

Abstract
Since the elucidation of the structure of double helical DNA, the construction of small molecules that recognize and react at specific DNA sites has been an area of considerable interest. In particular, the study of transition metal complexes that bind DNA with specificity has been a burgeoning field. This growth has been due in large part to the useful properties of metal complexes, which possess a wide array of photophysical properties and allow for the modular assembly of an ensemble of recognition elements. Here we review recent experiments in our laboratory aimed at the design and study of octahedral metal complexes that bind DNA non-covalently and target reactions to specific sites. Emphasis is placed both on the variety of methods employed to confer site-specificity and upon the many applications for these complexes. Particular attention is given to the family of complexes recently designed that target single base mismatches in duplex DNA through metalloinsertion.
Introduction
DNA is the library of the cell, simultaneously storing and dispensing the information required for life. Molecules that can bind and react with specific DNA sites provide a means to access this cellular information. Over the past few decades, small molecules that bind to DNA have shown significant promise as diagnostic probes, reactive agents and therapeutics. Much attention has focused on the design of organic, DNA-binding agents.1 However, over the past twenty five years, increasing interest has focused on another class of non-covalent DNA-binding agents: substitutionally inert, octahedral transition metal complexes.
At first glance, transition metal complexes seem an odd choice for DNA molecular recognition agents. Certainly, Nature herself offers very little precedent in this regard. With few exceptions, biological transition metals are confined to coordination sites in proteins or cofactors, not in discrete, free-standing coordination complexes.2 Further, the cell generally employs organic moieties for the binding and recognition of DNA. Yet despite the lack of many natural examples, transition metals complexes offer two singular advantages as DNA-binding agents. First and foremost, coordination complexes offer a uniquely modular system. The metal center acts in essence as an anchor, holding in place a rigid, three-dimensional scaffold of ligands that can, if desired, bear recognition elements. DNA-binding and recognition properties can thus be varied relatively easily via the facile interchange of ligands. Second, transition metal centers benefit from rich photophysical and electrochemical properties, thus extending their utility far beyond that of mere passive molecular recognition agents. Indeed, these characteristics have allowed metal complexes to be used in a wide range of capacities, from fluorescent markers to DNA foot-printing agents to electrochemical probes.3
With few exceptions, non-covalent, DNA-binding metal complexes share a few important characteristics. All are kinetically inert, a requisite trait due to the paramount importance of stability. Indeed, the vast majority of complexes are d6 octahedral or d8 square-planar. In addition, most exhibit a rigid or mostly rigid three-dimensional structure, an important facet considering that in many cases undue fluxionality could negate recognition. Moreover, the stereochemistry of the complex, if applicable, can provide specificity, an understandable notion given the chirality of the DNA target. Finally most of the complexes that have been prepared are, by design, photochemically or photophysically active, properties that confer tremendous utility in probing or effecting chemistry.
In this review, we do not strive to carry out an exhaustive survey of the field; instead, we seek to provide a discussion of more limited scope, highlighting important contributions from other researchers, yet concentrating principally on the work from our own laboratory. The early history of non-covalent, DNA-binding metal complexes is first addressed, followed by a more comprehensive look at the last two decades of research. In subsequent sections, complexes that bind DNA in each of three different non-covalent modes are discussed: groove binding, intercalation, and insertion (Figs. 1 and 2). Lastly, recent work on the development of therapeutic and diagnostic applications for some of these complexes is described. It should be noted that some of the most well-known work involving metal complexes and DNA has centered upon covalent interactions, most remarkably the work on platinum-based chemotherapeutics. Given the considerable breadth of this work, it is understandably outside the scope of this review. However, it has been extensively covered elsewhere.4
Fig. 1.

The three binding modes of metal complexes with DNA: (a) groove binding, (b) intercalation, and (c) insertion.
Fig. 2.

Geometries of (a) groove binder, (b) metallointercalator, (c) metalloinsertor.
Before embarking on our discussion of DNA binding and recognition, a brief description of the structure of DNA may be helpful. The most common form of DNA (and the form addressed almost exclusively in these pages) is the anti-parallel, right-handed double helix termed B-DNA, though the less common right-handed A-form and left-handed Z-form occasionally enter the discussion.5 Within the polynucleotide assembly, the heterocyclic bases – adenine (A), guanine (G), cytosine (C), and thymine (T) – are bonded to the sugars in an anti orientation with a disposition perpendicular to the helical axis. The base pairs collectively form a central, hydrogen-bonded π-stack that runs parallel to the helical axis between the two helical strands of the sugar-phosphate backbone. Each base forms hydrogen bonds with its complement on the opposite, anti-parallel strand, A with T and C with G. The rise per base is 3.4 Å, and there are ten base pairs per helical turn. Surrounding the central base stack, the polyanionic sugar-phosphate backbone forms two distinct grooves, a wide major groove and a narrow minor groove. All of these structural characteristics can and have been exploited for molecular recognition.
Early Work
The earliest research into the interactions between metals and DNA focused almost exclusively on the binding strength and location of metal-aquo ions, both those with and without biological significance.6 Perhaps as a result of these studies, the potential utility of metal-DNA interactions was realized early on. For example, melting temperature measurements for DNA in the presence of each of the first row transition metal ions were obtained to assess which metal ions stabilize or destabilize the duplex.7 The use of uranyl-bound nucleosides was investigated as a possible tool for electron microscopy-based DNA sequence determination.8 Further, studies of the binding of mercury to non-thiolated and thiolated guanosine residues also portended the growing interest in metals as useful DNA probes.9 Importantly, these studies all focused upon the coordination of metal ions to DNA and as such employed either aquo ions or complexes with open coordination sites. Our interest, however, is in the non-covalent binding of coordinatively saturated metal complexes to DNA. With respect to this area, clues suggesting the interaction of inert metal complexes and DNA were evident as early as the 1950s, most notably in F.P. Dwyer’s work on the biological activity of metal polypyridyl complexes.10 Simple tris(chelate) complexes of Ru(II) and Ni(II) were found to have antiviral and bacteriostatic activities. Quite remarkably, stereoselective biological activity was observed in some cases.
It was not until the mid-1970s, however, that a progenitor non-covalent DNA-binding complex was prepared by S. J. Lippard and coworkers.11 During their work on metal-binding to thiolated bases, it was observed that the planar complex [Pt(2,2′,2″-terpyridine)(Cl)]+ induced a spectral shift for 4-thiouridine in the presence of tRNA. Follow up work, this time using [Pt(terpyridine)(SCH2CH2OH)]+ to eliminate the labile coordination site, employed a variety of techniques to establish the intercalative binding mode. X-ray fiber diffraction patterns provided further evidence for intercalation, revealing a periodicity of one platinum unit every 10 angstroms (every other base-pair) and a partial unwinding of the phosphate backbone.12 Subsequent work expanded the family of intercalators to include other complexes with planar heterocyclic ligands, [Pt(bpy)(en)]2+ and [Pt(phen)(en)]2+, established binding constants in the realm of 104 – 105 M−1 for the family with DNA base pairs, and investigated the effects of sequence context and ionic strength on intercalation.13
Just as Lippard’s platinum complexes laid the groundwork for future work on intercalative binding, the study of another complex, [Cu(phen)2]+, in the lab of D.S. Sigman during the late 1970s and early 1980s unearthed the rich chemistry of groove-binding metal complexes.14 The complex was serendipitously discovered to degrade DNA during investigations into the inhibition of E. coli DNA polymerase by 1,10-phenanthroline, and it was soon learned that the DNA cleavage reaction was oxygen-dependent.15 Product isolation and analysis led to a proposed mechanism that suggested minor-groove binding by [Cu(phen)2]+ formed in situ, a hypothesis later confirmed through elegant labeling experiments.16 Additional reactivity studies have revealed that the complex cleaves not only B-form duplex DNA but also, though in some cases to a lesser extent, A-form DNA, RNA, and folded nucleic acid structures.17
Nature’s Example
Before moving on to our main discussion of synthetic complexes, it is important to address, at least briefly, nature’s lone example of a non-covalent DNA-binding metal complex: metallobleomycin. First isolated from Streptomyces verticillus in the late 1960s, bleomycins are a widely-studied family of glycopeptide antibiotics that have been used successfully in the treatment of some forms of cancer.18 The structure of bleomycins can be broken down into three domains: a metal-binding domain containing a pyrimidine moiety and five nitrogen atoms for octahedral metal coordination, a peptide linker region bearing a disaccharide side-chain, and a bithiazole unit with an appended, positively charged tail. While the metal-binding region can coordinate a variety of metals including Zn(II), Cu(II), and Co(III), the majority of research has focused on understanding the reactivity of Fe-bleomycin complexes.19 Significantly, exposure of the Fe bleomycin complex to oxygen and a reductant leads to the formation of activated bleomycin, a species that can, in turn, affect both single-stranded and double-stranded DNA cleavage via 4′-hydrogen atom abstraction by a high valent Fe-oxo species.
Metallobleomycins bind DNA via the minor groove, though neither affinity nor specificity is particularly high. Over the past twenty years, extensive synthetic and spectroscopic studies have helped to elucidate the contribution of each structural moiety to DNA-binding and reactivity.20 The bithiazole subunit and positively-charged tail are considered to play the most important roles in DNA-binding. The charge of the cationic tail is generally agreed to provide electrostatic impetus for binding. The role of the bithiazole, however, is subject to some debate. While the bulk of the evidence suggests that this moiety intercalates between base-pairs neighboring the binding site of the complex21, others have suggested that the bithiazole interacts with the DNA primarily in the minor groove.22 Hydrogen-bonding of the pyrimidine moiety in the metal-binding region is thought to help confer 5′-G-Py-3′ cleavage selectivity.19d,20b The definitive roles of the linker region and disaccharide have proven more subtle and elusive, with the linker region likely of conformational importance and the disaccharide having been given roles ranging from DNA binding to metal chelation to cellular uptake and localization.
