

Abstract
Fungi that cause brown rot of wood are essential biomass recyclers and also the principal agents of decay in wooden structures, but the extracellular mechanisms by which they degrade lignocellulose remain unknown. To test the hypothesis that brown-rot fungi use extracellular free radical oxidants as biodegradative tools, Gloeophyllum trabeum was examined for its ability to depolymerize an environmentally recalcitrant polyether, poly(ethylene oxide) (PEO), that cannot penetrate cell membranes. Analyses of degraded PEOs by gel permeation chromatography showed that the fungus cleaved PEO rapidly by an endo route. 13C NMR analyses of unlabeled and perdeuterated PEOs recovered from G. trabeum cultures showed that a major route for depolymerization was oxidative C—C bond cleavage, a reaction diagnostic for hydrogen abstraction from a PEO methylene group by a radical oxidant. Fenton reagent (Fe(II)/H2O2) oxidized PEO by the same route in vitro and therefore might account for PEO biodegradation if it is produced by the fungus, but the data do not rule out involvement of less reactive radicals. The reactivity and extrahyphal location of this PEO-degrading system suggest that its natural function is to participate in the brown rot of wood and that it may enable brown-rot fungi to degrade recalcitrant organopollutants.
Lignocellulose, the predominant form of terrestrial fixed carbon, is degraded efficiently only by certain filamentous fungi. Among the least-understood of these are the brown-rot basidiomycetes, which are essential contributors to biomass recycling and soil fertility in coniferous forest ecosystems (1, 2). Brown-rot fungi are also responsible for the most destructive type of decay in wooden structures. It has been estimated that ≈10% of all trees cut in the United States go to replace wood that decays in service, largely because of brown rot (3).
Brown-rot fungi have the singular ability to digest wood cellulose without removing the lignin that encases and normally protects it from microbial attack (4). Instead, the lignin is modified chemically by reactions that include side chain oxidation and cleavage of ether linkages (5, 6). These reactions are unusual because lignin and other polyethers are relatively unreactive and resist oxidation by most biochemical mechanisms (4, 7, 8). The cellulose in wood also is oxidized and depolymerized rapidly during brown rot, which may indicate cleavage of its acetal linkages by the same species that oxidizes lignin (9, 10).
Because sound wood is impermeable to enzymes (11, 12), it has been proposed that the agent responsible for brown rot is a low molecular weight one-electron oxidant such as the hydroxyl radical (⋅OH) or Fenton reagent (Fe(II)/H2O2) (13–19). These extremely reactive oxidants attack ethers and acetals by similar pathways that include β-scission of aliphatic C—C bonds (20, 21). However, the chemical changes that occur in lignin and cellulose during brown rot are complex and might reflect processes other than one-electron oxidation. Moreover, although brown-rot fungi do produce one-electron oxidants of some type (13–16, 18), it never has been demonstrated that these agents are sufficiently reactive to degrade ethers or that they have the extrahyphal location necessary for a role in lignocellulose breakdown.
To address these problems, we have examined the ability of the brown-rot fungus Gloeophyllum trabeum to degrade a structurally simple polyether by using extracellular reactions. The model substrate we used, poly(ethylene) oxide (PEO), meets several important criteria: It undergoes diagnostic cleavage reactions when oxidized by one electron (22–24), it is inert to hydrolysis under physiological conditions (8), and it is unable to penetrate cell membranes (25–27). PEOs resist biodegradation and have become widespread, persistent environmental contaminants (8). Our results show that G. trabeum produces an extracellular one-electron oxidant that cleaves PEO rapidly via β-scission reactions.
MATERIALS AND METHODS
Chemicals.
PEOs with Mrs under 5 × 104 generally are called poly(ethylene glycols), but we use the former term throughout for brevity. Unlabeled PEOs were obtained from Polysciences. PEO labeled with 14C in its terminal hydroxyethyl positions [19.2 mCi⋅g−1 (1 Ci = 37 GBq); Mr stated as 4 × 103] was from Amersham, and poly(ethylene-2H4 oxide) (99.8% perdeuterated, Mr = 1.4 × 104) was from CDN Isotopes (Quebec, Canada). Formate ester-terminated PEO was obtained by refluxing PEO overnight in 96% formic acid and was purified by gel permeation chromatography (GPC) on a 3 × 36-cm column of Sephadex G-25 (Pharmacia) in ethanol:water (1:1). This procedure resulted in ≈20% formylation. All other chemicals were reagent grade.
