

Abstract
It is well established that certain stress proteins or molecular chaperones are highly efficient in cross-presenting tumor-derived antigens, resulting in a potent antitumor immune response. In this study we demonstrate that genetic modification of weakly immunogenic murine prostate tumor cells (TRAMP-C2) by stable transfection with a secretable form of endoplasmic reticulum resident chaperone grp170 significantly enhances its immunogenicity in vivo. Generation of systemic antitumor immunity is indicated by the growth suppression of distant parental tumors, which is associated with increased tumor infiltration, elevated effector functions of CD8+ T-cells. Immunization with inactivated grp170-secreting C2 cells augments a CD8+ T-cell dependent, tumor-protective effect. Furthermore, infection of C2 tumor cells with a nonreplicating adenoviral vectors encoding secretable grp170 promotes tumor immunogenicity more effectively than plasmid transduction, as shown by the increased production of pro-inflammatory cytokine TNF-α by dendritice cells and enhanced therapeutic efficacy in treating pre-established tumors. Given a repertoire of undefined antigens in prostate tumor, manipulation of cellular compartmentalization of immuno-stimulatory chaperone grp170 to elicit systemic tumor immunity may be used to improve treatment outcomes for prostate cancer when combined with other treatment modalities.
Keywords: Prostate cancer, Stress protein, Chaperone, grp170, Immuogenicity
Introduction
Prostate cancer (CaP) is the most common noncutaneous malignancy among men in the US [1]. Although greater public awareness of CaP, early diagnosis using PSA screening and improvements in the treatment of localized disease have led to a modest decline in the mortality rate of patients with this disease, many men suffer recurrence of CaP after radical prostatectomy. There is no curative treatment for patients who develop recurrences or those who have metastatic disease at the time of diagnosis. Androgen deprivation therapy (ADT) is used to palliate locally advanced or metastatic CaP. However, virtually all patients eventually progress to hormone-refractory disease after a short duration of response. Given the profound medical impact of CaP and lack of adequate therapies for locally advanced or metastatic disease, there is an urgent need to develop new modalities of treatment that can extend the chance of cure at initial treatment.
Due to the ubiquitous presence of the immune system, immunotherapy offers one of the most promising strategies for cancer treatment, possessing the potential of fighting both the primary tumor and distant metastases. Despite cancer cells harboring a repertoire of tumor-specific antigens and shared self-antigens, these tumor cells, including CaP, generally are incapable of provoking an effective immune response [2]. Genetic modification of tumor cells with genes for co-stimulatory molecules or cytokines (e.g., GM-CSF) has been reported to improve the tumor immunogenicity [3]. Monotherapy using GM-CSF-transduced prostate cancer cells (i.e., GVAX) has shown moderate clinical efficacy [4, 5].
Stress proteins are one of most abundant intracellular proteins and function as molecular chaperones in a variety of cellular functions [6]. An extracellular localization of stress proteins due to release of cell content under pathological conditions or active secretion in response to cellular stress has been documented [7, 8]. Increasing evidence indicates that stress proteins play important roles in eliciting innate and adaptive immunity. The extracellular stress proteins, including grp170, are thought to mediate immune activation in that they deliver and facilitate their chaperoned protein or peptide antigens to antigen-presenting cells (APCs) for cross-presentation [9, 10] and concomitantly act as a ‘danger’ signal triggering the activation of APC and production of proinflammatory cytokines [11–14]. These unique properties make stress proteins attractive vehicles for the delivery of antigens into antigen-presenting pathways. Our earlier studies have shown that tumor-derived endoplasmic reticulum (ER) resident chaperone grp170 or grp170 complexed with tumor-associated antigens are highly effective in eliciting antigen and tumor specific antitumor immune responses [10, 15].
Using a poorly immunogenic murine prostate cancer model, in this study we examine the role of grp170 as an immune-stimulatory agent in modulation of tumor immunogenicity and promotion of a tumor-specific immune response. We show that TRAMP-C2 prostate tumor expressing a secretable form of grp170 is highly immunogenic as indicated by the induction of systemic T-cell response against the unmodified tumor cells and marked inhibition of progression of distant parental tumors. In addition, immunization with irradiated grp170-secreting tumor cells results in an effective control of C2 tumor growth in both prophylactic and therapeutic settings, suggesting the adjuvant-like properties of grp170 may be exploited to improve immunotherapeutic potency of whole-tumor cell vaccine against CaP.
Materials and methods
Mice and cell lines
Eight to 12-week-old male C57BL/6 mice, purchased from the National Institutes of Health animal facilities, were maintained in a pathogen-free facility at Roswell Park Cancer Institute. Animal care and experiments were approved by the Institutional Animal Care and Use Committee. TRAMP-C2 cell line derived from a prostate tumor that arose in a transgenic adenocarcinoma of mouse prostate (TRAMP) mouse [16] was maintained in DMEM containing 10% fetal bovine serum, 2 mM l-glutamine, and 100 U/ml penicillin/streptomycin.
Stable transfection of TRAMP-C2
Cells were transfected with pCMV-sgrp170 described previously [17] using FuGENE 6 (Roche, Indianapolis, IN). To distinguish the secretable grp170 from endogenous grp170, a His-tag was fused to the C-terminus of mouse grp170, in which the ER retention sequence (i.e., KNDEL) has been eliminated. Cells were then trypsinized and selected in media containing 1 mg/ml G418. To characterize tumor cell secreting grp170, culture media were collected and subjected to immunoblotting analysis using antibodies against His-tag [18].
Adenovirus infection and characterization
The adenovirus carrying a secretable form of the grp170 gene (Ad.sgrp170) was constructed using BD Adeno-X™ Adenoviral Expression System (BD Bioscience, Palo Alto, CA) as previously described [19]. Non-replicating adenoviral particles were produced in HEK293 cells. Infection titers were determined by plaque tittering on 293 cells. Viruses were concentrated and purified using AdenoPACK Maxi columns from Sartorius Stedim Biotech (Edgewood, NY) according to the procedure provided by the manufacturer. Cells were infected with different multiplicity of infection (MOI) under standard culture conditions.