Finally, it is also both interesting and important to note that metallobleomycins, unlike many of the metal complexes discussed below, are exquisitely sensitive to structural changes, for attempts to alter any of the domains have been met with dramatically reduced cleavage efficiencies.20
Tris(phenanthroline) Complexes
The earliest work on the DNA-binding of octahedral metal centers focused on tris(phenanthroline) complexes of ruthenium, cobalt, zinc, nickel, and cobalt. (Fig. 3).23 Extensive photophysical and NMR experiments suggested that these complexes bound to DNA via two distinct modes: (a) hydrophobic interactions in the minor groove and (b) partial intercalation of a phenanthroline ligand into the helix in the major groove. Perhaps more important than the discovery of these dual binding modes, however, was the revelation these complexes provided regarding the importance of chirality in DNA-binding.24 In the case of Ru(phen)32+, for example, the Δ-enantiomer is preferred in the intercalative binding mode, while the complementary Λ-enantiomer is favored in the minor groove binding mode. In subsequent years, it was discovered that metal centers bearing more sterically demanding phenanthroline ligand derivatives, such as diphenylphenanthroline (DIP), display even more dramatic chiral discrimination. Luminescence and hypochromism assays have revealed enantioselective binding on the part of [Ru(DIP)3]2+; the Δ-enantiomer binds enantiospecifically to right-handed B-DNA and the Λ-enantiomer binds only to left-handed Z-DNA.25 This enantiospecificity has been exploited to map left-handed Z-DNA sites in supercoiled plasmids using [Λ-Co(phen)3]3+.26 Indeed, this trend in enantiomeric selectivity for octahedral tris(chelate) complexes, matching the symmetry of the complex to that of DNA helix, has repeatedly and consistently been observed for non-covalent DNA-binding complexes developed in the years since these initial discoveries.3, 27
Fig. 3.

Λ– and Δ-enantiomers of [Rh(phen)3]3+.
These earliest tris(phenanthroline) complexes do not, of course, represent the only examples of complexes that bind DNA via the minor or major grooves. For instance, the extensively studied [Cu(phen)2]+, has been shown to bind DNA via the minor groove. Indeed, these groove-binding complexes not only bind DNA but also cleave the macromolecule in the presence of hydrogen peroxide.28 Metal complexes that bind in the groove have come a long way since these first studies and are now quite sophisticated. Turro, for instance, developed an artificial photonuclease by linking the metallogroove-binder [Ru(bpy)3]2+ to an electron-acceptor chain containing two viologen units.29 Interestingly, the chemistry of metallogroove-binders also extends to supramolecular self-assembly. Following the initial work of Lehn on the interaction and cleavage of DNA with a cuprous double-helicate,30 Hannon and coworkers designed a triple-helicate capable of recognizing three-way junctions in DNA. This intricate recognition has recently been characterized by single crystal X-ray crystallography.31
Metallointercalators
General architecture and binding mode
Intercalators are small organic molecules or metal complexes that unwind DNA in order to π-stack between two base pairs (Fig. 1). Metallointercalators, it then follows, are metal complexes that bear at least one intercalating ligand (Fig. 2). As their name suggests, these ligands, oriented parallel to the base pairs and protruding away from the metal center, can readily π-stack in the DNA duplex. Further, upon binding, the ligands behave as a stable anchor for the metal complex with respect to the double helix and direct the orientation of the ancillary ligands with respect to the DNA duplex. Two well-known examples of intercalating ligands are phi (9,10-phenanthrenequinone diimine) and dppz (dipyrido[3,2-a:2′,3′-c]phenazine) (Fig. 4).3
Fig. 4.

Chemical structure of two common metallointercalators: (a) Δ-[Rh(phen)2phi]3+ and Δ-[Ru(bpy)2dppz]2+. The intercalating ligands are highlighted in blue, the ancillary ligands in yellow.
Ligand intercalation was first demonstrated by photophysical studies.23, 32 However, it was not until extensive NMR studies33 and high resolution crystal structures had been performed that the structural details of this binding mode were properly illuminated (Fig. 5).34 Metallointercalators enter the double helix via the major groove, with the intercalating ligand acting in effect as a new base pair. No bases are ejected from the duplex. Further, intercalation results in a doubling of the rise and a widening of the major groove at the binding site. Importantly, this interaction distorts only minimally the structure of DNA. In the case of B-DNA, for example, the sugars and bases all maintain their original C2′ endo and anti conformations, respectively. Indeed, only the opening of the phosphate angles, not any base or sugar perturbations, is necessary for intercalation.
Fig. 5.

Crystal structure of the metallointercalator Δ–α-[Rh[(R,R)-Me2trien]phi]3+ bound to its target sequence, 5′-TGCA-3′.
The three crystal structures of a metal complex intercalated within a duplex, two containing an octahedral rhodium complex inserted in an oligonucleotide and one a square-planar platinum complex inserted into a paired dinucleotide, each demonstrated that intercalation occurs via the major groove.34–36 Yet this may not always be the case. NMR studies indicate that metal complexes bearing dpq (dipyrido[2,2-d:2′,3′-f]quinoxaline), a close analogue of dppz lacking the terminal aromatic ring, favors binding via the minor groove.37 Whether this binding by the more hydrophobic complex involves one or two binding modes, groove-binding from the minor groove and intercalation, still needs to be confirmed.
Exploiting the photophysical and photochemical properties of metallointercalators
By design, metallointercalators are coordinatively saturated and substitutionally inert such that no direct coordination with DNA bases occurs. Nonetheless, they often possess rich photochemistry and photophysics that have been advantageously exploited both to probe their interaction with DNA and to understand further various aspects of nucleic acid chemistry. The most studied example is probably the molecular light switch complex, [Ru(phen)2dppz]2+. This ruthenium complex shows solvatochromic luminescence in organic solutions. In aqueous solutions, however, it does not luminesce because water deactivates the excited state through hydrogen-bonding with the endocyclic nitrogen atoms of the intercalating ligand. Remarkably, however, the complex luminesces brightly upon the addition of duplex DNA. In this case, the metal complex intercalates into the DNA, and the surrounding duplex prevents water from gaining access to the intercalated ligand; thus, the DNA has created a local region of organic ‘solvent’ in which the metal complex, now free of any hydrogen bonds, can display its characteristic luminescence. (Fig. 6).38
Fig. 6.
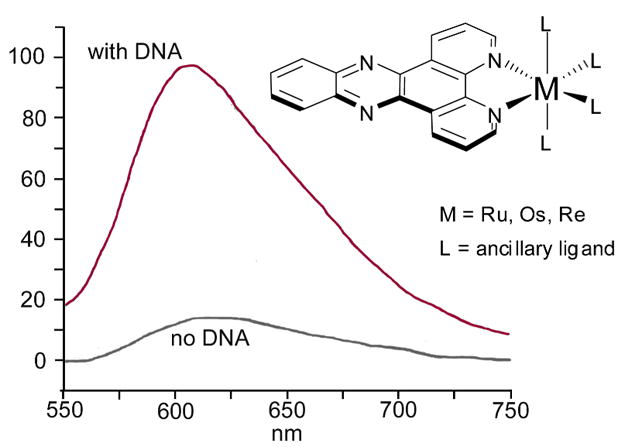
The light-switch effect of dppz-based metallointercalators..
Although there has been some debate over the binding orientation of [Ru(phen)2dppz]2+,39 it has now been established that it intercalates via the major groove.32b,c Direct competition titrations against both a minor groove binder (distamycin) and a well-characterized major groove intercalator (Δ–α-[Rh[(R,R)-Me2trien]phi]3+, vide infra)33b,24 clearly demonstrate that the molecular light switch intercalates via the major groove with a slight preference for poly-d(AT) regions over poly-d(GC).40 This conclusion is further supported by detailed NMR studies performed with complexes bearing selectively deuterated dppz ligands. The latter investigations, together with the observed biexponential decay of the luminescence of [Ru(phen)2dppz]2+, further stipulated the presence of two populations with slightly different intercalation geometries.32b,c Many analogues of the popular molecular light switch, such as Nordén’s threading ruthenium bis-intercalators,41 have been synthesized and their photophysics have been extensively studied and reviewed.42
While ruthenium and dppz-based metallointercalators have proven to be powerful molecular light switches for the detection of DNA, rhodium intercalators have been shown to be efficient agents for photoactivated DNA strand cleavage. Importantly, this reactivity enables us to mark directly the site of intercalation and to characterize the recognition properties of each metallointercalator. In this case, the most well studied examples are rhodium complexes employing the phi ligand as the intercalator, such as [Rh(bpy)2(phi)]3+, [Rh(phen)2(phi)]3+, and [Rh(phi)2(bpy)]3+ (Fig. 4).43
In many cases, the irradiation of the intercalated metal complex with short wavelength light (313–325 nm) initiates strand scission near the binding site. More specifically, this irradiation prompts the formation of an intercalating ligand-based radical that abstracts a hydrogen atom from an adjacent deoxyribose ring.43 Subsequent degradation of the resultant sugar radical then leads to direct DNA strand scission. In the absence of dioxygen, the photolysis of intercalated rhodium complexes leads to the formation of 3′- and 5′-phosphate terminated strands as well as a free base. To contrast, in the presence of dioxygen, direct strand cleavage still occurs but instead produces a 5′-phosphate terminated strand, a 3′-phosphoglycaldehyde terminated strand, and a base propenoic acid. These observations are consistent with previously observed chemistry at the C3′ position of the sugar. However, since both an atomic resolution crystal structure and a solution NMR study of a metal complex intercalated in the major groove of DNA indicate that the C2′ hydrogen of the neighbouring sugar is closer to the intercalating ligand than the C3′ hydrogen of said sugar, we propose that the photoactivated intercalator initially abstracts the C2′ hydrogen of the sugar. This step is immediately followed by hydrogen atom migration to form the more stable tertiary C3′ radical. Degradation of the sugar ring completes the process.
