the inhibitors after 24 h incubation at a concentration of 20 μM. In contrast, compounds SA1-5 significantly inhibited the spontaneous proliferation of HeLa cells, compared to a DMSO control, with SA3 being the most effective one, reducing HeLa cell numbers to less than 50% (Figure 4A). In a second assay, we measured proliferation of HeLa and Caski cells by following the incorporation of tritium (3H) labeled thymidine into the DNA (Figure 4B/C). A clear antiproliferative effect was found for all compounds, though more pronounced in CaSki compared to HeLa cells. In HeLa cells, SA3 revealed the best antiproliferative effect, reducing cell growth by as much as 58% compared to DMSO alone. For all other compounds, the inhibition did not exceed 30% (Figure 4B). In CaSki cells, however, all inhibitors reduced the spontaneous proliferation by more then 50%, with SA3 being most effective, decreasing the proliferation by as much as 74% (Figure 4C). To exclude that this decrease in cell proliferation was due to apoptosis, we incubated HeLa and Caski cells with 20 μM SA3 or SA2 for 24 h, stained cells with AnnexinV and propidium iodide (PI), and used flow cytometry to determine the percentage of apoptotic cells. As Figure 4D clearly shows, there was no difference between the percentages of AnnexinV/PI negative cells in samples treated with the inhibitors or treated with the vehicle, clearly indicating that the observed antiproliferative effect of the compounds was not due to apoptosis.
Figure 4.

Effects of VHR inhibitors on proliferation and survival of cervix cancer cells. A. Cell numbers of HeLa cervix carcinoma cells and normal keratinocytes (NK) after 24 h of treatment with 20 μM inhibitors, given as percentage of the DMSO control. B/C. [3H]-thymidine incorporation assay with 20 μM inhibitors or vehicle (DMSO) in HeLa (B) or Caski (C) cervix cancer carcinoma cells. D. Survival/death of HeLa and CasKi cells in presence of 20 μM inhibitors or DMSO after 24 h, evaluated by AnnexinV-FITC/PI flow cytometry assay (FACSVantage, BD). Percentage of live cells (Annexin V negative and PI negative cells on flow cytometry dot plots) is given. All data are reported as mean ± SD from three independent experiments.
Discussion and Conclusions
In this study we used high-throughput chemical library screening in a first step to generate hits for VHR, which were further prioritized through Michaelis-Menten kinetic studies and counter screens against a number of structurally related phosphatases. The resulting lead compound 1 competitively inhibited VHR with a Ki value of 0.81 μM and exhibited a promising degree of selectivity for VHR among other PTPs. In silico docking of 1 into the active site of VHR and SAR analysis suggested a binding mode, in which the sulfonic acid moiety of 1 binds through a network of hydrogen bond interactions into the phosphate binding pocket, whereas the thiazolidine heterocycle interacts with the rim of the pocket and the phenyl-allyl moiety makes van der Waals interactions with side chains of hydrophobic amino acids that flank the active site.
The sulfonic acid head group is an ideal chemical structure to mimic a phosphate group, without undergoing any hydrolysis reaction. Phosphorus and sulfur atoms have similar atomic and van der Waals radii, and are coordinated by 4 (phosphate) or 3 (sulfonate) oxygen atoms, respectively. However, under physiological pH conditions, a phosphate group carries two negative charges whereas the sulfonate has only one, making it easier for compounds carrying the latter to penetrate cell membranes. We therefore decided to keep the oxo-thioxothiazolidinyl-ethanesulfonic acid moiety of 1 as pharmacophore and applied a structure-based approach to optimize 1 in terms of both potency and selectivity.
Examination of the surface area that surrounds the active site in VHR revealed several distinct hydrophobic regions that were exploited in the design of more potent and selective inhibitors. A search for compounds that would still bind to the catalytic pocket, and additionally could interact with not just one but multiple hydrophobic areas in a multidentate fashion resulted in five molecules with significant lower IC50 values than lead structure 1. These compounds contain a thioxothiazolidinyl-ethanesulfonic acid moiety, representing the defined pharmacophore in 1. In addition, a pyrazole ring functions as a linker from which two hydrophobic entities branch off – a phenyl ring in 1-position and a more variable group at 3-position. Co-crystallization of VHR with one of these compounds, SA3, yielded a crystal structure in which clear electron density of SA3 was observed for the thioxothiazolidinyl-ethanesulfonic acid moiety. As predicted by the docking studies, the sulfonic acid group was found to bind tightly into the catalytic pocket, mimicking the phosphate group of the natural substrate. No clear electron density was observed for the two hydrophobic entities of SA3, which is most likely due to some degree of flexibility in the interactions between these parts of the molecule and the hydrophobic amino acid residues of the protein. Nonetheless, modeling of SA3 into the visible electron density leaves no doubt about the bidentate fashion that SA3 employs to bind to distinct hydrophobic patches at the surface that flanks the catalytic pocket in VHR. Besides highly improved potency against VHR with IC50 values as low as 18 nM, these multidentate inhibitors were at least one order of magnitude less potent for any other PTP tested, including HePTP and MKP-1, which share the same physiological substrate with VHR. This result provides a good example for a general applicable concept, in which targeting unique surface features outside of the catalytic pocket can generate selective small-molecule inhibitors for individual members of the PTP family.