Culture Conditions.
G. trabeum (ATCC 11539) was grown at 30°C under air in 125-ml Erlenmeyer flasks that contained 2.5 g of perlite and 15 ml of basal growth medium with 10 g/liter glucose as the carbon source (28). The cultures were inoculated at a rate of 1% with homogenized potato dextrose agar plates of the fungus.
In experiments to measure PEO degradation, the polymer was added to 7-day-old cultures at a final concentration of 3.3 g/liter in 1.0 ml of sterile H2O. This initial concentration of PEO was high enough to ensure that the proportion of polymer mineralized was negligible (3% in 9 days), leaving most of the degradation products as oligomers for analysis. When GPC of the degraded PEO was done, unlabeled PEO with a Mr of 4 × 103 was used in combination with [14C]PEO [1.1 × 105 dpm per culture (1 Bq = 60 dpm)]. For 13C NMR experiments, unlabeled PEO with a Mr of 2 × 105 or perdeuterated PEO with a Mr of 1.4 × 104 was used.
In experiments to measure glucose mineralization, 1.1 × 105 dpm of 6-[14C]glucose (45 mCi⋅mmol−1; Research Products International) was added with the unlabeled glucose at the time of culture inoculation. The 14CO2 evolved by these cultures and by cultures that received [14C]PEO was determined as described (29).
Mr Analysis of Degraded PEO.
Cultures were harvested in triplicate 0, 5, 7, and 9 days after [14C]PEO was added. Each culture was extracted by shaking it for 1 h with 15 ml of ethanol, and the mycelium was removed by filtration through a 0.45-μm pore-size nylon membrane. This procedure gave >90% recovery of the 14C present in each culture. The extracts from each set of triplicates then were pooled, and 1.5 ml of the total was fractionated by GPC on a 1.3 × 24.5-cm column of Sephadex LH-60 in ethanol:water (1:1). Fractions (0.9 ml) were collected and analyzed for 14C by scintillation counting. PEO standards with Mrs of 7.5 × 103, 3.4 × 103, 1.5 × 103, 1.0 × 103, 4.0 × 102, and 1.5 × 102 were used to calibrate the GPC column and were detected in the eluate by refractometry. Number-average Mr (Mn) values and weight-average Mr (Mw) values were calculated by using the standard equations (30).
The rate of PEO scission was calculated from the time course of Mn decrease in PEO recovered from the cultures. The number of times a polymer is cleaved is one less than the ratio of its Mn before cleavage to its Mn after cleavage (31). Accordingly, the rate of PEO cleavage, r, was calculated from the equation
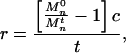 |
where Mn0 equals the Mn of the polymer at time zero, Mnt equals its Mn at time t, and c equals the concentration of polymer chains at time zero. Values for Mn and t used in the calculations are given in the legend to Fig. 1. The value for c (0.67 mM) was based on an initial Mn of 5.0 × 103.
Figure 1.

GPC analyses of [14C]PEO degraded by G. trabeum. Day 0 after PEO addition (····): Mn = 5.0 × 103, Mw = 6.2 × 103. Day 5 (- - -): Mn = 9.7 × 102, Mw = 3.0 × 103. Day 7 (not shown): Mn = 6.9 × 102, Mw = 2.3 × 103. Day 9 (—): Mn = 5.9 × 102, Mw = 1.9 × 103. Mr values and elution positions are shown for some of the PEO Mr standards used to calibrate the GPC column. (Inset) Time course of PEO scission as calculated in Materials and Methods.
13C NMR Spectrometry of Degraded PEO.
Cultures (five replicates) were harvested 13 days after the polymer was added. The pooled medium and mycelium were stirred for 30 min with two volumes of water, the mycelium was removed by filtration through a 0.45-μm pore-size nylon membrane, and the combined aqueous extracts were extracted four times with 0.25 volume of CH2Cl2. The organic phase was dried over Na2SO4, was evaporated under reduced pressure to dryness, was redissolved in 10 ml of ethanol:water (1:1), and was fractionated by GPC on a 3 × 36-cm column of Sephadex G-25 in ethanol:water (1:1). The excluded portion of the eluate (Mr > 5 × 102) was collected and evaporated to dryness under reduced pressure. The residue, containing 60–90 mg of PEO, was redissolved in ≈0.5 ml of C2HCl3 and was transferred to a 5-mm NMR tube.