Although rhodium complexes efficiently cleave DNA upon photoactivation, many research laboratories find more convenient the use of DNA cleavage agents that cut without irradiation (Fig. 7).44 This is achieved with the use of a bifunctional metallointercalator – peptide chimera in which a metal-coordinating peptide is covalently attached to [Rh(phi)2bpy′]3+. The metallointercalator acts as a targeting vector that delivers the metallopeptide to the sugar-phosphate backbone. The latter then promotes hydrolytic DNA strand cleavage.
Fig. 7.

Chemical structures of (a) an artificial nuclease and (b) a luminescent cross-linking agent.
In a similar approach, luminescent DNA cross-linking probes were achieved using bifunctional ruthenium intercalators conjugated to short peptides (Fig. 7).45 In the presence of an oxidative quencher, irradiation of the intercalated [Ru(phen)(bpy′)(dppz)]2+ oxidizes the oligonucleotide. The nearby tethered peptide then crosslinks with the oxidized site of the DNA. Although delivery of the peptide by the metallointercalator is not essential for cross-linking, this technique advantageously yields cross-linking adducts that are luminescent and are thus easily detectable. Furthermore, these cross-links may resemble those found in vivo under conditions of oxidative stress.
Shape-selective recognition
On the whole, metallointercalators are structurally rigid molecules with well-defined symmetry, making them particularly well suited for selective molecular recognition of specific DNA sequences. Importantly, because of the general rigidity of the complexes, the overall shape and ancillary ligands of these complexes can also be exploited in the development of useful compounds.
Perhaps not surprisingly, stereochemistry is of utmost importance in the construction of site-specific recognition agents. Indeed, one of the earliest findings of this chemistry is the necessity of matching the chirality of the metallointercalator with that of the double helix: the Δ-enantiomer of the metal complex preferentially binds to right-handed B-DNA. This enantioselective discrimination is primarily steric in nature and depends on the size of the ancillary ligands relative to that of the DNA groove. For instance, poor enantioselectivity is observed with metallointercalators bearing small ancillary ligands such as phenanthroline and bipyridine, whereas complete enantiospecificity is achieved with bulkier ancillary ligands such as DPB (4,4′-diphenyl-bipyridine).46 The Δ-enantiomer of [Rh(phi)(DPB)2]3+ (Fig. 8), for example, readily cleaves the sequence 5′-CTCTAGAG-3′ upon photoactivation, but no intercalation or cleavage is observed with the Λ-enantiomer, even with a thousand fold excess of metallointercalator. For Z-DNA, which is a left-handed helix, there is little enantioselectivity of the chiral metal complexes because of the very shallow, almost convex major groove; 25 hence the Λisomer, which cannot bind at all to B-form DNA becomes a probe for Z-DNA.
Fig. 8.

Sequence-specific metallointercalators and their target sequences. The intercalation sites are marked with grey ovals.
As a monomer, Δ-[Rh(phi)(DPB)2]3+ is geometrically capable of spanning only six base pairs; however, the metallointercalator is able to recognize a palindromic sequence eight base pairs long by dimerizing. The target sequence 5′-CTCTAGAG-3′ can be considered as two overlapping 5′-CTCTAG-3′ intercalation sites. Concomitant intercalation of two of the metal complexes, each at a central 5′-CT-3′ of the 6-mer, favors stacking of the ancillary phenyls from both complexes over the central 5′-TA-3′ step. This binding cooperativity, more common with DNA binding proteins, enhances the binding affinity of the second intercalator by 2 kcal. As a result, irradiation of the metallointercalators/DNA adduct cleaves both DNA strands with three base pairs separating the two cleavage sites.
The remarkable specificity and intricate binding mode of Δ-[Rh(phi)(DPB)2]3+ enables it to inhibit efficiently the activity of XbaI restriction endonuclease at the palindromic site.46 Notably, no comparable inhibition of XbaI has been achieved with any other metallointercalators, and Δ-[Rh(phi)(DPB)2]3+ cannot inhibit restriction enzymes that bind different sites. Thus, metallointercalators have found use not only as probes for nucleic acid structures but also as mimics, probes and, perhaps, inhibitors of DNA-binding proteins.
Interestingly, more moderate shape-based site recognition can be achieved even with sterically smaller ancillary ligands like phenanthroline. [Rh(phen)2phi]3+, for instance, preferentially intercalates at sites with high propeller twisting toward the major groove.43,47 This intercalator preferentially photocleaves 5′-Py-Py-Pu-3′ sites and occasionally 5′-Pu-Py-Pu-3′ sites but not 5′-Pu-Pu-Py-3′ sites. Comparison of photocleavage experiments with the crystal structures of several B-form oligonucleotides revealed a direct correlation between the binding preference of [Rh(phen)2phi]3+ and the increased propeller twisting at the site of intercalation. Opening of the major groove in the 5′-Py-Py-Pu-3′ sequence produces more steric leeway for the hydrogens of the ancillary phenanthroline ligands, thus enabling deeper intercalation by the metal complex. In the case of a 5′-Pu-Pu-Py-3′ site, however, reduced propeller twisting creates a more sterically confining major grove at the intercalation site; in this instance, then, increased steric hindrance between the groove and the phenanthroline ligands pushes the intercalating phi ligand farther away from the DNA helical axis, thereby reducing the binding affinity of the complex.
Due to its unique properties, [Rh(phen)2(phi)]3+ has also been employed as a probe for RNA tertiary structure.48 As discussed above, the complex can only intercalate from the major groove side of DNA, a property which prevents it from binding via the sterically-altered groove of duplex RNA and binding instead preferentially to triplex RNA. In this capacity, the rhodium complex is able to compete for binding at the TAT protein binding site in the immunodeficiency virus TAR RNA.49 [Rh(phen)2phi]3+ efficiently binds and photocleaves the U24 base involved in the base-triplex of the RNA hairpin essential to TAT binding. The metal complex similarly competes with and inhibits the binding of the bovine BIV-TAT peptide to its RNA site. Mutants of the RNA oligomer lacking the base triplex and which could therefore no longer bind the TAT peptide were likewise no longer targeted by the metallointercalator.
Sequence recognition based on functionality
Selective recognition of a DNA sequence by a metallointercalator can also be achieved by matching the functionality of the ancillary ligands positioned in the major groove with that of the targeted base pairs. Specific targeting of the sequence 5′-CG-3′, for instance, is achieved with the complexes [Rh(NH3)4phi]3+, [Rh([12]aneN4)phi]3+ and Δ- [Rh(en)2phi]3+ (Fig. 8).50 In these examples, recognition is ensured both by the C2 symmetry of the metal complexes and hydrogen bonding between the axial amines of the metallointercalators and the O6 of the guanine. The Λ–enantiomer of [Rh(en)2phi]3+, instead, recognizes the sequence 5′-TA-3′ due to van der Waals contact between the methylene groups on the backbone of the complex and the thymine methyl.
The predictive design of sequence specific metallointercalators was expanded with Δ–α-[Rh[(R,R)-Me2trien]phi]3+, a complex that directly recognizes and photocleaves the sequence 5′-TGCA-3′ (Fig. 8).51 The rhodium complex was designed to recognize this sequence via hydrogen bonding contacts between the axial amines and the O6 of guanine, as well as potential van der Waals contacts between the pendant methyl groups on the metal complex and the methyl groups on the flanking thymines (Fig. 5). A high resolution NMR solution structure33b followed by the first crystal structure34 of a metallointercalator-DNA adduct later revealed at atomic resolution the details of the intercalation and recognition. In fact, it is because of the high sequence-specificity of this intercalator that a high resolution view of intercalation within a long DNA duplex could be obtained. In the DNA octamer containing the central 5′-TGCA-3′ site, the DNA unwinds to enable deep and complete intercalation of the phi ligand of the metal complex via the major groove. This results in a doubling of the rise at the intercalation site without any base ejection. The metallointercalator thus behaves as a newly added base pair that causes only minimal structural perturbation to the DNA. Furthermore, both the NMR study and crystal structure confirmed that the sequence-specific recognition was, indeed, based on the anticipated hydrogen bonding and van der Waals interactions.
Sequence recognition based on shape and functionality
Yet another metallointercalator provides an interesting example of sequence-specific recognition predicated on both shape and functionality. 1-[Rh(MGP)2phi]5+, a derivative of [Rh(phen)2phi]3+ containing pendant guanidinium groups on the ancillary phenanthroline ligands, was designed to bind a subset of the sequences recognized by the latter, specifically those 5′-Py-Py-Pu-3′ triplets flanked by two G•C base pairs. Hydrogen bonding between the guanidinium groups on the ancillary ligands and the O6 atoms of the flanking guanines was expected to confer this selectivity. (Fig. 8).52 As predicted, NMR studies demonstrated that the Δ-enantiomer recognizes the sequence 5′-CATCTG-3′ specifically.
Surprisingly, in spite of the large size of the ancillary ligands, the Λ-enantiomer also binds DNA and recognizes a different sequence, 5′-CATATG-3′. The large MGP ligands certainly prevent the left-handed isomer from passively entering the major groove of right-handed DNA. However, plasmid unwinding assays and NMR studies established that the Λ-enantiomer of the metallointercalator binds DNA by unwinding it up to 70°.33a It is in this conformation that the complex can span the entire six-base pair binding site and contact the N7 position of the flanking guanines with the pendant guanidinium groups. Replacing these flanking guanines with deazaguanines demonstrated that the absence of the N7 nitrogen atoms eliminated any site selectivity. Therefore, we can conclude that the guanidinium functionalities of the ancillary ligands are responsible for the recognition of the flanking guanines, whereas the shape of the metallointercalator enables the recognition of the “twistable” central 5′-ATAT-3′ sequence.