Tumor challenge studies
2 × 106 C2-sgrp170 tumor cells suspended in 100 μl sterile PBS were injected into the left dorsal flank of mice. Tumor growth was monitored by measuring perpendicular tumor diameters. To determine the impact of grp170 secretion on distant un-modified tumors, mice were established with C2, C2-neo, or C2-sgrp170 tumor in left flank, and parental C2 tumor in right flank. Growth of contralateral parental tumors was followed to determine systemic antitumor effect.
Immunohistochemical staining
Tumors were fixed in zinc fixative, dehydrated, and embedded in paraffin wax. Sections (5 μm) were prepared using a rotary microtome and placed on polylysinecoated slides. Endogenous peroxidase activity was quenched with 3% H2O2, and sections were blocked with Superblock (Pierce, Rockford, IL). Sections were incubated overnight at 4°C with the primary antibody directed against CD8 at a dilution of 1:100 (Pharmingen, San Diego, CA). Sections were rinsed with PBS, followed by incubation with biotinylated secondary antibody for 45 min at room temperature. The bound antibody was visualized by the peroxidase ABC method using diaminobenzidine (Sigma Chemical Co., St Louis, MO). Slides were counterstained with hematoxylin. Controls were performed by replacing the primary antibody with 10% nonimmune serum or IgG were always negative.
Cell-based vaccination
Mice (5 mice per group) were immunized s.c. with 1 × 106 irradiated tumor cells. Two immunizations at 2-week intervals were carried out. One week later, mice were challenged by inoculation of parental TRAMP-C2 tumor cells. For immunization of mice with cells infected with adenoviral virus encoding secretable grp170, C2 tumor cells were incubated with Ad.sgrp170 or Ad.vector for 24 h at an MOI of 100 or left untreated. Cells were extensively washed and irradiated (1.2 × 104 rad) before injection into mice. For treatment of tumor-bearing mice, animals were inoculated with 1 × 106 C2 cells on day 0, and received plasmid or adenovirus-encoding sgrp170-modified C2 cells on days 3, 8 and 13. Depletion of T-cell subsets or NK cell was accomplished by i.p. injection of mAb as previously described [17]. Effective depletion of cell subsets was maintained by the antibody injections once a week for the duration of experiment.
Enzyme-linked immunosorbent spot (ELISPOT) and CTL assays
Splenocytes were isolated from immunized mice 2 weeks after immunization and stimulated with mitomycin C-treated TRAMP-C2 cells to determine tumor-specific IFN-γ production as previously described [18]. For CTL assay, splenocytes were stimulated with mitomycin C-treated tumor cells in the presence of IL-2 (20 U/ml) for 5 days. CD8 T-cells were isolated by negative selection using immunocolumn kit from CERARLANE (Burlington, NC) and used as effector cells in chromium release assays to determine its killing activity [18]. Cells were routinely to be greater than 90% pure as analyzed by flow cytometry.
Enzyme-linked Immunosorbent assays (ELISA)
Microtiter plates were coated with purified grp170 protein (1 μg/ml) in 50 mM carbonate buffer (pH 9.0) overnight at 4°C covered with parafilm. Plates were then blocked with PBS containing 1% BSA and 0.02% azide, and incubated with serially diluted sera for 1 h at room temperature. After washes with PBS containing 0.05% Tween-20, plates were incubated with alkaline phosphatase conjugated secondary antibody. Plates were developed using 1 mg/ml substrate PNPP (Santa Cruz Biotech, Santa Cruz, CA) in diethanolamine buffer and read on microtiter plate reader at OD 450.
BM-DC and cytokine assay
DCs were generated from mouse bone marrow in the presence of GM-CSF (20 ng/ml) and IL-4 (10 ng/ml). 1 × 106 day-7 DCs were cultured with irradiated tumor cells at a ratio of 1:2 for 24 h. Irradiated tumor cells were cultured in serum-free medium for 24 h prior to use. The supernatants were harvested for measuring TNF-α using ELISA kits (eBioscience, San Diego, CA).
Statistical analysis
Statistical analysis was performed using paired or unpaired Student’s t test. Ps < 0.05 were considered statistically significant.
Results
Scretable grp170 reduces tumorigenicity of TRAMP-C2 prostate tumor cells in vivo
To achieve targeting of grp170 to extracellular environment, the ER retention sequence, i.e., KNDEL, at its COOH-terminus was removed followed by sequential fusion in frame to a His tag, which allows the distinction between secreted and endogenous grp170. The fusion gene of sgrp170-His was under the control of CMV promoter for constitutive and effective gene expression (i.e., pCMV-sgrp170) (Fig. 1a).
Fig. 1.

Expression of secretable grp170 reduces the tumorigenicity of the TRAMP-C2 tumor cell. a Schematic elucidation of a construct encoding a secretable form of grp170 (pCMV-sgrp170). His-tagged, ER retention sequence ‘KNDEL’ deleted grp170 gene was under the control of a constitutively active cytomegalovirus promoter/enhancer (CMV). b Supernatants were collected from parental, pCMV-neo or pCMV-sgrp170 stably transduced C2 cells and analyzed for the expression of sgrp170 using antibodies against His-tag. c Mice (n = 5) were inoculated subcutaneously with 2 × 106 parental C2, C2-neo or C2-sgrp170 tumor cells. Tumor development was followed by measuring the size of tumors. Results are representative of three separate experiments
The TRAMP-C2 cell line established from a spontaneous tumor of the autochthonous TRAMP model was chosen for our studies and it has been shown to be nonimmunogenic or poorly immunogenic in vivo [16]. Stably transfected C2 cells were obtained by G418 selection and confirmed by immunoblotting analysis using His-tag-specific antibodies (Fig. 1b). C2 cells transduced with KNDEL depleted grp170 cDNA (C2-sgrp170) consistently secreted grp170 to the supernatant, whereas parental C2 cells and mock-transduced cells (i.e., C2-neo) did not. The grp170 secretion did not appear to affect the viability and growth kinetics of TRAMP-C2 cells under culture conditions (data not shown). In addition, the levels of MHC I expression in C2 tumor cells were not altered by the grp170 transduction (data not shown).