13C NMR analyses were obtained in C2HCl3 at 62.9 MHz and 300 K on a Bruker DPX250 spectrometer (Bruker, Billerica, MA) with a 5-mm conventional geometry four-nucleus probe. Proton-decoupled 13C spectra were acquired with a 30° pulse and a relaxation delay of 1.0 s for a total of at least 2 × 104 scans. Chemical shifts were referenced to internal tetramethylsilane. Semiquantitative signal integrations were based on the assumptions that the relaxation times for all PEO methylenes were equivalent and that the relaxation times for PEO formate carbonyls and aldehyde carbonyls were equivalent.
Distortionless enhancement with polarization transfer (DEPT) spectra (1.4 × 104 scans with a 135° editing pulse) were obtained with a standard Bruker microprogram. The times for relaxation delay and polarization transfer were 0.5 s and 3.57 ms, respectively, resulting in a coupling constant of 140 Hz.
Inverse z-gradient-selected two-dimensional heteronuclear multiple bond correlation experiments (g-HMBC) and heteronuclear single quantum coherence experiments (g-HSQC) were obtained using standard Bruker microprograms. Coupling delays for the formate ester-terminated PEO were optimized such that the one-bond and three-bond coupling constants (1JCH and 3JCH) between the formate ester proton and neighboring carbons were 205 Hz and 5 Hz, respectively. All two-dimensional experiments consisted of 256 increments of 64 scans each and were acquired by using a relaxation delay of 1.5 s.
Fenton Oxidation of PEO.
PEO (400 mg; Mr = 2 × 105) and FeCl2⋅4H2O (200 mg) were dissolved in 250 ml of H2O. H2O2 (5 mmol) then was added dropwise with stirring over 30 min. The final pH of the reaction mixture was ≈2. The products were extracted into CH2Cl2 and were fractionated by GPC as described for PEO from G. trabeum cultures.
RESULTS AND DISCUSSION
Rate of PEO Cleavage.
GPC analyses of [14C]PEO recovered from G. trabeum cultures showed that the fungus cleaved the polymer rapidly. The cultures mineralized their carbon source, glucose, at a linear rate of 1.6 mM per day (data not shown) and cleaved [14C]PEO at a linear rate of 0.6 mM scissions per day (Fig. 1 Inset). This PEO scission rate is an underestimate because it neglects contributions from PEO fragments with Mrs <150, which were not resolved by our GPC procedure. High performance liquid chromatography of the metabolized PEO confirmed that it contained at least two such low-Mr products, ethylene glycol and diethylene glycol (data not shown).
Possible Routes for PEO Oxidation.
The Mr distribution of [14C]PEO in G. trabeum cultures spread unimodally to lower values as degradation progressed (Fig. 1). Because the PEO was labeled with 14C only at its termini, this result establishes that depolymerization followed an endo rather than an exo route. That is, exo degradation would have released all of the 14C from the PEO as low-Mr material at the outset of degradation. There are two likely routes for endo PEO oxidation by G. trabeum: oxygen insertion between carbon and hydrogen in a methylene group or hydrogen abstraction from a methylene carbon. These two mechanisms are expected to yield different product profiles.
Oxygen insertion into a PEO methylene group would yield a hemiacetal, which would undergo acid-catalyzed C—O cleavage to generate fragments terminated by new alcohol (I) and aldehyde (II) end groups (Fig. 2). This is the mechanism thought to operate in intracellular monooxygenase-catalyzed oxidations of certain low-Mr ethers (8).
Figure 2.

Predicted pathways of PEO scission after oxygen insertion or hydrogen abstraction. Labeled structures are those identified in 13C NMR experiments (see Fig. 3). Mn indicates a transition metal ion.
Hydrogen abstraction from a PEO methylene group would yield a carbon-centered radical that would rapidly add O2 to give a peroxyl radical. PEO peroxyl radicals have been proposed to fragment unimolecularly via hexagonal intermediates (22–24), but it appears equally possible that they would decompose via alkoxyl radical intermediates in reactions analogous to those that occur during fatty acid peroxidation (32) (Fig. 2). In any event, the unimolecular pathway proposed earlier and the alkoxyl radical pathway shown here lead to PEO fragments that carry the same end groups: alcohols (I) and aldehydes (II) via hemiacetal cleavage and other reactions and formate esters (III) via β-scission of a C—C bond. Products I–III are formed when PEO is cleaved by radiolytically generated ⋅OH (22–24).