Due to its high site-specificity, the Λ-enantiomer of this complex has found biological application as an inhibitor of transcription factor binding.53 In a manner similar to [Rh(phen)2(phi)]3+,Λ-1-[Rh(MGP)2phi]5+ can site-specifically inhibit the binding of a transcription factor to its modified activator recognition region. In competition experiments with yeast Activator Protein 1 (yAP-1), the metal complex was able to compete with the protein for a domain that included both the binding region of yAP-1 and that of Λ-1-[Rh(MGP)2phi]5+ at concentrations as low as 120 nM. This result represents one of the first hints at the therapeutic potential of rhodium intercalators, a notion strongly supported by subsequent investigations illustrating that Rh(phi)2(phen)3+ and other rhodium bis(quinone diimine) complexes inhibit transcription in vitro.54
Metalloinsertors
Without a doubt, the vast majority of non-covalent, DNA-binding metal complexes are either groove-binders or intercalators. However, the dearth of complexes that bind DNA via other means does not necessarily exclude the existence of alternative modes. Indeed, L.S. Lerman, in his seminal article proposing intercalation as the DNA-binding mode for organic dyes, presciently proposed a third non-covalent binding mode: insertion.55 A molecule, he posited, may bind “a DNA helix with separation and displacement of a base-pair.” While Lerman was addressing organic moieties, we can apply this thinking to metal complexes quite easily. Metalloinsertors, like metallointercalators, contain a planar aromatic ligand that extends into the base-stack upon DNA-binding. However, while metallointercalators unwind the DNA and insert their planar ligand between two intact base-pairs, metalloinsertors eject the bases of a single base-pair, with their planar ligand acting as a π-stacking replacement in the DNA base stack.
Until very recently, no examples of DNA-binding insertors, neither metal-based nor organic, had been reported. However, our research into mismatch-specific DNA-binding agents has led to the discovery of a family of rhodium complexes that bind DNA via this unique mode. These novel complexes have been dubbed metalloinsertors.
Background
Over the past ten years, much of our work in molecular recognition has been focused on the design, synthesis, and study of metal complexes that selectively bind mismatched sites in DNA. Mismatched DNA not only represents a very important target but also presents a unique challenge from the perspective of molecular recognition. DNA base mismatches, for example adenine-cytosine or cytosine-cytosine, occur in the cell as a result of errors during replication or exposure to genotoxic agents.56 Left uncorrected, these mismatches ultimately lead to single nucleotide polymorphisms (SNPs), single base mutations that lead (among other things) to variations in disposition to disease.57 To preserve the fidelity of its genome, the cell has developed a complex mismatch repair (MMR) machinery to locate and repair these mismatches.58 However, mismatches, and thus mutations, can accumulate if this machinery is somehow damaged or disabled, increasing the likelihood of cancerous transformations in the genome. Indeed, mutations in MMR genes have been identified in almost 80% of hereditary non-polyposis colon cancers. Further, 15–20% of biopsied solid tumors show some evidence of somatic mutations in MMR genes.59 Clearly, a selective mismatch detection agent could prove a significant development in the diagnosis of MMR deficiency and, in turn, cancer.
Rational Design
When compared to the sequence-specific metallointercalators, the design of mismatch-specific complexes presents a peculiar challenge. In this case, the recognition target is not a unique sequence but rather a type of site, specifically a thermodynamically destabilized region in the duplex created by the mismatch’s imperfect hydrogen-bonding. Indeed, the ideal mismatch recognition agent would bind any mismatched site (CC, CA, AG, etc.) without regard to the sequence context surrounding the mismatch. Taken together, these requirements dictate that the recognition elements of our mismatch-selective complexes must move from the ancillary ligands to the intercalating/inserting ligand.
Somewhat surprisingly, mismatch-specificity was achieved simply by replacing the non-specific phi ligand with the similar but more sterically expansive 5,6-chrysene quinone diimine (chrysi) ligand. Specifically, the chrysi ligand is 0.5 Å wider than the span of matched DNA and 2.1 Å wider than its parent phi ligand (Fig. 2). Unlike the phi ligand, which is the ideal size for intercalation into matched DNA, the chrysi ligand, with its additional fused ring, is too bulky to intercalate at stable, matched sites due to inevitable steric clash with the sugar rings of the DNA. Thermodynamically destabilized mismatch sites, it was proposed, would be a different story altogether, for at these locales, the added energetic benefit of the π-stacking ligand would outweigh the energetic cost of any steric clash. When synthesizing the complex, rhodium was again chosen as the metal primarily due its photophysical properties, most notably the ability of the non-specific rhodium complexes to promote strand scission upon irradiation.
Recognition Experiments
The first generation complex, [Rh(bpy)2(chrysi)]3+ was synthesized from [Rh(bpy)2(NH3)2]3+ and 5,6-chrysene quinone via base-mediated condensation of the quinone onto the ammine ligands of the metal ion (Fig. 9a).60 Initial photocleavage experiments showed that the complex does, indeed, bind mismatched sites and, upon photoactivation with UV-light, promotes direct strand cleavage of the DNA backbone adjacent to the mismatch site.61 The compound also proved to be remarkably selective; mismatches are bound at least 1000 times tighter than matched base-pairs. A dramatic enantiomeric effect is also observed, with the Δ-enantiomer binding and cleaving extremely well and the Λ-enantiomer almost completely inactive. While the preference for the Δ-isomer binding to right-handed DNA was expected, the remarkably high enantioselectivity even with a bpy complex was unexpected. Further experiments were performed to test the specificity of the complex. Photocleavage experiments employing alkaline agarose and denaturing polyacrylamide gels revealed that [Rh(bpy)2(chrysi)]3+ cleaves at, and only at, the single mismatch site incorporated into a linearized 2725 base-pair plasmid.51
Fig. 9.

Chemical structures of mismatch-specific metalloinsertors.
Subsequent experimentation estabished that [Rh(bpy)2(chrysi)]3+ binds and cleaves 80% of mismatch sites in all possible sequence contexts.62 Furthermore, correlating cleavage intensity against independent measurements of mismatch destabilization revealed a clear relationship between mismatch stability and metal complex binding and cleavage: in general, the more destabilized the mismatch, the tighter the binding. For example, the mismatch-selective binding constants range from 3 × 107 M−1 for the dramatically destabilized CC mismatch to 2.9 × 105 M−1 for the far more stable AA mismatch.63 Consistent with this relationship, [Rh(bpy)2(chrysi)]3+ almost completely fails to target the most stable mismatches, specifically those containing guanine nucleotides. In essence, the less destabilized mismatched sites “look” just like well-matched base-pairs to the chrysi complex.
More recently, higher mismatch binding affinities were obtained using [Rh(bpy)2(phzi)]3+, a second-generation complex bearing a similar expansive intercalating ligand, benzo[a]phenazin-5,6-quinone diimine (Fig. 9b).64 For example, the binding constants of this complex for CA and CC mismatches were measured to be 0.3 and 1 × 107 M−1, respectively, affinities that allow for mismatch recognition and photocleavage at nanomolar concentrations. Importantly, the higher binding affinities are not accompanied by a concomitant decrease in selectivity, which remains at 1000-fold or greater. The increased affinity, however, is not sufficient to facilitate binding to the more stable G-containing mismatches.
Structural Information
While the above experiments provide comprehensive information on the range, strength, and specificity of the mismatch recognition exhibited by [Rh(bpy)2(chrysi)]3+, they yield little, if any, information on the structure of the complex and DNA upon binding. Previous NMR and crystal structures of phi-bearing metallointercalators clearly indicated that these complexes bind by classical intercalation via the major groove.65 There was, however, no guarantee that a mismatch recognition complex would bind DNA in a similar manner. Thus, the elucidation of the structure of [Rh(bpy)2(chrysi)]3+ became a project of significant importance.
[Δ-Rh(bpy)2(chrysi)]3+ was co-crystallized with a self-complementary oligonucleotide containing two AC mismatches (5′-CGGAAATTCCCG-3′). The structure was subsequently solved at atomic resolution (1.1 Å) using the single anomalous diffraction technique (Figure 10).25 Quite surprisingly, the structure revealed two binding modes for [Rh(bpy)2(chrysi)]3+. In the crystal, not only is the complex bound to both mismatched sites as expected, but it is also intercalated at a matched site at the center of the oligonucleotide. However, a large volume of evidence, including a second crystal structure, supports the idea that the binding observed at the matched sites results entirely from crystal packing forces.66
Fig. 10.
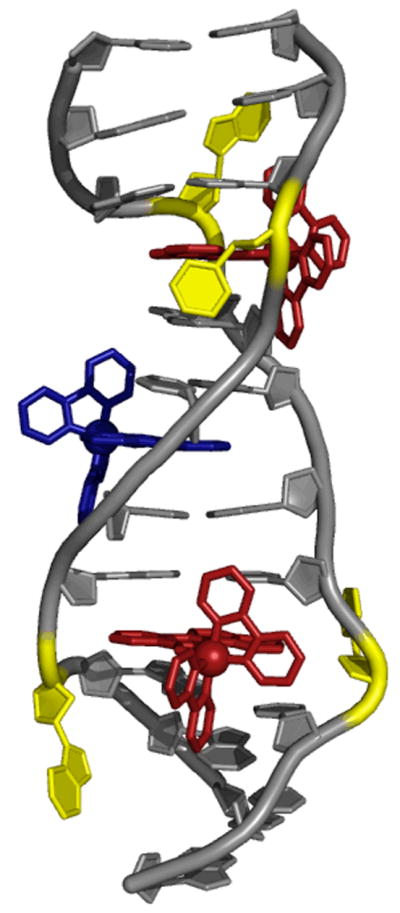
Crystal structure of the metalloinsertor (red) bound to a target CA mismatch.