We also present several lines of evidence strongly suggesting that our VHR inhibitors are able to pass cell membrane barriers and target VHR in cultured cells. In particular, we tested our compounds in cervical cancer cells, which were shown earlier to express higher levels of endogenous VHR compared to non-cancerous cells of the cervix. Indeed, incubation of the cancer cell lines HeLa and CaSki with the inhibitors at 20 μM induced a very significant inhibition of cell proliferation after an incubation period as short as 24 h. This inhibition of cell growth was not due to cell death, and is also in accordance with our previous work, demonstrating that loss of VHR using RNAi induces a dramatic decrease of HeLa cell numbers and proliferation. It is interesting to note that SA3 was more effective in inhibiting proliferation than SA1, although its IC50 value is 4times higher in vitro. SA3 also exhibited greater antiproliferative effects than SA2 and SA4, both of which share similar IC50 values with SA3 in vitro. These results suggest a beneficial role for the fluorine substituent (which is only present in SA3), maybe by facilitating better membrane permeability. The latter could be a limiting factor for these compounds, considering the substantially higher concentration that is needed to see clear effects in cells vs. inhibition of recombinant protein. Nonetheless, the fact that our inhibitors are not toxic to cells with low levels of endogenous VHR, such as primary normal keratinocytes, these compounds may well be a starting point to develop drugs for the treatment of cervical cancer and perhaps other cancers. Indeed, our results provide first evidence that pharmacological inhibition of VHR could be beneficial in treating such diseases. However, additional studies will be necessary to get better insights into the role of VHR phosphatase in cell cycle regulation and cancer, and to test the activity of these compounds in vivo, using mouse models.
Materials and Methods
Reagents
p-nitrophenyl phosphate (pNPP) was purchased from Sigma-Aldrich (St. Louis, MO). BIOMOL GREEN™ Reagent was from BIOMOL® Research Laboratories, Inc. (Plymouth Meeting, PA). 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP) was from Invitrogen Inc. (Carlsbad, CA). The 56 hit compounds were purchased from ChemBridge Inc., the sulfonic acid compounds were from Sigma; all compounds had a purity of >95%. All other chemicals and reagents were of the highest grade available commercially. The anti-VHR monoclonal antibody was from Transduction Laboratories (Los Angeles, CA). Anti-Erk and anti-p38 were from Cell Signaling Technology Inc. (Beverly, MA); anti-phospho-Erk and anti-phospho-p38 were from Promega (Madison, WI).
Recombinant PTPs
Recombinant VHR, HePTP, and YopH were expressed in E. coli and purified as described previously (xxvii). Recombinant CD45, PTP1B, MKP-1, and Cdc25A were purchased from BIOMOL® Research Laboratories, Inc. (Plymouth Meeting, PA). Shp2 was generously provided by Dr. Gen-Sheng Feng.
Chemical Library Screening
A set of 50,000 drug-like molecules from the DIVERSet™ library (ChemBridge, Inc.) was screened in a 96-well format in vitro phosphatase assay. Compounds had a working concentration of 0.15 mg/ml in 10% DMSO. Each reaction contained 180 nM VHR, 4 mM pNPP, and 0.02 mg/mL compound in 0.1 M Bis-Tris pH 6.0 reaction buffer with 1 mM dithiothreitol (DTT) present. The final volume amounted to 70 μL and contained 1.4% DMSO. The reaction was initiated by addition of pNPP after a preincubation of the enzyme with compounds for 10 min at room temperature. After 15 min, the reaction was quenched by addition of 140 μL BIOMOL GREEN™ Reagent, and the pNPP hydrolysis was determined by measuring the absorbance of the complexed free phosphate at 620 nm. The nonenzymatic hydrolysis of the substrate was corrected by measuring the negative control without addition of enzyme. Other controls included a positive control (no inhibitor added), a background control (no substrate added), and a control with 200 μM of the general PTP inhibitor sodium orthovanadate. To quantitate the inhibitory efficacy of the library compounds, we determined the ratio of inhibition in comparison to the positive control. Every compound with >60% inhibition was cherry-picked and rescreened to confirm it as a hit.