Identification of New End Groups in Degraded PEO.
To distinguish between oxygen insertion and hydrogen abstraction, we obtained 13C NMR spectra of the products generated by G. trabeum from a 2 × 105 Mr unlabeled PEO. Unchanged PEO was recovered from uninoculated cultures after 13 days of incubation. The only significant NMR signal observed was caused by methylenes in internal repeating ethoxyl units (70.5 ppm). The two carbons in terminal hydroxyethyl groups (Ia, 61.4 ppm and Ib, 72.6 ppm) were undetectable because of their low frequency in this high-Mr polymer (Fig. 3A).
Figure 3.

13C NMR analyses of PEO. (A) PEO recovered from an uninoculated culture. (B) PEO recovered from a G. trabeum culture. (Inset) The spectrum obtained in the formate ester carbonyl region when perdeuterated PEO rather than natural abundance PEO was supplied. (C) PEO recovered after Fenton oxidation. Structures corresponding to the labeled NMR signals are shown in Fig. 2. The spectra have been normalized by setting the amplitude of the 70.5-ppm signals (which are truncated here) to a constant height. The signal for structure IIb was superimposed on the C2HCl3 signals and is not shown.
Prominent new end group signals appeared in PEO that had been degraded by G. trabeum for 13 days (Fig. 3B). The major terminal structures were alcohols (Ia, 61.4 ppm, ≈60% of total), formate esters (IIIa, 161.0 ppm, ≈30% of total), and aldehydes (IIa, 200.8 ppm, ≈10% of total). The other major NMR signals in the spectrum came from methylenes adjacent to the terminal alcohols (Ib, 72.6 ppm), aldehydes (IIb, 76.8 ppm), and formate esters (IIIb, 62.9 ppm and IIIc, 68.8 ppm). Integration of the end group and internal 13C signals indicated that the material subjected to analysis had been depolymerized to a Mr of ≈2 × 103. Similar results were obtained when PEO was oxidized in vitro with Fenton reagent (Fig. 3C).
The identity of the formate ester was confirmed in several ways: (i) An authentic standard of PEO formate also exhibited signals III a–c. (ii) Distortionless enhancement with polarization transfer spectra of the degraded sample and of PEO formate showed that the carbon responsible for the 161.0-ppm signal (IIIa) carried a single proton. (iii) The degraded sample and PEO formate both exhibited a 1H NMR signal characteristic of a formate ester proton at 8.1 ppm. A heteronuclear correlation experiment confirmed this assignment by giving a cross-peak between the 8.1-ppm proton signal and the 161.0-ppm carbon signal. (iv) When the degraded sample was hydrolyzed with aqueous NaOH, the 161.0-ppm ester carbonyl signal disappeared, the amplitude of the 61.4 ppm alcohol signal increased, and a new signal attributable to formic acid appeared at 162.3 ppm.
These data show that terminal formate esters were major products of PEO degradation by G. trabeum, but they do not exclude the possibility that these groups were derived from an esterification reaction between exogenous formate and new —CH2OH termini on the cleaved polymer. To address this question, we repeated the biodegradation experiment with a 1.4 × 104 Mr perdeuterated PEO. 13C NMR analysis of this sample showed a three-line signal centered at 160.8 ppm (coupling constant = 34 Hz), as expected for a deuterated formate ester carbonyl when the spectrum is not decoupled for deuterium (Fig. 3B Inset). This result proves that the formate ester was derived from the original PEO polymer and consequently that oxidative C—C bond cleavage is a major reaction in PEO degradation by G. trabeum.
Conclusions.
These experiments show that G. trabeum produces a strong extracellular oxidant that leads to extensive PEO depolymerization by abstracting hydrogens from the polymer’s internal methylene groups. One possibility is that this oxidant is Fenton reagent, which we found to cleave PEO by the same route in vitro. Previous work has shown that some brown-rot fungi produce extracellular Fe oxidoreductases (17) and Fe chelators (13, 15, 16, 18) that might reduce Fe3+ to Fe2+ as required for a Fenton mechanism, and it has been suggested that the necessary H2O2 might be produced via Fe2+ autooxidation (17).