In stark contrast to other known metallointercalators, [Rh(bpy)2(chrysi)]3+ is bound to the mismatched DNA via the minor groove. Further, and perhaps more remarkably, the complex does not bind via classical intercalation but rather the previously unreported mode of insertion. Rather than stacking an intercalating ligand between base-pairs, thereby prompting an increase in the rise of the DNA, the complex completely ejects the mismatched nucleotides from the base-stack and replaces the ejected bases with its own chrysi ligand. Despite this insertion, the complex does not significantly distort the DNA, with all sugars maintaining a C2′-endo puckering and all bases remaining in the anti-configuration. Instead, the DNA accommodates the bulky ligand by opening its phosphate backbone slightly. The chrysi ligand is inserted quite deeply into base stack, so much so that the rhodium is only 4.7 Å from the center of the helical axis, and the ligand itself is solvent accessible from the opposite major groove.67 Interestingly, the complex itself is perturbed very little, though some flattening of the chrysi ligand (perhaps to augment π-stacking) is observed. These structural observations have been independently verified in a recent NMR investigation.68
The details of the crystal structures and the NMR study help to explain three observations about which we could previously only hypothesize. First, the binding of the complex to the sterically smaller minor groove without an increase in rise explains the observed enantiomeric effect on affinity. Second, the minor groove insertion of the complex explains the different cleavage products created by [Rh(bpy)2(chrysi)]3+ and [Rh(bpy)2(phi)]3+ as observed via mass spectrometry.69 The major groove binding mode for the metallointercalator positions it to cleave the DNA via abstraction of the deoxyribose ring C2′H. Because it binds via the minor groove, [Rh(bpy)2(chrysi)]3+ is positioned to abstract preferentially the C1′H of the sugar adjacent to the mismatch site, and in this case we see products consistent with C1′H abstraction. Finally, while we had previously hypothesized that the thermodynamically destabilized site created by the mismatched base-pairs somehow allowed for [Rh(bpy)2(chrysi)]3+ binding, the ejected bases observed in the structure point to the concrete explanation. Since [Rh(bpy)2(chrysi)]3+ readily displaces the bases of destabilized mismatch sites to bind to the DNA, it follows that the more destabilized the site, the more easily the complex can eject the mismatched bases, and the tighter it can bind. Conversely, the complex cannot eject matched bases (or even more stable mismatched bases) because their hydrogen bonding interactions are too strong to allow for it.
Diagnostic Applications
Given the important role of mismatches and mismatch repair deficieny in cancer susceptibility, the development of our unique recognition technology for diagnostic and therapeutic applications has also been a focus of our laboratory.
Fluorescence is a particularly attractive reporter in diagnostic applications and could be very useful as a sensitive early diagnostic in detecting the presence of mismatches in genomic DNA. As a result, we have developed two different mismatch-specific fluorophores as potential diagnostics. The first probe, [Ru(bpy)2(tactp)]2+, sought to combine the DNA light-switch character of [Ru(dppz)(L)2]2+ complexes and the mismatch-specificity of the chrysi ligand in a single complex bearing a bulky chrysi/dppz hybrid ligand (Fig. 11a).70 However, while the complex does exhibit some light-switch behavior and mismatch-specific binding, the avid dimerization of the large aromatic ligand leads to non-specific fluorescence and thus dramatically limits its diagnostic potential.
Fig. 11.

Luminescent probes for mismatch detection.
A second, somewhat more efficient probe for mismatched DNA was achieved by tethering a negatively charged fluorophore to a trisheteroleptic, mismatch-specific rhodium complex bearing a linker-modified bipyridine ligand (Fig. 11b).71 In free solution and in the presence of matched DNA, ion-pairing between the cationic rhodium complex and the anionic fluorophore dramatically quenches the fluorescence of the conjugate. In the presence of mismatched DNA, the bulky metalloinsertor binds the polyanionic DNA, and the anionic fluorophore is consequently electrostatically repelled away from the rhodium moiety, thereby attenuating the intramolecular quenching and increasing fluorescence. In this manner, the fluorescence of the conjugate is increased over 300% in the presence of mismatched oligonucleotide DNA. This probe, like the Ru complex, has its limitations, however, chief among them being that even when “turned on” in the presence of mismatched DNA, the fluorescence of the conjugate is still significantly quenched with respect to free, equimolar fluorophore.
In an alternative strategy, the site-specific photocleavage capability of both [Rh(bpy)2(chrysi)]3+and [Rh(bpy)2(phzi)]3+ may also be exploited for diagnostic mismatch detection. Of course, the detection of mismatches in (labeled) oligonucleotides and synthetic plasmids does not hold particular diagnostic utility. Rather, the ideal system would allow for the quantification of the number of cleavage events (and thus mismatches) in the DNA from a particular cell sample or biopsy, thus indicating whether the tissue in question is MMR-deficient. [Rh(bpy)2(phzi)]2+, for example, has been used in conjunction with alkaline agarose electrophoresis to illustrate differences in site-specific cleavage frequencies in the DNA from MMR-proficient and -deficient cell lines. Further development of such a cleavage-based, whole-genome mismatch detection methodology using fluorescence is currently underway.
Mismatch-specific metalloinsertors have also been applied to the discovery of single nucleotide polymorphisms (SNPs).72 SNPs are single base mutations that constitute the largest source of genetic variation in humans and can lead to variations in disposition to disease or response to pharmaceuticals.57,73 While other methodologies for SNP discovery exist, detection remains expensive and false positive rates high.74 In this application, a region of the genome suspected to contain an SNP is amplified via PCR, denatured, and then reannealed in the presence of a pooled sample. If the region of interest had indeed contained an SNP, the re-annealing process statistically generates a mismatch at the polymorphic site. The resultant mismatch-containing duplexes are then selectively cleaved via irradiation in the presence of [Rh(bpy)2(chrysi)] 3+ or [Rh(bpy)2(phzi)]3+, fluorescently end-labeled, and analyzed with capillary gel electrophoresis (Fig. 12). This new methodology allows for the rapid identification of SNP sites with single-base resolution. The methodology is further made useful by its sensitivity, for it allows for the detection of SNPs with allele frequencies as low as 5%.
Fig. 12.

Single nucleotide polymorphism detection using mismatch-directed photocleavage.
Therapeutic Applications
The application of mismatch-specific metalloinsertors as the basis for designing new chemotherapeutics has also been of interest, especially considering that MMR-deficiency not only increases the likelihood of cancerous transformations but also decreases the efficacy of many common chemotherapeutic agents.75 Recently, it was discovered that both [Rh(bpy)2(chrysi)]3+ and [Rh(bpy)2(phzi)]3+ selectively inhibit cellular proliferation in MMR-deficient cells when compared to cells that are MMR-proficient. Few small molecules have shown a similar cell-selective effect. Interestingly, enantiomeric differences are also observed associated with this inhibition.76 While the mismatch-bindingΔ-enantiomer of [Rh(bpy)2(chrysi)]3+ shows a high level of differential anti-proliferative effect, no such difference is seen using the non-binding Λ-enantiomer. This observation is important for two reasons. First, the mere presence of an enantiomeric difference strongly suggests that the causative agent is the intact complex, not some unknown degradation product or metabolite. Second, the observation that the DNA-binding [Δ-Rh(bpy)2(phzi)]3+ and [Δ-Rh(bpy)2(chrysi)]3+ are the active enantiomers suggests that DNA mismatch binding plays at least some role in the anti-proliferative effect of these complexes. Furthermore, the anti-proliferative response is enhanced by irradiation, hinting that rhodium-mediated DNA photocleavage may also be involved. Considering these complexes bind DNA only non-covalently, the presence of any cytotoxic effect, especially without irradiation, was surprise enough. Currently, work is underway to understand the mechanism of cytotoxicity more fully and to maximize the differential effect of these complexes. The results observed, however, suggest a completely new MMR-deficient, cell-selective strategy for chemotherapeutic design.
Several bifunctional, mismatch-specific conjugates have also been developed with a potential for chemotherapeutic application. In each, the rhodium moieties serve as the targeting vectors, delivering a cytotoxic cargo to mismatched DNA or, more generally, cells containing mismatched DNA, thereby tuning the reactivity of otherwise non-specific agents. Unlike [Rh(bpy)2(chrysi)]3+ or [Rh(bpy)2(phzi)]3+, these conjugates are trisheteroleptic, employing a tether-modified bipyridine to establish the link between the two moieties. For example, in one conjugate the metalloinsertor is linked to an aniline mustard known to form covalent adducts at 5′-GXC-3′ sites (Fig. 13a).77 PAGE experiments with radiolabeled oligonucleotides confirmed that the rhodium moiety successfully confers mismatch-selectivity on the alkylating agent. The two moieties neither abrogate nor attenuate function. Significantly, independent of any chemotherapeutic application, this conjugate may also prove useful due to its ability to “mark” mismatch sites covalently.
Fig. 13.

Mismatch-specific conjugates for therapeutic applications.