To determine whether grp170 secretion impacts tumor growth in vivo, syngeneic C57BL6 mice were inoculated with viable C2, C2-neo or C2-sgrp170 tumor cells. It was seen that mice challenged with parental or mock transduced C2 cells developed aggressively growing tumors. However, mice inoculated with grp170-secreting C2 cells developed small-size tumors with a significantly slower growth rate (Fig. 1c).
The reduced tumor growth is associated with an enhanced tumor-specific CTL response
We next determined whether the secretable grp170 facilitates tumor-specific immune responses. Splenocytes were isolated from mice inoculated with parental C2 tumors, mock transduced-C2 tumors, or sgrp170-expressing tumors, and subjected to an ELISPOT assay (Fig. 2a). Compared with those from C2 or C2-neo tumor-bearing mice, a significant elevation in the level of IFN-γ was observed in cells from animals bearing C2-sgrp170 tumors, when stimulated with mitomycin C-treated TRAMP-C2 tumors.
Fig. 2.
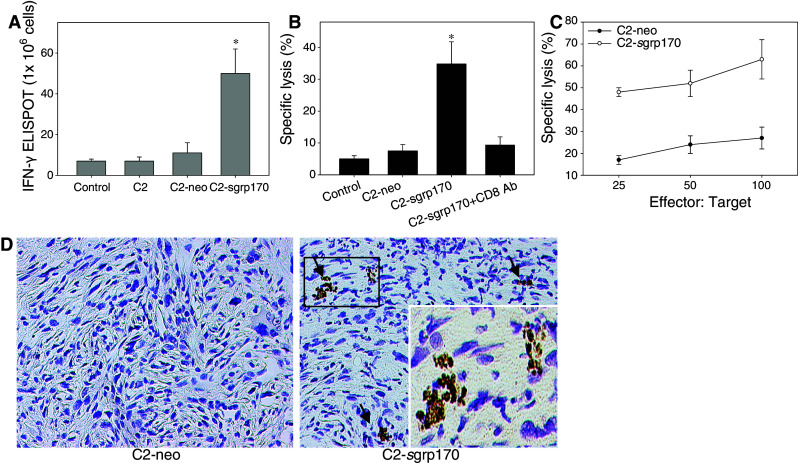
The reduced growth of C2-sgrp170 tumor is associated with an enhanced tumor-specific CTL response. a Extracellular grp170 promotes tumor-specific IFN-γ production. Splenocytes were isolated from mice one week after inoculation of C2, C2-neo or C2-sgrp170 tumor cells. Cells were stimulated with mitomycin C-treated C2 cells at a ratio of 20:1. IFN-γ production was measured using an ELISPOT assay (*P < 0.01, C2.sgrp170 vs. C2.neo). Data (mean ± SD) are representative of two experiments in which three mice of each group were analyzed. b CD8+ T-cells mediates the enhanced cytolytic activity in mice inoculated with C2-sgrp170 cells. Splenocytes were harvested from mice injected with C2, C2-neo, or C2-sgrp170 tumors. Cell were re-stimulated with C2 cells in vitro for 5 days, and analyzed for cytotoxic activities using 51Cr-labeled C2 as targets (E:T, 60:1). In one case, CD8+ cells were removed by 2.43 mAb before the CTL assay was carried out. c CD8+ T-cells were isolated from re-stimulated splenocytes by negative selection, and analyzed for tumor-lytic activities at different E:T ratios. Data are representative of three experiments. d Zinc fixed, paraffin embedded tumor tissues were stained with anti-CD8 antibodies (brown). Representative ×10 images of TRAMP-C2 tumors are shown. Insert in the right panel shows a magnified view of CD8+ cells. No positive staining was detected in IgG controls stained sections
Effector cell function was assessed by chromium release assays using C2 tumor cells as targets. Although unstimulated splenocytes from all mice failed to show significant cytolytic activities against tumor cell targets (data not shown), restimulated cells from C2-sgrp170 tumor-challenged mice displayed significantly increased cytolytic activities than those from naïve mice or C2-neo tumor-challenged mice (Fig. 2b). Elimination of CD8+ cells from splenocytes disrupted the observed killing effect, suggesting that CD8+ T-cells might mediate cytolytic activities (Fig. 2b). To confirm the contribution of CD8+ T-cell, CD8+ cells were isolated from re-stimulated splenocytes by negative selection. At different effector-target (E:T) ratios, CD8+ T-cells from C2-sgrp170 cell inoculated mice were more efficient in lysing target cells than those from C2-neo tumor-bearing mice (Fig. 2c). However, no cytotoxic activity was detectable when syngeneic B16 melanoma cells were used as targets, indicating the specificity of the tumor killing (data not shown).
Tumor tissues were examined 3 weeks after inoculation of tumor cells by immunohistochemistry staining for immune infiltration. A significantly increased infiltration of CD8+ cells was seen in C2-sgrp170 tumors compared to C2-neo tumors, in which CD8 cells were not detectable (Fig. 2d). There was minimal infiltration of CD4 cells in both tumors. However, no statistic difference was reached (data not shown). NK cell staining was negative in both C2-neo and C2-sgrp170 tumors (data not shown).
The increased immunogenicity of C2-sgrp170 cells leads to growth control of distant C2 tumors
We next tested whether the tumor-specific immune response associated with the enhanced immunogenicity of grp170-secreting C2 tumor results in a systemic antitumor effect. The parental C2, C2-neo, or C2-sgrp170 tumors were inoculated into the left flank of mice, while parental TRAMP-C2 tumors were inoculated simultaneously into the right flank (Fig. 3a). A significant inhibition in tumor growth on the contralateral side was shown in mice inoculated with C2-sgrp170 cells. However, progression of contralateral tumors was essentially unaffected in mice bearing C2 or C2-neo tumors.
Fig. 3.