Alternatively, the G. trabeum oxidant may be a less reactive free radical. The C—H bond dissociation energy for ether methylenes such as those in PEO is ≈90 kcal⋅mol−1 (33), which is approximately the same as the O—H bond dissociation energy for alkyl hydroperoxides (34). Therefore, the oxidant that initiates PEO cleavage could be a peroxyl radical or a similarly reactive species, and PEO peroxyl radicals could propagate the reaction. It is accordingly possible that the amount of initiating oxidant produced by the fungus is small relative to the amount of PEO cleaved, but, at present, we cannot distinguish between initiation and propagation reactions in this system.
We consider it likely that the G. trabeum oxidant’s natural function is to participate in the brown rot of wood because the reactivity of C—H bonds in cellulose toward radical oxidants is expected to be similar to that of C—H bonds in PEO (33). However, a major difference between hydrogen abstraction from PEO and hydrogen abstraction from cellulose is that the latter process cannot yield chain-propagating peroxyl radicals on every carbon. Although α-alkoxyalkylperoxyl radicals can be formed at C1, C4, and C5 of each glucosyl unit in cellulose, α-hydroxyalkylperoxyl radicals will be produced instead at C2, C3, and C6, and will eliminate perhydroxyl radicals (⋅OOH) (21). However, at the high extracellular acidities (pH 2–4) generated by brown-rot fungi (17, 35), this ⋅OOH either could initiate new H-abstraction reactions from cellulose or could dismutate to provide H2O2 for Fenton oxidation of the polymer (32).
G. trabeum is unusual in its ability to degrade an aliphatic polyether via extracellular one-electron oxidation. Even white-rot basidiomycetes, which use radical chemistry to oxidize lignin and many other organic chemicals (4, 7, 36), are unable to depolymerize PEO significantly (28, 37). Our results and recent data from Wetzstein et al. (38) suggest that brown-rot fungi may be useful for organopollutant bioremediation as a consequence of the one-electron oxidants they produce.
Acknowledgments
We are grateful to K. Hirth for the NMR analyses, to K. A. Jensen, Jr. and D. J. Litwin for GPC analyses, and to T. K. Kirk, W. H. McClain, J. Ralph, J. Stubbe, and J. J. Worrall for helpful discussions. This work was supported by U.S. Department of Energy Grant FG02-94-ER20140 (to K.E.H.) and by Postdoctoral Award No. FI-233-96 from the United States–Israel Binational Agricultural Research and Development Fund (to Z.K.).
ABBREVIATIONS
- GPC
gel permeation chromatography
- PEO
poly(ethylene) oxide
References
- 1.McFee W W, Stone E L. Soil Sci Soc Am Proc. 1966;30:513–516. [Google Scholar]
- 2.Gilbertson R L, Ryvarden L. North American Polypores. Oslo: Fungiflora; 1986. [Google Scholar]
- 3.Zabel R A, Morell J J. Wood Microbiology: Decay and Its Prevention. San Diego: Academic; 1992. [Google Scholar]
- 4.Eriksson K-E L, Blanchette R A, Ander P. Microbial and Enzymatic Degradation of Wood and Wood Components. Berlin: Springer; 1990. [Google Scholar]
- 5.Kirk T K. Holzforschung. 1975;29:99–107. [Google Scholar]
- 6.Jin L, Schultz T P, Nicholas D D. Holzforschung. 1990;44:133–138. [Google Scholar]
- 7.Kirk T K, Farrell R L. Annu Rev Microbiol. 1987;41:465–505. doi: 10.1146/annurev.mi.41.100187.002341. [DOI] [PubMed] [Google Scholar]
- 8.White G F, Russell N J, Tidswell E C. Microbiol Rev. 1996;60:216–232. doi: 10.1128/mr.60.1.216-232.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cowling, E. B. (1961) U. S. Dept. Agric. Tech. Bull. 1258.