Another bifunctional conjugate was created by linking the rhodium moiety to an analogue of the well-known anticancer drug cisplatin, a metal complex that coordinates to single- and double-guanine sites in DNA and subsequently inhibits both transcription and replication (Fig. 13b).78 Like its alkylator cousin, this conjugate succeeds in tuning the reactivity of the platinum subunit; upon binding a mismatched site, the platinum moiety then forms a covalent adduct with a nearby site. It is clear that it is the mismatch-selective Rh complex dictates binding; the Pt moiety is seen to form interstrand as well as intrastrand crosslinks to the DNA, even though without linkage to the Rh center, cisplatin substantially prefers forming intrastrand crosslinks. Clearly, it is hoped that imparting mismatch-selectivity on such a potent anti-cancer drug may lead to a therapeutic agent against MMR-deficient cell lines.
Most recently, a third conjugate has sought to create a mismatch-specific DNA cleavage agent by tethering a [Cu(phen)2]2+ analogue to a selective metalloinsertor.79 Preliminary data suggest that this conjugate, like the others, successfully directs the reactivity of the copper oxidant. Upon the addition of a stoichiometric reductant to convert Cu(II) to the active Cu(I), light-independent DNA backbone cleavage is observed near the mismatch site at concentrations for which no cleavage is seen with untethered [Cu(phen)2]2+ alone. Irrespective of potential chemotherapeutic applications, a mismatch-directed DNA-cleaving conjugate could prove very useful, for it eliminates the need for a light source when cleaving mismatched sites.
The antiproliferative effects of all three of these conjugates are currently being investigated, and the design and synthesis of other reactive conjugates are being explored. Building upon the mismatch-selective binding through bifunctional conjugates certainly offers new tools to probe MMR deficiencies in biological contexts.
Cellular Uptake
Whether for diagnostic or therapeutic applications, establishing the rapid and efficient cellular uptake of metal complexes is of fundamental importance. Cellular (and nuclear) delivery was first achieved through the conjugation of a D-octaarginine cell-penetrating peptide to the mismatch-binding rhodium complex (Fig. 14).80 The pendant peptide does not impair the ability of the rhodium moiety to bind and cleave mismatched sites; however, it increases the non-specific binding by the complex, an effect easily attributed to the strongly cationic character of the peptide. Confocal microscopy images of a similar trifunctional conjugate (this time containing a fluorophore in addition to rhodium and peptide) provide visual evidence for the rapid uptake of the conjugate into the nuclei of HeLa cells.
Fig. 14.

A mismatch-specific conjugate for nuclear uptake.
Despite the success of the peptide conjugate, it is becoming increasingly apparent that the cellular uptake properties of these metal complexes can be altered more simply by exploiting the modularity of their ancillary ligands.81 Using [Ru(L)2(dppz)]2+ as a scaffold, it has been shown that increasing the lipophilicity of the ancillary ligands of the complex can dramatically increase their uptake by HeLa cells. For example, data from both fluorescent cell sorting experiments and confocal microscopy confirm that [Ru(phen)2(dppz)]2+ is more readily taken up than [Ru(bpy)2(dppz)]2+, while the extremely lipophilic [Ru(DIP)2(dppz)]2+ is taken up far better than the preceding two (Fig. 15). Currently, work is in progress on both the expansion of the library of compounds tested and the elucidation of the cellular uptake mechanism. Flow cytometry experiments have also been recently carried out using dibenzo-dppz complexes of Ru(II) as a probe of cell viability.82 In general systematic variation of the ligands on these Ru complexes offers a means to learn the characteristics of the metal complex that are essential to facilitate uptake. Furthermore, the lessons learned here by varying the ancillary ligands of the complex may be exploited directly to increase the absolute and differential anti-proliferative effects of [Rh(bpy)2(chrysi)]3+ and [Rh(bpy)2(phzi)]3+.
Fig. 15.
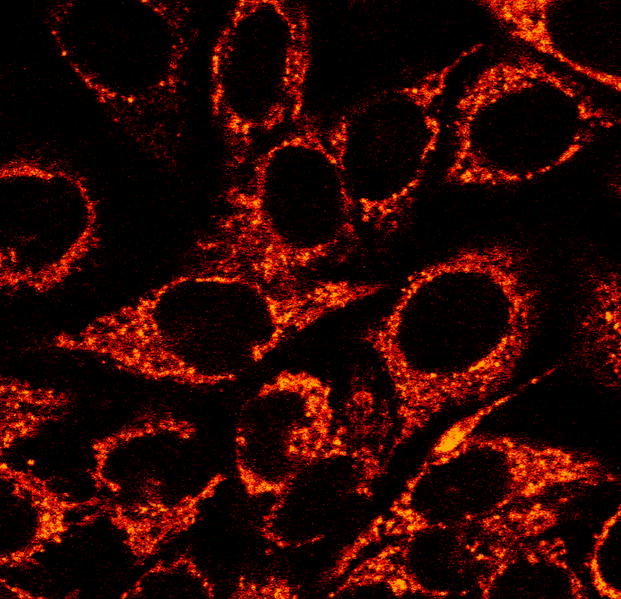
Confocal microscopy of HeLa cells incubated with Ru(DIP)2dppz2+.
Outlook
One clear conclusion from the work described here is the explosive growth and advancement of the field over the years, from Lerman’s initial suggestion of non-covalent binding modes, to the first platinum metallointercalator, then to expansion into three dimensions with octahedral complexes of ever increasing complexity and specificity, and finally, the design of bifunctional mismatch-specific conjugates. Yet surely, much remains to be done. From a design and synthesis standpoint, myriad possibilities exist, including the exploitation of different metals for their unique characteristics, the recognition of more complex and varied sites, and the expansion of the nascent metalloinsertor family. However, the intersection of this field with biology holds the greatest potential for growth. Despite some significant strides, the employment of these complexes in biological systems as probes, diagnostics, or therapeutics, represents a largely untapped area of potentially tremendous value. Doubtless, this topic, along with many others in the field, will be investigated thoroughly and creatively in years to come.
Acknowledgments
The authors would like to thank laboratory members past and present, along with many collaborators, for their efforts. We also thank the National Institutes of Health for their financial support (GM33309).
Notes and references
- 1.Demeunynck MC, Bailly WD. Small Molecule DNA and RNA Binders: From Synthesis to Nucleic Acid Complexes. Weinheim: Wiley-VCH; 2002. [Google Scholar]
- 2.Lippard SJ, Berg JM. Principles of Bioinorganic Chemistry. Mill Valley: University Science Books; 1994. [Google Scholar]; Bertini I, Gray HB, Lippard SJ. Bioinorganic Chemistry. Mill Valley: University Science Books; 1994. [Google Scholar]
- 3.Erkilla KE, Odom DT, Barton JK. Chem Rev. 1999;99:2777. doi: 10.1021/cr9804341. [DOI] [PubMed] [Google Scholar]
- 4.Jamieson ER, Lippard SJ. Chem Rev. 1999;99:2467. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]
- 5.Watson JD, Crick FH. Nature. 1953;171:737. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]; Saegner W. Principles of Nucleic Acid Structure. New York: Springer-Verlag; 1984. [Google Scholar]
- 6.Izatt RM, Christensen JJ, Rytting JW. Chem Rev. 1971;71:439. doi: 10.1021/cr60273a002. [DOI] [PubMed] [Google Scholar]
- 7.Eichhorn GA, Shin YA. J Am Chem Soc. 1968;90:7323. doi: 10.1021/ja01028a024. [DOI] [PubMed] [Google Scholar]
- 8.Beer M, Moudrianakis EN. J Am Chem Soc. 1962;48:409. [Google Scholar]
- 9.Kan LS, Li NC. J Am Chem Soc. 1970;92:4823. doi: 10.1021/ja00719a010. [DOI] [PubMed] [Google Scholar]; Heitner HI, Lippard SJ, Vasiliades GA, Bauer WR. Proc Nat Acad Sci USA. 1972;94:8936. [Google Scholar]
- 10.Brandt WW, Dwyer FP, Gyarfas EC. Chem Rev. 1954;54:959. [Google Scholar]; Dwyer FP, Gyarfas EC, Rogers WP, Koch JH. Nature. 1952;170:190. doi: 10.1038/170190a0. [DOI] [PubMed] [Google Scholar]