Secretable grp170 promotes a systemic antitumor response. Mice (n = 5) were inoculated with parental C2, C2-neo or C2-sgrp170 tumor cells in the left flank. At the same time, 2 × 106 parental C2 tumor cells were injected on contralateral side (i.e., right flank). Tumor progression was followed by measuring perpendicular tumor diameters (*P < 0.01, C2.sgrp170 vs. C2.neo). Representative results of three separate experiments are shown. b Inoculation of grp170-secreting C2 cells does not induce a humoral response against grp170. Sera were collected from tumor-bearing mice, and subjected to ELISA assays to measure grp170-specific IgG. Grp170-specific IgG was used as a positive control. Results represent two independent experiments
To determine whether grp170-secreting C2 cells elicit a grp170-specific humoral response, sera were collected from mice bearing C2-neo, or C2-sgrp170 tumors. No significant increase in the levels of IgG specific for grp170 was detected following inoculation of C2-sgrp170 cells (Fig. 3b).
Immunization with inactivated C2-sgrp170 cells elicits a CD8+ T cell-dependent antitumor response
To evaluate the potential of using grp170-modified tumor cell as a cell-based cancer vaccine, we immunized mice with irradiated C2, C2-neo or C2-sgrp170 cells, followed by tumor challenge with parental C2 cells. Mice immunized with C2-sgrp170 cells effectively delayed tumor growth upon tumor challenge, whereas immunization with parental C2 or C2-neo cells provided no significant protection (Fig. 4a).
Fig. 4.

Immunization with C2-sgrp170 cells generates CD8+ T-cell dependent antitumor immunity. a C2-sgrp170 cell-based vaccination inhibits tumor growth. C57BL/6 mice (n = 5) were immunized twice with 2 × 106 irradiated C2 cells, C2-neo cells or C2-sgrp170 cells. Two weeks after the second immunization, mice were challenged with 2 × 106 parental TRAMP-C2 tumor cells (*P < 0.01, IR.C2-sgrp170 vs. IR.C2-neo). Data are representative of two independent experiments. b CD8+ T-cells are required for the C2-sgrp170 cell vaccination-mediated antitumor activity. Prior to tumor challenge, immunized mice (n = 8) were depleted of CD4+, CD8+ T-cells by i.p. injection of GK1.5, 2.43 mAb, respectively. Isotype-matched antibodies were used as controls. The results shown are from a representative two experiments (*P < 0.01, CD8 depletion vs. IgG control; **P > 0.05, CD4 depletion vs. IgG control). c NK cells are not involved in the C2-sgrp170 cell-augmented antitumor response. CD8+ T-cells or NK cells were depleted in immunized mice (n = 5) by i.p. injection of 2.43 and PK136 mAbs, respectively, before tumor challenge (*P < 0.01, CD8 depletion vs. IgG control; **P > 0.05, NK depletion vs. IgG control). Data are representative of two independent experiments
To examine the immune effector cells involved in the tumor-inhibitory activities, antibody depletion of T-cell subsets was performed during the challenge phase. Mice were immunized with irradiated C2-sgrp170 cells, followed by depletion of CD4+ or CD8+ T-cells (Fig. 4b). Upon challenging with parental C2 tumor cells, it was seen that depletion of CD8+ T cell abrogated the tumor preventive effect. However, elimination of CD4+ T cell did not affect the vaccine activity in immunized animals. To determine the contribution of NK cell to the tumor protection, NK cells were depleted by i.p. injection of immunized mice with PK136 antibodies. While CD8 depletion effectively abolished C2-sgrp170 cell-mediated antitumor effect, C2-sgrp170 cell generated antitumor immunity in NK-cell depleted mice remained largely intact (Fig. 4b).
C2 tumor cells infected with sgrp170-encoding adenovirus augment a potent antitumor response
We next evaluated whether antitumor efficacy can be achieved using sgrp170-expressing adenovirus (i.e., Ad.sgrp170) infected TRAMP-C2 cells rather than stably transduced grp170-secreting cells. Following infection of C2 tumor cells with Ad.sgrp170 at different MOIs, expression of the secretable grp170 gene was examined in the supernatants of the infected cells by immunoblotting using anti-His-tag antibodies. An increase in the level of secreted grp170 was seen in culture media derived from cells infected with Ad.sgrp170 at a higher MOI (Fig. 5a). In addition, the infection of C2 cells with Ad.sgrp170 at a MOI of up to 500 had no observable cytotoxic effect on C2 cells in vitro (data not shown).
Fig. 5.

TRAMP-C2 tumor cells modified with adenovirus encoding secretable grp170 exhibits therapeutic efficacy in treating established tumors. a Active secretion of grp170 following non-replicating Ad.sgrp170 infection. C2 cells were incubated with or without Ad.sgrp170 at different MOIs. Supernatants were collected and analyzed for the presence of grp170 using antibodies against His-tag. b C2 cells were infected for 24 h with or without Ad.vector, Ad.sgrp170 at a MOI of 300 or left untreated. Cells were extensively washed, irradiated and injected s.c. into mice (n = 8). One week after the second immunization, mice were challenged with 2 × 106 C2 tumor cells (*P < 0.01, Ad.sgrp170 vs. Ad.vector or untreated control). c Ad.sgrp170 modified C2 cells are more effective than sgrp170 plasmid transduced cells in inhibiting established tumors. 3 days after inoculation of C2 tumor cells, mice (n = 8) received treatment s.c. on days 3, 8 and 13 with irradiated C2-sgrp170, Ad.vector or Ad.sgrp170 infected C2 cells (*P < 0.02, Ad.sgrp170 vs. C2-sgrp170; **P < 0.01, C2-sgrp170 vs. untreated control). d Adenovirus modified C2 cells are more efficient than plasmid transduced cells in stimulating DCs to produce pro-inflammatory cytokine. BM-DCs were co-cultured with irradiated C2-neo, C2-sgrp170 or Ad.sgrp170-modified C2 cells for 24 h. Levels of TNF-α in the supernatants were measured by ELISA (*P < 0.01, C2-sgrp170 vs. untreated control; **P < 0.01, Ad.sgrp170 vs. C2-sgrp170). Data are representative of two separate experiments
Mice were immunized with irradiated C2 cells that had been infected with Ad.vector, Ad.sgrp170 or left untreated, followed by tumor challenge with parental C2 tumor cells (Fig. 5b). Mice receiving C2 cells, or C2 cells infected with control empty adenovirus developed tumors at a growth rate that was comparable to that of tumors in untreated animals after tumor challenge. However, mice vaccinated with Ad.sgrp170 infected C2 cells were protected against subsequently inoculated C2 tumors.