IC50 Measurements
The PTP-catalyzed hydrolysis of 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP) in the presence of compound was assayed at 30°C in a 60 μL 96-well format reaction system in 150 mM Bis-Tris, pH 6.0 assay buffer having an ionic strength of 150 mM (adjusted with NaCl) and containing 1 mM DTT and 5% DMSO. At various concentrations of the compound, the initial rate at fixed DiFMUP concentration (equal to the corresponding Km value for each PTP) was determined using a FLx800 micro plate reader (Bio-Tek Instruments, Inc.), an excitation wave length of 360 nm and measuring the emission of the fluorescent reaction product 6,8-difluoro-7- hydroxy-4-methylcoumarin (DiFMU) at 460 nm. The nonenzymatic hydrolysis of the substrate was corrected by measuring the control without addition of enzyme. The IC50 value was determined by plotting the relative activity versus inhibitor concentration and fitting to Equation 1 using the software GraphPad Prism® (GraphPad Software, Inc.).
| (Eq. 1) |
In this case, Vi is the reaction velocity when the inhibitor concentration is [I], V0 is the reaction velocity with no inhibitor, and IC50 = Ki + Ki[S]/Km.
Ki Determination
Using the same format and buffer system as for IC50 measurements, the enzyme was preincubated with various fixed concentrations of inhibitors for 10 min. The reaction was initiated by addition of various concentrations of pNPP (ranging from 0.2 to 10 Km) to the reaction mixture. The initial rate was determined using an ELx808 micro plate reader (Bio-Tek Instruments, Inc.), reading the absorbance of the product p-nitrophenol at 404 nm. The nonenzymatic hydrolysis of the substrate was corrected by measuring the control without addition of enzyme. The inhibition constant and inhibition pattern was evaluated by fitting the data to the Michaelis-Menten equations for either competitive, uncompetitive or mixed inhibition as described before (xxvii), using nonlinear regression and the program GraphPad Prism® (GraphPad Software, Inc.). For a comparison of the fitting results the second-order Akaike’s Information Criterion (AICc) was calculated as previously described (xxvii).
Molecular Modeling and In Silico Docking
Molecular modeling studies were conducted on a Linux workstation with the software package SYBYL version 7.1 (TRIPOS). Energy-minimized molecular models of the compounds were generated by the SYBYL/CONCORD routine. Flexible ligand docking calculations were performed with FlexX as implemented in Sybyl. For each compound, 30 solutions were generated and ranked by their FlexX score and CSCORE. The coordinates of the three-dimensional structure of the catalytic domain of VHR (PDB code 1J4X, ref. xxiv) were used in the docking studies, and the binding pocket was defined as an 8 Å radius within the catalytic cysteine 124. Molecular surfaces were generated with MOLCAD as implemented in Sybyl. The chemical clustering uses Tanimoto distance as calculated with the program ICM Pro (Molsoft, Inc.), which, by default, uses binary fingerprints of the molecular properties.