- 11.Jennette KW, Lippard SJ, Vasiliades GA, Lippard SJ. Proc Nat Acad Sci USA. 1974;71:3839. doi: 10.1073/pnas.71.10.3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bond PJ, Langridge R, Jennette KW, Lippard SJ. Proc Nat Acad Sci USA. 1975;72:4825. doi: 10.1073/pnas.72.12.4825. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lippard SJ, Bond PJ, Wu KC, Bauer WR. Science. 1976;194:726. doi: 10.1126/science.982037. [DOI] [PubMed] [Google Scholar]
- 13.Howe-Grant M, Lippard SJ. Biochemistry. 1979;18:5762. doi: 10.1021/bi00593a003. [DOI] [PubMed] [Google Scholar]
- 14.Sigman DS, Landgraf R, Perrin DM, Pearson L. Metal Ions in Biological Systems. 1996;33:485. [PubMed] [Google Scholar]
- 15.Sigman DS, Graham DR, D’Aurora V, Stern AM. J Biol Chem. 1979;254:12279. [PubMed] [Google Scholar]
- 16.Goyne TE, Sigman DS. J Am Chem Soc. 1987;109:2848. [Google Scholar]; Sigman DS. Acc Chem Res. 1986;19:180. [Google Scholar]; Meijler M, Zelenko O, Sigman DS. J Am Chem Soc. 1997;119:1135. [Google Scholar]; Thederahn TB, Juwabara MD, Larsen TA, Sigman DS. J Am Chem Soc. 1989;111:4941. [Google Scholar]; Zelenko O, Gallagher J, Xu Y, Sigman DS. 1998;37:2198. doi: 10.1021/ic971154r. [DOI] [PubMed] [Google Scholar]
- 17.Pope L, Sigman DS. Proc Nat Acad Sci USA. 1984;81:3. doi: 10.1073/pnas.81.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]; Spassky A, Sigman DS. Biochemistry. 1985;24:8050. doi: 10.1021/bi00348a032. [DOI] [PubMed] [Google Scholar]; Murakawa GJ, Chen CHB, Kuwabara MD, Nierlich D, Sigman DS. Nucleic Acids Res. 1989;17:5361. doi: 10.1093/nar/17.13.5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Umezawa H, Maeda K, Takeuchi T, Okami Y. J Antibiotiocs (Tokyo) 1966;19:200. [PubMed] [Google Scholar]; Chen J, Stubbe J. Nat Rev Cancer. 2005;5:102. doi: 10.1038/nrc1547. [DOI] [PubMed] [Google Scholar]
- 19.Stubbe J, Kozarich JW. Chem Rev. 1987;87:1107. [Google Scholar]; Absalon MJ, Kozarich JW, Stubbe J. Biochemistry. 1995;34:2065. doi: 10.1021/bi00006a029. [DOI] [PubMed] [Google Scholar]; Absalon MJ, Wu W, Kozarich JW, Stubbe J. Biochemistry. 1995;34:2076. doi: 10.1021/bi00006a030. [DOI] [PubMed] [Google Scholar]; D’Andrea AD, Haseltine WA. Proc Nat Acad Sci U S A. 1978;75:3608. doi: 10.1073/pnas.75.8.3608. [DOI] [PMC free article] [PubMed] [Google Scholar]; Decker A, Chow MS, Kemsley JN, Lehnert N, Solomon EI. J Am Chem Soc U S A. 2006;128:4719. doi: 10.1021/ja057378n. [DOI] [PubMed] [Google Scholar]
- 20.Stubbe J, Kozarich JW, Wu W, Vanderwall DE. Acc of Chem Res. 1996;29:322. [Google Scholar]; Boger DL, Cai H. Agnew Chem Int Ed. 1999;38:448. doi: 10.1002/(SICI)1521-3773(19990215)38:4<448::AID-ANIE448>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]; Burger RM. Chem Rev. 1998;98:1153. doi: 10.1021/cr960438a. [DOI] [PubMed] [Google Scholar]
- 21.Wu W, Vanderwall DE, Teramoto S, Lui SM, Hoehn ST, Tang XJ, Turner CJ, Boger DL, Kozarich JW, Stubbe J. J Am Chem Soc U S A. 1998;120:2239. [Google Scholar]; Wu W, Vanderwall DE, Lui DM, Tang XJ, Turner CJ, Kozarich JW, Stubbe J. J Am Chem Soc U S A. 1996;118:1268. [Google Scholar]; Wu W, Vanderwall DE, Stubbe J, Kozarich JW, Turner CJ. J Am Chem Soc. 1994;116:10843. [Google Scholar]
- 22.Keck MV, Manderville RA, Hecht SM. J Am Chem Soc U S A. 2001;123:8690. doi: 10.1021/ja003795i. [DOI] [PubMed] [Google Scholar]; Manderville RA, Ellena JF, Hecht SM. J Am Chem Soc U S A. 1995;117:7891. [Google Scholar]; Manderville RA, Ellena JF, Hecht SM. J Am Chem Soc U S A. 1994;116:10851. [Google Scholar]
- 23.Barton JK, Dannenberg JJ, Raphael AL. J Am Chem Soc. 1982;104:4967. [Google Scholar]; Barton JK, Danishefsky AT, Goldberg JM. J Am Chem Soc. 1984;106:2172. [Google Scholar]; Kumar CV, Barton JK, Turro NJ. J Am Chem Soc. 1985;107:5518. [Google Scholar]; Barton JK, Goldberg JM, Kumar CV, Turro NJ. J Am Chem Soc. 1986;108:2081. [Google Scholar]; Barton JK, Raphael AL. Proc Natl Acad Sci USA. 1985;82:6460. doi: 10.1073/pnas.82.19.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]; Rehmann JP, Barton JK. Biochemistry. 1990;29:1701. doi: 10.1021/bi00459a006. [DOI] [PubMed] [Google Scholar]; Rehmann JP, Barton JK. Biochemistry. 1990;29:1710. doi: 10.1021/bi00459a007. [DOI] [PubMed] [Google Scholar]
- 24.Barton JK. Science. 1986;233:727. doi: 10.1126/science.3016894. [DOI] [PubMed] [Google Scholar]
- 25.Barton JK, Basile LA, Danishefsky AT, Alexandrescu A. Proc Nat Acad Sci U S A. 1984;81:1961. doi: 10.1073/pnas.81.7.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barton JK, Raphael AL. Proc Nat Acad Sci U S A. 1985;82:6460. doi: 10.1073/pnas.82.19.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Satyanaraana S, Dabrowiak JC, Chaires JB. Biochemistry. 1992;32:2573. doi: 10.1021/bi00061a015. [DOI] [PubMed] [Google Scholar]; Eriksson M, Leijon M, Hiort C, Norden B, Graslund A. Biochemistry. 1994;33:5031. doi: 10.1021/bi00183a005. [DOI] [PubMed] [Google Scholar]
- 28.Chen CHB, Milne L, Landgraf R, Perrin DM, Sigman DS. ChemBioChem. 2001;2:735. doi: 10.1002/1439-7633(20011001)2:10<735::aid-cbic735>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]; Sigman DS. Acc Chem Res. 1986;19:180. [Google Scholar]; Sigman DS, Bruice TW, Mazumder A, Sutton CL. Acc Chem Res. 1993;26:98. [Google Scholar]
- 29.Fu PKL, Bradley AM, van Loyen D, Durr H, Bossmann SH, Turro C. Inorg Chem. 2002;41:3808. doi: 10.1021/ic020136t. [DOI] [PubMed] [Google Scholar]
- 30.Schoentjes B, Lehn JM. Helv Chim Acta. 1995;78:1. [Google Scholar]
- 31.Hannon MJ. Chem Soc Rev. 2007;36:280. doi: 10.1039/b606046n. [DOI] [PubMed] [Google Scholar]; Childs LJ, Malina J, Rolfsnes BE, Pascu M, Prieto MJ, Broome MJ, Rodger PM, Sletten E, Moreno V, Rodger A, Hannon MJ. Chem Eur J. 2006;12:4919. doi: 10.1002/chem.200600060. [DOI] [PubMed] [Google Scholar]; Oleksi A, Blanco AG, Boer R, Uson I, Aymami J, Rodger A, Hannon MJ, Coll M. Angew Chem, Int Ed. 2006;45:1227. doi: 10.1002/anie.200503822. [DOI] [PubMed] [Google Scholar]; Uerpmann C, Malina J, Pascu M, Clarkson GJ, Moreno V, Rodger A, Grandas A, Hannon MJ. Chem-Eur J. 2005;11:1750. doi: 10.1002/chem.200401054. [DOI] [PubMed] [Google Scholar]
- 32.Friedman AE, Chambron JC, Sauvage JP, Turro NJ, Barton JK. J Am Chem Soc. 1990;112:4960. [Google Scholar]; Hartshorn RM, Barton JK. J Am Chem Soc. 1992;114:5919. [Google Scholar]; Jenkins Y, Friedman AE, Turro NJ, Barton JK. Biochemistry. 1992;31:10809. doi: 10.1021/bi00159a023. [DOI] [PubMed] [Google Scholar]; Tuite E, Lincoln P, Nordén B. J Am Chem Soc. 1997;119:239. [Google Scholar]; Hiort C, Lincoln P, Nordén B. J Am Chem Soc. 1993;115:3448. [Google Scholar]; Lincoln P, Broo A, Nordén B. J Am Chem Soc. 1996;118:2644. [Google Scholar]
- 33.Franklin SJ, Barton JK. Biochemistry. 1998;37:16093. doi: 10.1021/bi981798q. [DOI] [PubMed] [Google Scholar]; Hudson BP, Barton JK. J Am Chem Soc. 1998;120:6877. [Google Scholar]; Dupureur CM, Barton JK. J Am Chem Soc. 1994;116:10286. [Google Scholar]; Collins JG, Shields TP, Barton JK. J Am Chem Soc. 1994;116:9840. [Google Scholar]
- 34.Kielkopf CL, Erkkila KE, Hudson BP, Barton JK, Rees DC. Nat Struc Biol. 2000;7:117. doi: 10.1038/72385. [DOI] [PubMed] [Google Scholar]
- 35.Pierre VC, Kaiser JT, Barton JK. Proc Natl Acad Sci U S A. 2007;104:429. doi: 10.1073/pnas.0610170104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang AHJ, Nathans J, van der Marel G, van Boom JH, Rich A. Nature. 1978;276:471. doi: 10.1038/276471a0. [DOI] [PubMed] [Google Scholar]
- 37.Greguric I, Aldrichwright JR, Collins JG. J Am Chem Soc. 1997;119:3621. [Google Scholar]; Fry JV, Collins JG. Inorg Chem. 1997;36:2919. doi: 10.1021/ic960937u. [DOI] [PubMed] [Google Scholar]; Collins JG, Sleeman AD, Aldrich-Wright JR, Greguric I, Hambley TW. Inorg Chem. 1998;37:3133. [Google Scholar]