We also compared immunogenicity and therapeutic efficacy of C2 cells genetically modified with secretable grp170-encoding plasmid and adenovirus. Mice were established with C2 tumors 3 days before the treatment. Mice received irradiated C2-sgrp170 cells, Ad.vector or Ad.sgrp170 infected C2 cells on days 3, 8 and 13. Treatment with C2-sgrp170 cells resulted in a modest tumor growth delay in this therapeutic setting, whereas treatment with C2 cells infected with Ad.sgrp170 led to highly effective tumor suppression (Fig. 5c).
To examine whether dendritic cell, a key player in immune initiation, was more responsive to C2 cells modified with adenovirus compared to plasmid construct-transduced cells, BM-DCs were incubated with irradiated C2-neo cells, C2-sgrp170 cells, C2 cells infected with Ad.vector or Ad.sgrp170. No TNF-α was detectable in the culture of C2-sgrp170 cells alone or cells infected with Ad.sgrp170 (data not shown). It was shown that C2-sgrp170 promoted DCs to produce more pro-inflammatory cytokine TNF-α, which is consistent with the documented role of grp170 as a ‘danger’ signal. However, a significantly higher level of TNF-α was observed when DCs were stimulated with adenovirus modified C2 cells than plasmid stably transduced cells (Fig. 5d).
Discussion
Despite a wide range of available treatments that have resulted in statistically significant improvements in survival, as well as in quality of life, CaP remains the second leading cause of cancer-related death among American men, emphasizing a need for alternative complementary approaches. Harnessing the immune system to control cancer is an attractive idea. However, stimulation of an effective immune response that can eradicate cancer cells has been a challenge. Poor immune response to tumor or tumor-associated antigens is often believed to be the basis for tumor progression in immunocompetent hosts, thus presenting a major hurdle for successful immunotherapy. In this study, we demonstrate that molecular engineering of TRAMP-C2 mouse prostate tumor cells to produce secretable ER chaperone grp170 significantly enhances tumor immunogenicity, as indicated by a significant increase in tumor-specific CD8+ T-cell frequency, cytolytic activity and tumor infiltration of CD8+ cells, and control of distant tumors. Furthermore, we have shown that grp170-secreting TRAMP-C2 tumor cells derived from genetic modification by plasmid transduction or adenovirus infection can be utilized as vaccines to promote systemic antitumor immunity in both prophylactic and therapeutic settings.
Chaperone proteins have long been recognized to be involved in numerous cellular processes via interaction with different client proteins. Therefore, immunostimulatory properties of stress proteins have been attributed to their capability to chaperone and present associated antigens to professional APCs, such as dendritic cells. The structural studies [20–23] and considerable immunological evidence [24–27] suggest that the peptide or protein antigens are bound to these endogenous chaperones. Earlier studies have shown that grp170 interacts with transporter associated with antigen processing (TAP) translocated peptides and may participate in polypeptide trafficking [28, 29]. Recently we demonstrated that tumor-derived grp170 or grp170 complexed with tumor-associated antigens are highly effective in eliciting antigen and tumor specific immune responses [10, 15, 30]. Based on these findings, we speculate that TRAMP-C2 cell-secreted grp170 in our experimental setting is likely to carry the prostate tumor-associated antigens. The secreted grp170-antigen complexes are then captured and internalized by APCs via interaction with grp170-binding receptors (e.g., certain scavenger receptors), leading to antigen cross-presentation and tumor-specific immunity [10, 31]. In addition to transferring tumor-associated antigens to host APCs, the secreted grp170 itself may serve as a ‘danger’ molecule, stimulating innate immune cells to produce inflammatory cytokines or chemokines favorable for the initiation of antigen-specific immune response [14]. Although molecular basis for secretable grp170-mediated immune activation needs to be defined, it is possible that constitutive secretion of grp170 to tumor microenvironment may change the way the tumor cells are perceived by immune sentinel cells such as antigen sampling APCs, or make tumor cells appear ‘dangerous’ to host immune system, leading to efficient T-cell priming [32].
The results derived from the current study are in line with other reports suggesting a positive correlation between stress protein expression and tumor immunogenicity in rodent models [33–36]. In addition, intentional manipulation of localization of stress proteins has been shown to significantly influence host response to tumor cells. Forced expression of stress proteins on cell surface of tumor cell [37, 38] or secretion of endogenous stress proteins [39, 40] strongly promotes a tumor-specific immune response. Moreover, the results are consistent with our previous findings showing that extracellular targeting of grp170 promotes immune responses against murine melanoma [17], confirming the enhanced tumor immunogenicity by secretable grp170 in two different model systems. Grp170, one of the largest ER chaperone, is a highly ‘diverged’ relative of the hsp70 family [41]. Earlier studies have indicated that grp170 is much more efficient in binding polypeptide chains than other chaperones such as hsp70 [17, 42]. Our studies of various structural depletion mutants of grp170 suggest that the chaperoning activity of grp170 is essential for its immunologic properties, e.g., receptor binding on APCs and antigen cross-presentation [15]. Several reports support the notion that ability of chaperone-antigen complexes to generate a CTL response correlates with affinity with which the chaperone binds antigen [43–45]. In view of the fact that secretion of hsp70 by TRAMP-C2 tumor cells also facilitates immune protection against tumor growth [46], it would be interesting to compare the efficacy of these two hsp70 superfamily members in stimulating tumor immunity in vivo using the same system.