Co-crystallization of VHR with Compound SA3
VHR (amino acids 3–185) was subcloned into pGEX vector (GE Healthcare Life Sciences), overexpressed in E. coli BL21 (DE3), and purified as GST-fusion protein by affinity chromatography (glutathione sepharose 4B). After cleavage of GST by thrombin digestion and purification by gel filtration, the purified enzyme was stored at −20°C in 50 mM Tris pH7.4, 1mM EDTA, and 10 mM DTT. The recombinant protein was confirmed by mass spectroscopy, finding a molecular weight of 20259.3 Da compared to the predicted molecular weight of 20260.0 Da. For crystallization, the stocked VHR was mixed with inhibitor SA3 to a final concentration of 0.37 mM VHR and 1.0 mM SA3, respectively. Using hanging-drop vapor diffusion technique, VHR-SA3 was crystallized in the hanging drop of 2 mL of protein solution mixed with 2 mL of reservoir solution (100 mM Tris pH 7.4, 15% polyethylene glycol 8000, 60 mM NaF) at 24°C. The crystals for data collection were obtained by repeated macroseeding. The crystal was flash frozen in a stream of nitrogen gas to 140 K. The diffraction data was collected using a R-AXIS IV detector with a wavelength of 1.5418 Angstroms and oscillation of 1 degree. The data were processed using the HKL2000 program package (xxviii). The structure of the VHR-SA3 complex was solved with molecular replacement using a reported VHR crystal structure (PDB code 1VHR, ref. viii) as search model and the program Molrep as implemented in CCP4 (xxix). An iterative process of refinement in CCP4 and model building using the program Coot (xxx) was used to construct the model of the complex. The SA3 ligand dictionary file used in CCP4 and Coot was generated with PRODRG (xxxi). After refinement, difference maps (Fo − Fc and 2Fo − Fc) were used to fit the inhibitor. Clear electron density was visible for only the partial inhibitor in the VHR active site. Refinement statistics for the complex are presented in Table 2. For the final structure, R factor and Rfree values were 17.8 and 21.9%, respectively. Graphic images presented in the figures were prepared with PyMol (DeLano Scientific, Palo Alto, CA).
Cell Lines and Primary Keratinocytes
HeLa and CaSki cells were grown in DMEM medium (Dulbecco’s modified Eagle’s medium; ICN; low Laboratories) complemented with 10% (v/v) heat inactivated fetal calf serum (FCS), 30 units/mL of penicillin, 30 μg/mL of streptomycin, 1 mM sodium pyruvate, 1% (v/v) non-essential amino acids (100×) and 2 mM of L-glutamine. Normal keratinocytes were prepared from hysterectomies as described previously (xxii). Briefly, fragments were plunged in a solution containing gentamycin, fungizon, and anti-mycoplasm. These fragments were cut in smaller pieces, then incubated in trypsin-EDTA (Invitrogen) at 37°C under agitation for 1–2 h. The epithelium was scraped and cells were recovered in FCS. After centrifugation, the cells were resuspended in K-SFM medium (Serum Free Media; Invitrogen) complemented with EGF (0.1 ng/mL), pituitary hormone (20–30 μg/mL), and gentamycin (5 μg/mL).
Cell Proliferation, Survival, and Apoptosis Assays
To evaluate the effect of the inhibitors on viability and apoptosis of HeLa and CaSki cells and primary normal keratinocytes, 105 cells were incubated for 24 h with 20 μM of the different inhibitors or with the vehicle (DMSO). Cells were then trypsinized and counted on a thomacytometer plate using trypan blue. Both death (blues cells) and live cells were counted. Half of the collected cells were stained with AnnexinV and propidium iodide and analyzed by flow cytometry to evaluate the percentage of apoptotic and death cells. Proliferation assays were performed by incubation of 5×104 HeLa or CaSki cells in 200 mL of DMEM complete medium in flat-bottomed 96-well microtiter plates. Proliferation was analyzed after incubating cells for 24 h with 20 μM inhibitors or DMSO, adding [3H]thymidine (0.4 mCi) for the last 4 h, before collecting cells with a cell harvester (Skatron, Sterling, VA, U.S.A.) onto glass fiber filters, and counting radioactivity using a scintillation analyzer (Tri-Carb, Packard, Meriden, CT, USA).
Acknowledgments
This work was supported by grant 1R21CA132121 (to LT) from the National Institutes of Health and by a grant from the Fond de la Recherche Scientifique (FRS-FNRS) and Télévie (to SR).
Abbreviations
- VHR
Vaccinia H1-related
- PTP
protein tyrosine phosphatase
- pTyr
phospho-tyrosine
- pThr
phospho-threonine
- Erk
extracellular signal-regulated kinase
- Jnk
c-Jun N-terminal kinase
- MAP
mitogen-activated protein
- MKP
MAP kinase phosphatase
- HPV
human papillomavirus
- SAR
structure activity relationship
- pNPP
p-nitrophenyl phosphate
- DiFMUP
6,8-difluoro-4-methylumbelliferyl phosphate
- DiFMU
6,8-difluoro-7-hydroxy-4-methylcoumarin
- DTT
dithiothreitol
- R.m.s.d
root mean square deviation
Footnotes
Data deposition: Xray crystallography data was deposited at the PDB database and has the code 3F81.
The data for the primary and confirmatory screen as well as for SAR support and MKP-1 and HePTP counterscreens have been deposited at PubChem (AIDs 1992, 2004, 2074, 2082, 2083).