- 38.Friedman AE, Chambron J-C, Sauvage J-P, Turro NJ, Barton JK. J Am Chem Soc. 1990;112:4960. [Google Scholar]; Turro C, Bossmann SH, Jenkins Y, Barton JK, Turro NJ. J Am Chem Soc. 1995;117:9026. [Google Scholar]; Olson EJC, Hu D, Hormann A, Jonkman AM, Arkin MR, Stemp EDA, Barton JK, Barbara PF. J Am Chem Soc. 1997;119:11458. [Google Scholar]
- 39.Tuite E, Lincoln P, Nordén B. J Am Chem Soc. 1997;119:239–240. [Google Scholar]; Lincoln P, Broo A, Nordén B. J Am Chem Soc. 1996;118:2644–2653. [Google Scholar]; Greguric A, Greguric ID, Hambley TW, Aldrich-Wright J, Collins JG. Dalton Transactions. 2002:849. [Google Scholar]
- 40.Holmlin RE, Stemp EDA, Barton JK. Inorg Chem. 1998;37:29. doi: 10.1021/ic970869r. [DOI] [PubMed] [Google Scholar]
- 41.Onfelt B, Lincoln P, Nordén B. J Am Chem Soc. 1999;121:10846. doi: 10.1021/ja003624d. [DOI] [PubMed] [Google Scholar]; Onfelt B, Lincoln P, Nordén B. J Am Chem Soc. 2001;123:3630. doi: 10.1021/ja003624d. [DOI] [PubMed] [Google Scholar]
- 42.Elias B, Kirsch-De Mesmaeker A. Coord Chem Rev. 2006;250:1627. [Google Scholar]; Kirsch-De Mesmaeker A, Moucheron C, Boutonnet N. J Phys Org Chem. 1998;11:566. [Google Scholar]; Zou XH, Xiao-Hua JLN. Trends Inorg Chem. 2001;7:99. [Google Scholar]; Xiong Y, Ji LN. Coord Chem Rev. 1999;185–186:711. [Google Scholar]
- 43.Sitlani A, Long EC, Pyle AM, Barton JK. J Am Chem Soc. 1992;114:2303. [Google Scholar]
- 44.Fitzsimons MP, Barton JK. J Am Chem Soc. 1997;119:3379. [Google Scholar]
- 45.Copeland KD, Lueras AMK, Stemp EDA, Barton JK. Biochemistry. 2002;41:12785. doi: 10.1021/bi020407b. [DOI] [PubMed] [Google Scholar]
- 46.Sitlani A, Dupureur CM, Barton JK. J Am Chem Soc. 1993;115:12589. [Google Scholar]; Sitlani A, Barton JK. Biochemistry. 1994;33:12100. doi: 10.1021/bi00206a013. [DOI] [PubMed] [Google Scholar]
- 47.Pyle AM, Long EC, Barton JK. J Am Chem Soc. 1989;111:4520. [Google Scholar]; Pyle AM, Morii T, Barton JK. J Am Chem Soc. 1990;112:9432. [Google Scholar]; Campisi D, Morii T, Barton JK. Biochemistry. 1994;33:4130. doi: 10.1021/bi00180a005. [DOI] [PubMed] [Google Scholar]
- 48.Chow CS, Barton JK. J Am Chem Soc. 1990;112:2839. [Google Scholar]; Jenkins Y, Friedman AE, Turro NJ, Barton JK. Biochemistry. 1992;31:10809. doi: 10.1021/bi00159a023. [DOI] [PubMed] [Google Scholar]; Chow CS, Behlen LS, Uhlenbeck OC, Barton JK. Biochemistry. 1992;31:97. doi: 10.1021/bi00119a005. [DOI] [PubMed] [Google Scholar]; Lim AC, Barton JK. Biochemistry. 1993;32:11029. doi: 10.1021/bi00092a012. [DOI] [PubMed] [Google Scholar]
- 49.Chow CS, Hartmann KM, Rawlins SL, Huber PW, Barton JK. Biochemistry. 1992;31:3534. doi: 10.1021/bi00128a030. [DOI] [PubMed] [Google Scholar]
- 50.Krotz AH, Kuo LY, Shields TP, Barton JK. J Am Chem Soc. 1993;115:3877. [Google Scholar]; Krotz AH, Kuo LY, Barton JK. Inorg Chem. 1993;32:5963. [Google Scholar]; Shields TP, Barton JK. Biochemistry. 1995;34:15037. doi: 10.1021/bi00046a009. [DOI] [PubMed] [Google Scholar]; Shields TP, Barton JK. Biochemistry. 1995;34:15049. doi: 10.1021/bi00046a010. [DOI] [PubMed] [Google Scholar]
- 51.Krotz AH, Hudson BP, Barton JK. J Am Chem Soc. 1993;115:12577. [Google Scholar]
- 52.Terbrueggen RH, Barton JK. Biochemistry. 1995;34:8227. doi: 10.1021/bi00026a003. [DOI] [PubMed] [Google Scholar]; Terbrueggen RH, Johann TW, Barton JK. Inorg Chem. 1998;37:6874. doi: 10.1021/ic980837j. [DOI] [PubMed] [Google Scholar]
- 53.Odom DT, Parker CS, Barton JK. Biochemistry. 1999;38:5155. doi: 10.1021/bi9827969. [DOI] [PubMed] [Google Scholar]
- 54.Fu PKL, Turro C. Chem Comm. 2001;3:279. [Google Scholar]; Fu PKL, Bradley PM, Turro C. Inorg Chem. 2003;42:878. doi: 10.1021/ic020338p. [DOI] [PubMed] [Google Scholar]
- 55.Lerman LS. J Mol Biol. 1961;3:18. doi: 10.1016/s0022-2836(61)80004-1. [DOI] [PubMed] [Google Scholar]
- 56.Modrich PL. Annu Rev Genet. 1991;25:228. doi: 10.1146/annurev.ge.25.120191.001305. [DOI] [PubMed] [Google Scholar]; Kolodner R. Genes Dev. 1996;10:1433. doi: 10.1101/gad.10.12.1433. [DOI] [PubMed] [Google Scholar]
- 57.Sylvanen A. Nat Rev Genet. 2001;2:930–932. doi: 10.1038/35103535. [DOI] [PubMed] [Google Scholar]
- 58.Iyer RR, Pluciennik A, Burdett V, Modrich PL. Chem Rev. 2006;106:302. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]; Modrich PL. J Biol Chem. 2006;281:30305. doi: 10.1074/jbc.R600022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kolodner R. Trends Biochem Sci. 1995;20:297. doi: 10.1016/s0968-0004(00)89087-8. [DOI] [PubMed] [Google Scholar]; Arizamoglou II, Gilbert F, Barber HR. Cancer. 1998;82:1808. doi: 10.1002/(sici)1097-0142(19980515)82:10<1808::aid-cncr2>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 60.Murner H, Jackson BA, Barton JK. Inorg Chem. 1998;37:3007. [Google Scholar]; Zeglis BM, Barton JK. Nat Prot. 2007;2:357. doi: 10.1038/nprot.2007.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jackson BA, Barton JK. J Am Chem Soc. 1997;119:12986. [Google Scholar]
- 62.Jackson BA, Alekseyev VY, Barton JK. Biochemistry. 1999;38:4655. doi: 10.1021/bi990255t. [DOI] [PubMed] [Google Scholar]
- 63.Jackson BA, Barton JK. Biochemistry. 2000;39:6176. doi: 10.1021/bi9927033. [DOI] [PubMed] [Google Scholar]
- 64.Junicke H, Hart JR, Kisko J, Glebov O, Kirsch IR, Barton JK. Proc Nat Acad Sci U S A. 2003;100:3737. doi: 10.1073/pnas.0537194100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hudson BP, Dupureur CM, Barton JK. J Am Chem Soc. 1995;117:9379. [Google Scholar]
- 66.Pierre VC, Barton JK. unpublished results. [Google Scholar]
- 67.Compared to the 10 Å of B-DNA
- 68.Cordier C, Pierre VC, Barton JK. submitted. [Google Scholar]
- 69.Brunner J, Barton JK. J Am Chem Soc. 2006;128:6772. doi: 10.1021/ja0612753. [DOI] [PubMed] [Google Scholar]
- 70.Ruba E, Hart JR, Barton JK. Inorg Chem. 2004;43:4570. doi: 10.1021/ic0499291. [DOI] [PubMed] [Google Scholar]
- 71.Zeglis BM, Barton JK. J Am Chem Soc. 2006;128:5654. doi: 10.1021/ja061409c. [DOI] [PubMed] [Google Scholar]
- 72.Hart JR, Johnson MD, Barton JK. Proc Nat Acad Sci U S A. 2004;101:14040. doi: 10.1073/pnas.0406169101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shastry BS. J Hum Genet. 2002;47:561. doi: 10.1007/s100380200086. [DOI] [PubMed] [Google Scholar]
- 74.Rider MJ, Taylor SL, Tobe VO, Nickerson DA. Nucleic Acids Res. 1998;26:967. doi: 10.1093/nar/26.4.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fink D, Aebi S, Howell SD. Clin Cancer Res. 1998;4:1. [PubMed] [Google Scholar]
- 76.Hart JR, Glebov O, Ernst RJ, Kirsch IR, Barton JK. Proc Nat Acad Sci U S A. 2006;103:15359. doi: 10.1073/pnas.0607576103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schatzschneider U, Barton JK. J Am Chem Soc. 2004;126:8630. doi: 10.1021/ja048543m. [DOI] [PubMed] [Google Scholar]
- 78.Petitjean A, Barton JK. J Am Chem Soc. 2004;126:14728. doi: 10.1021/ja047235l. [DOI] [PubMed] [Google Scholar]
- 79.Lim MH, Barton JK. unpublished results. [Google Scholar]
- 80.Brunner J, Barton JK. Biochemistry. 2006;45:12295. doi: 10.1021/bi061198o. [DOI] [PubMed] [Google Scholar]
- 81.Puckett CA, Barton JK. J Am Chem Soc. 2007;129:46. doi: 10.1021/ja0677564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jimenez-Hernandez ME, Orellana G, Montero F, Portoles MT. Photochem Photobiol. 2000;72:28. doi: 10.1562/0031-8655(2000)072<0028:arpfcv>2.0.co;2. [DOI] [PubMed] [Google Scholar]