In addition to plasmid transduced C2 cells secreting grp170, we have made similar observations in tumor cells infected with replication-incompetent adenovirus encoding the secretabe grp170. Immunization with sgrp170-expressing adenovirus modified C2 cells can also achieve a tumor protective effect in a prophylactic model. In pre-established tumor models, treatment with adenovirus infected C2 tumor cells were more potent in inhibiting tumor growth than plasmid stably transfected cell line. Moreover, the adenovirus-modified cells were more efficient than plasmid-transduced cells in promoting TNF-α production by DCs, suggesting that the adenovirus vector was able to provide an additional innate stimulating ‘danger’ signal in addition to the grp170-mediated immuno-enhancing activities. Although adenovirus empty vector-infected cells could promote a pro-inflammatory response in DCs, it was not sufficient for generation of effective tumor-protective immunity. Our studies indicate that immuno-stimulatory adjuvant properties (i.e., antigen-chaperoning/presenting) of grp170 are critical for therapeutic efficacy of genetically modified C2 cells. Nonetheless, the use of adenoviral vector to deliver secretable grp170 can further enhance sgrp170-mediated vaccine potency by driving a more robust inflammatory response. Compared to stable transfection procedure, adenoviral approach with high infection efficiency would be more clinically applicable in terms of ease of vaccine production, especially in the case of CaP, for which procuring tissue and establishing patient-specific cell lines is difficult. Therefore, in this sense ex vivo modification of allogenic prostate tumor cells instead of autologous cells may be more feasible because of several added advantages, including ease of large-scale production, storage and administration. Furthermore, vaccine using CaP cells genetically modified to secrete GM-CSF has been shown to be generally well tolerated and demonstrated anti-tumor activity in phase I/II studies [4, 5]. Given that immunochaperone grp170 facilitates antigen-cross-presentation while GM-CSF can help overcome tumor-induced immune suppression and promotes the recruitment and maturation of specialized APCs, it is expected that double transfection of the prostate tumor cells with these two therapeutic molecules should further enhance tumor-specific immunity and improve clinical efficacy.
It is conceivable that ex vivo modification of prostate tumor cells by secretable grp170 has a unique capacity to induce individual tumor-specific immune responses against a broad array of CaP-associated antigens. Given the potent immunostimulatory efficacy of grp170 in mouse models and the well documented minimal toxic side effects in clinical trials with stress protein-based cancer vaccines [47], the approach described here by genetic modification of CaP cells to actively release chaperone molecule grp170 with associated tumor antigens for augmentation of tumor-specific immunity warrants further evaluation. As is the case with other standard cancer therapies (e.g., chemotherapy), it is doubtful that maximal survival benefit will be achieved with single-agent immunotherapy. However, treatments combining immunotherapy with other mechanistically distinct approaches (e.g., chemotherapy or hormonal therapy) should provide more clinical benefits to CaP patients.
Acknowledgments
This work was supported by National Cancer Institute (NCI) Grant CA121848, CA129111, American Cancer Society Grant RSG-08-187-01-LIB, Roswell Park Alliance Foundation, and NCI Cancer Center Support Grant to the Roswell Park Cancer Institute CA016056.
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Sanda MG, Restifo NP, Walsh JC, Kawakami Y, Nelson WG, Pardoll DM, Simons JW. Molecular characterization of defective antigen processing in human prostate cancer. J Natl Cancer Inst. 1995;87:280–285. doi: 10.1093/jnci/87.4.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dranoff G. GM-CSF-based cancer vaccines. Immunol Rev. 2002;188:147–154. doi: 10.1034/j.1600-065X.2002.18813.x. [DOI] [PubMed] [Google Scholar]
- 4.Simons JW, Carducci MA, Mikhak B, Lim M, Biedrzycki B, Borellini F, Clift SM, Hege KM, Ando DG, Piantadosi S, Mulligan R, Nelson WG. Phase I/II trial of an allogeneic cellular immunotherapy in hormone-naive prostate cancer. Clin Cancer Res. 2006;12:3394–3401. doi: 10.1158/1078-0432.CCR-06-0145. [DOI] [PubMed] [Google Scholar]
- 5.Small EJ, Sacks N, Nemunaitis J, Urba WJ, Dula E, Centeno AS, Nelson WG, Ando D, Howard C, Borellini F, Nguyen M, Hege K, Simons JW. Granulocyte macrophage colony-stimulating factor-secreting allogeneic cellular immunotherapy for hormone-refractory prostate cancer. Clin Cancer Res. 2007;13:3883–3891. doi: 10.1158/1078-0432.CCR-06-2937. [DOI] [PubMed] [Google Scholar]
- 6.Craig EA, Gambill BD, Nelson RJ. Heat shock proteins: molecular chaperones of protein biogenesis. Microbiol Rev. 1993;57:402–414. doi: 10.1128/mr.57.2.402-414.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calderwood SK, Theriault JR, Gong J. Message in a bottle: role of the 70-kDa heat shock protein family in anti-tumor immunity. Eur J Immunol. 2005;35:2518–2527. doi: 10.1002/eji.200535002. [DOI] [PubMed] [Google Scholar]