References
- i.Bialy, Waldmann Angew Chem Int Ed Engl. 2005;44:3814–39. doi: 10.1002/anie.200461517. [DOI] [PubMed] [Google Scholar]
- ii.Tonks Nat Rev Mol Cell Biol. 2006;7:833–46. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- iii.Ostman, et al. Nat Rev Cancer. 2006;6:307–20. doi: 10.1038/nrc1837. [DOI] [PubMed] [Google Scholar]
- iv.Tautz, et al. Expert Opin Ther Targets. 2006;10:157–77. doi: 10.1517/14728222.10.1.157. [DOI] [PubMed] [Google Scholar]
- v.Jiang, Zhang Cancer Metastasis Rev. 2008;27:263–72. doi: 10.1007/s10555-008-9113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vi.Alonso, et al. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- vii.Ishibashi, et al. Proc Natl Acad Sci U S A. 1992;89:12170–4. doi: 10.1073/pnas.89.24.12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- viii.Yuvaniyama, et al. Science. 1996;272:1328–31. doi: 10.1126/science.272.5266.1328. [DOI] [PubMed] [Google Scholar]
- ix.Alonso, et al. J Biol Chem. 2001;276:4766–71. doi: 10.1074/jbc.M006497200. [DOI] [PubMed] [Google Scholar]
- x.Todd, et al. Oncogene. 2002;21:2573–83. doi: 10.1038/sj.onc.1205344. [DOI] [PubMed] [Google Scholar]
- xi.Todd, et al. J Biol Chem. 1999;274:13271–80. doi: 10.1074/jbc.274.19.13271. [DOI] [PubMed] [Google Scholar]
- xii.Waskiewicz, Cooper Curr Opin Cell Biol. 1995;7:798–805. doi: 10.1016/0955-0674(95)80063-8. [DOI] [PubMed] [Google Scholar]
- xiii.Robinson, Cobb Curr Opin Cell Biol. 1997;9:180–6. doi: 10.1016/s0955-0674(97)80061-0. [DOI] [PubMed] [Google Scholar]
- xiv.Ip, Davis Curr Opin Cell Biol. 1998;10:205–19. doi: 10.1016/s0955-0674(98)80143-9. [DOI] [PubMed] [Google Scholar]
- xv.Canagarajah, et al. Cell. 1997;90:859–69. doi: 10.1016/s0092-8674(00)80351-7. [DOI] [PubMed] [Google Scholar]
- xvi.Rahmouni, et al. Nat Cell Biol. 2006;8:524–31. doi: 10.1038/ncb1398. [DOI] [PubMed] [Google Scholar]
- xvii.Woods, et al. Mol Cell Biol. 1997;17:5598–611. doi: 10.1128/mcb.17.9.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xviii.Sewing, et al. Mol Cell Biol. 1997;17:5588–97. doi: 10.1128/mcb.17.9.5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xix.Serrano, et al. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- xx.Pumiglia, Decker Proc Natl Acad Sci U S A. 1997;94:448–52. doi: 10.1073/pnas.94.2.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxi.Wang, et al. Mol Cell Biol. 2002;22:3389–403. doi: 10.1128/MCB.22.10.3389-3403.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxii.Henkens, et al. BMC Cancer. 2008;8:147. doi: 10.1186/1471-2407-8-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxiii.Willett Drug Discov Today. 2006;11:1046–53. doi: 10.1016/j.drudis.2006.10.005. [DOI] [PubMed] [Google Scholar]
- xxiv.Schumacher, et al. Biochemistry. 2002;41:3009–17. doi: 10.1021/bi015799l. [DOI] [PubMed] [Google Scholar]
- xxv.Tautz, Mustelin Methods. 2007;42:250–60. doi: 10.1016/j.ymeth.2007.02.014. [DOI] [PubMed] [Google Scholar]
- xxvi.Barr, et al. Cell. 2009;136:352–63. doi: 10.1016/j.cell.2008.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxvii.Tautz, et al. J Biol Chem. 2005;280:9400–8. doi: 10.1074/jbc.M413122200. [DOI] [PubMed] [Google Scholar]
- xxviii.Otwinowski, Minor Methods in Enzymology. 1997;276:307–26. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- xxix.Collaborative Computational Project. Acta Crystallogr D Biol Crystallogr. 1994;50:760–3. [Google Scholar]
- xxx.Emsley, Cowtan Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- xxxi.Schuttelkopf, van Aalten Acta Crystallogr D Biol Crystallogr. 2004;60:1355–63. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]