- 8.Lancaster GI, Febbraio MA. Exosome-dependent trafficking of HSP70: a novel secretory pathway for cellular stress proteins. J Biol Chem. 2005;280:23349–23355. doi: 10.1074/jbc.M502017200. [DOI] [PubMed] [Google Scholar]
- 9.Binder RJ, Srivastava PK. Peptides chaperoned by heat-shock proteins are a necessary and sufficient source of antigen in the cross-priming of CD8+ T cells. Nat Immunol. 2005;6:593–599. doi: 10.1038/ni1201. [DOI] [PubMed] [Google Scholar]
- 10.Wang XY, Kazim L, Repasky EA, Subjeck JR. Characterization of heat shock protein 110 and glucose-regulated protein 170 as cancer vaccines and the effect of fever-range hyperthermia on vaccine activity. J Immunol. 2001;166:490–497. doi: 10.4049/jimmunol.166.1.490. [DOI] [PubMed] [Google Scholar]
- 11.Asea A, Kraeft SK, Kurt-Jones EA, Stevenson MA, Chen LB, Finberg RW, Koo GC, Calderwood SK. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med. 2000;6:435–442. doi: 10.1038/74697. [DOI] [PubMed] [Google Scholar]
- 12.Singh-Jasuja H, Scherer HU, Hilf N, Arnold-Schild D, Rammensee HG, Toes RE, Schild H. The heat shock protein gp96 induces maturation of dendritic cells and down-regulation of its receptor. Eur J Immunol. 2000;30:2211–2215. doi: 10.1002/1521-4141(2000)30:8<2211::AID-IMMU2211>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Kelly CG, Singh M, McGowan EG, Carrara AS, Bergmeier LA, Lehner T. Stimulation of Th1-polarizing cytokines, C–C chemokines, maturation of dendritic cells, and adjuvant function by the peptide binding fragment of heat shock protein 70. J Immunol. 2002;169:2422–2429. doi: 10.4049/jimmunol.169.5.2422. [DOI] [PubMed] [Google Scholar]
- 14.Manjili MH, Park J, Facciponte JG, Wang X-Y, Subjeck JR. Immunoadjuvant chaperone, GRP170, induces “danger signals” upon interaction with dendritic cells. Immmunol Cell Biol. 2006;84:203–208. doi: 10.1111/j.1440-1711.2006.01418.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park J, Facciponte JG, Chen X, MacDonald IJ, Repasky E, Manjili MH, Wang XY, Subjeck JR. Chaperoning function of stress protein grp170, a member of the hsp70 superfamily, is responsible for its immunoadjuvant activity. Cancer Res. 2006;66:1161–1168. doi: 10.1158/0008-5472.CAN-05-2609. [DOI] [PubMed] [Google Scholar]
- 16.Foster BA, Gingrich JR, Kwon ED, Madias C, Greenberg NM. Characterization of prostatic epithelial cell lines derived from transgenic adenocarcinoma of the mouse prostate (TRAMP) model. Cancer Res. 1997;57:3325–3330. [PubMed] [Google Scholar]
- 17.Wang XY, Arnouk H, Chen X, Kazim L, Repasky EA, Subjeck JR. Extracellular targeting of endoplasmic reticulum chaperone glucose-regulated protein 170 enhances tumor immunity to a poorly immunogenic melanoma. J Immunol. 2006;177:1543–1551. doi: 10.4049/jimmunol.177.3.1543. [DOI] [PubMed] [Google Scholar]
- 18.Wang XY, Chen X, Manjili MH, Repasky E, Henderson R, Subjeck JR. Targeted immunotherapy using reconstituted chaperone complexes of heat shock protein 110 and melanoma-associated antigen gp100. Cancer Res. 2003;63:2553–2560. [PubMed] [Google Scholar]
- 19.Gao P, Sun X, Chen X, Wang Y, Foster BA, Subjeck J, Fisher PB, Wang XY. Secretable chaperone Grp170 enhances therapeutic activity of a novel tumor suppressor, mda-7/IL-24. Cancer Res. 2008;68:3890–3898. doi: 10.1158/0008-5472.CAN-08-0156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishii T, Udono H, Yamano T, Ohta H, Uenaka A, Ono T, Hizuta A, Tanaka N, Srivastava PK, Nakayama E. Isolation of MHC class I-restricted tumor antigen peptide and its precursors associated with heat shock proteins hsp70, hsp90, and gp96. J Immunol. 1999;162:1303–1309. [PubMed] [Google Scholar]
- 21.Breloer M, Marti T, Fleischer B, von Bonin A. Isolation of processed, H-2 Kb-binding ovalbumin-derived peptides associated with the stress proteins HSP70 and gp96. Eur J Immunol. 1998;28:1016–1021. doi: 10.1002/(SICI)1521-4141(199803)28:03<1016::AID-IMMU1016>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 22.Grossmann ME, Madden BJ, Gao F, Pang YP, Carpenter JE, McCormick D, Young CY. Proteomics shows Hsp70 does not bind peptide sequences indiscriminately in vivo. Exp Cell Res. 2004;297:108–117. doi: 10.1016/j.yexcr.2004.02.030. [DOI] [PubMed] [Google Scholar]
- 23.Kunisawa J, Shastri N. Hsp90alpha chaperones large C-terminally extended proteolytic intermediates in the MHC class I antigen processing pathway. Immunity. 2006;24:523–534. doi: 10.1016/j.immuni.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 24.Srivastava P. Interaction of heat shock proteins with peptides and antigen presenting cells: chaperoning of the innate and adaptive immune responses. Annu Rev Immunol. 2002;20:395–425. doi: 10.1146/annurev.immunol.20.100301.064801. [DOI] [PubMed] [Google Scholar]
- 25.Noessner E, Gastpar R, Milani V, Brandl A, Hutzler PJ, Kuppner MC, Roos M, Kremmer E, Asea A, Calderwood SK, Issels RD. Tumor-derived heat shock protein 70 peptide complexes are cross-presented by human dendritic cells. J Immunol. 2002;169:5424–5432. doi: 10.4049/jimmunol.169.10.5424. [DOI] [PubMed] [Google Scholar]
- 26.Rivoltini L, Castelli C, Carrabba M, Mazzaferro V, Pilla L, Huber V, Coppa J, Gallino G, Scheibenbogen C, Squarcina P, Cova A, Camerini R, Lewis JJ, Srivastava PK, Parmiani G. Human tumor-derived heat shock protein 96 mediates in vitro activation and in vivo expansion of melanoma- and colon carcinoma-specific T cells. J Immunol. 2003;171:3467–3474. doi: 10.4049/jimmunol.171.7.3467. [DOI] [PubMed] [Google Scholar]
- 27.Callahan MK, Garg M, Srivastava PK. Heat-shock protein 90 associates with N-terminal extended peptides and is required for direct and indirect antigen presentation. Proc Natl Acad Sci USA. 2008;105:1662–1667. doi: 10.1073/pnas.0711365105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dierks T, Volkmer J, Schlenstedt G, Jung C, Sandholzer U, Zachmann K, Schlotterhose P, Neifer K, Schmidt B, Zimmermann R. A microsomal ATP-binding protein involved in efficient protein transport into the mammalian endoplasmic reticulum. EMBO J. 1996;15:6931–6942. [PMC free article] [PubMed] [Google Scholar]
- 29.Spee P, Subjeck J, Neefjes J. Identification of novel peptide binding proteins in the endoplasmic reticulum: ERp72, calnexin, and grp170. Biochemistry. 1999;38:10559–10566. doi: 10.1021/bi990321r. [DOI] [PubMed] [Google Scholar]
- 30.Wang XY, Kazim L, Repasky EA, Subjeck JR. Immunization with tumor-derived ER chaperone grp170 elicits tumor-specific CD8+ T-cell responses and reduces pulmonary metastatic disease. Int J Cancer. 2003;105:226–231. doi: 10.1002/ijc.11058. [DOI] [PubMed] [Google Scholar]
- 31.Facciponte JG, Wang XY, Subjeck JR. Hsp110 and Grp170, members of the Hsp70 superfamily, bind to scavenger receptor-A and scavenger receptor expressed by endothelial cells-I. Eur J Immunol. 2007;37:2268–2279. doi: 10.1002/eji.200737127. [DOI] [PubMed] [Google Scholar]
- 32.Todryk SM, Melcher AA, Dalgleish AG, Vile RG. Heat shock proteins refine the danger theory. Immunology. 2000;99:334–337. doi: 10.1046/j.1365-2567.2000.00002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lukacs KV, Lowrie DB, Stokes RW, Colston MJ. Tumor cells transfected with a bacterial heat-shock gene lose tumorigenicity and induce protection against tumors. J Exp Med. 1993;178:343–348. doi: 10.1084/jem.178.1.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Menoret A, Patry Y, Burg C, Le Pendu J. Co-segregation of tumor immunogenicity with expression of inducible but not constitutive hsp70 in rat colon carcinomas. J Immunol. 1995;155:740–747. [PubMed] [Google Scholar]
- 35.Melcher A, Todryk S, Hardwick N, Ford M, Jacobson M, Vile RG. Tumor immunogenicity is determined by the mechanism of cell death via induction of heat shock protein expression. Nat Med. 1998;4:581–587. doi: 10.1038/nm0598-581. [DOI] [PubMed] [Google Scholar]
- 36.Wang XY, Li Y, Manjili MH, Repasky EA, Pardoll DM, Subjeck JR. Hsp110 over-expression increases the immunogenicity of the murine CT26 colon tumor. Cancer Immunol Immunother. 2002;51:311–319. doi: 10.1007/s00262-002-0287-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng H, Dai J, Stoilova D, Li Z. Cell surface targeting of heat shock protein gp96 induces dendritic cell maturation and antitumor immunity. J Immunol. 2001;167:6731–6735. doi: 10.4049/jimmunol.167.12.6731. [DOI] [PubMed] [Google Scholar]
- 38.Chen X, Tao Q, Yu H, Zhang L, Cao X. Tumor cell membrane-bound heat shock protein 70 elicits antitumor immunity. Immunol Lett. 2002;84:81–87. doi: 10.1016/S0165-2478(02)00042-1. [DOI] [PubMed] [Google Scholar]
- 39.Yamazaki K, Nguyen T, Podack ER. Cutting edge: tumor secreted heat shock-fusion protein elicits CD8 cells for rejection. J Immunol. 1999;163:5178–5182. [PubMed] [Google Scholar]
- 40.Massa C, Guiducci C, Arioli I, Parenza M, Colombo MP, Melani C. Enhanced efficacy of tumor cell vaccines transfected with secretable hsp70. Cancer Res. 2004;64:1502–1508. doi: 10.1158/0008-5472.CAN-03-2936. [DOI] [PubMed] [Google Scholar]
- 41.Easton DP, Kaneko Y, Subjeck JR. The hsp110 and Grp1 70 stress proteins: newly recognized relatives of the Hsp70 s. Cell Stress Chaperones. 2000;5:276–290. doi: 10.1379/1466-1268(2000)005<0276:THAGSP>2.0.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park J, Easton DP, Chen X, MacDonald IJ, Wang XY, Subjeck JR. The chaperoning properties of mouse grp170, a member of the third family of hsp70 related proteins. Biochemistry. 2003;42:14893–14902. doi: 10.1021/bi030122e. [DOI] [PubMed] [Google Scholar]
- 43.Moroi Y, Mayhew M, Trcka J, Hoe MH, Takechi Y, Hartl FU, Rothman JE, Houghton AN. Induction of cellular immunity by immunization with novel hybrid peptides complexed to heat shock protein 70. Proc Natl Acad Sci USA. 2000;97:3485–3490. doi: 10.1073/pnas.070550797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.MacAry PA, Javid B, Floto RA, Smith KG, Oehlmann W, Singh M, Lehner PJ. HSP70 peptide binding mutants separate antigen delivery from dendritic cell stimulation. Immunity. 2004;20:95–106. doi: 10.1016/S1074-7613(03)00357-1. [DOI] [PubMed] [Google Scholar]
- 45.Tobian AA, Canaday DH, Harding CV. Bacterial heat shock proteins enhance class II MHC antigen processing and presentation of chaperoned peptides to CD4+ T cells. J Immunol. 2004;173:5130–5137. doi: 10.4049/jimmunol.173.8.5130. [DOI] [PubMed] [Google Scholar]
- 46.Wang MH, Grossmann ME, Young CY. Forced expression of heat-shock protein 70 increases the secretion of Hsp70 and provides protection against tumour growth. Br J Cancer. 2004;90:926–931. doi: 10.1038/sj.bjc.6601583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang XY, Facciponte JG, Subjeck JR. Molecular chaperones and cancer immunotherapy. Handb Exp Pharmacol. 2006;172:305–329. doi: 10.1007/3-540-29717-0_13. [DOI] [PubMed] [Google Scholar]