- 10.Kirk T K, Ibach R, Mozuch M D, Conner A H, Highley T L. Holzforschung. 1991;45:239–244. [Google Scholar]
- 11.Blanchette R A, Krueger E W, Haight J E, Akhtar M, Akin D E. J Biotechnol. 1996;53:203–213. [Google Scholar]
- 12.Fluornoy D S, Kirk T K, Highley T L. Holzforschung. 1991;45:383–388. [Google Scholar]
- 13.Chandhoke V, Goodell B, Jellison J, Fekete F A. FEMS Microbiol Lett. 1992;90:263–266. [Google Scholar]
- 14.Backa S, Gierer J, Reitberger T, Nilsson T. Holzforschung. 1992;46:61–67. [Google Scholar]
- 15.Hirano T, Tanaka H, Enoki A. Mokuzai Gakkaishi. 1995;41:334–341. [Google Scholar]
- 16.Hirano T, Tanaka H, Enoki A. Holzforschung. 1997;51:389–395. [Google Scholar]
- 17.Hyde S M, Wood P. Microbiology. 1997;143:259–266. doi: 10.1099/00221287-143-1-259. [DOI] [PubMed] [Google Scholar]
- 18.Goodell B, Jellison J, Liu J, Daniel G, Paszczynski A, Fekete F, Krishnamurthy S, Jun L, Xu G. J Biotechnol. 1997;53:133–162. [Google Scholar]
- 19.Koenigs J W. Wood Fiber. 1974;6:66–79. [Google Scholar]
- 20.Sonntag C V, Schuchmann H-P. In: The Chemistry of Ethers, Crown Ethers, Hydroxyl Groups, and their Sulfur Analogues: Part 2. Patai S, editor. New York: Wiley; 1980. pp. 935–970. [Google Scholar]
- 21.Sonntag C v. Adv Carbohydr Chem Biochem. 1980;37:7–77. [Google Scholar]
- 22.Gugumus, G. & Marchal, J. (1968) J. Polym. Sci. Part C: Polym. Symp. 3963–3972.
- 23.Gondet J C, Crouzet C, Marchal J. Kinetics and Mechanism of Polyreactions. Vol. 5. Budapest: Akadémiai Kiado; 1969. pp. 249–254. [Google Scholar]
- 24.Decker C, Marchal J. Kinetics and Mechanism of Polyreactions. Vol. 5. Budapest: Akadémiai Kiado; 1969. pp. 231–237. [Google Scholar]
- 25.Money N P. Exp Mycol. 1990;14:234–242. [Google Scholar]
- 26.Samuni A, Carmichael A J, Russo A, Mitchell J B, Riesz P. Proc Natl Acad Sci USA. 1986;83:7593–7597. doi: 10.1073/pnas.83.20.7593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scherrer R, Louden L, Gerhardt P. J Bacteriol. 1974;118:534–540. doi: 10.1128/jb.118.2.534-540.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawai S, Jensen K A, Jr, Bao W, Hammel K E. Appl Environ Microbiol. 1995;61:3407–3414. doi: 10.1128/aem.61.9.3407-3414.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirk T K, Connors W J, Bleam R D, Hackett W F, Zeikus J G. Proc Natl Acad Sci USA. 1975;72:2515–2519. doi: 10.1073/pnas.72.7.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yau W W, Kirkland J J, Bly D D. Modern Size-Exclusion Liquid Chromatography. New York: Wiley; 1979. [Google Scholar]
- 31.Hammel K E, Jensen K A, Jr, Mozuch M D, Landucci L L, Tien M, Pease E A. J Biol Chem. 1993;268:12274–12281. [PubMed] [Google Scholar]
- 32.Dix T A, Aikens J. Chem Res Toxicol. 1993;6:2–18. doi: 10.1021/tx00031a001. [DOI] [PubMed] [Google Scholar]
- 33.O’Neal H E, Benson S W. In: Free Radicals. Kochi J K, editor. Vol. 2. New York: Wiley Interscience; 1973. pp. 275–359. [Google Scholar]
- 34.Koppenol W H. FEBS Lett. 1990;264:165–167. doi: 10.1016/0014-5793(90)80239-f. [DOI] [PubMed] [Google Scholar]
- 35.Espejo E, Agosin E. Appl Environ Microbiol. 1991;57:1980–1986. doi: 10.1128/aem.57.7.1980-1986.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hammel K E. In: Microbial Transformation and Degradation of Toxic Organic Chemicals. Young L Y, Cerniglia C E, editors. New York: Wiley–Liss; 1995. pp. 331–346. [Google Scholar]
- 37.Kay-Schoemake J L, Watwood M E. Appl Microbiol Biotechnol. 1996;46:438–442. [Google Scholar]
- 38.Wetzstein H-G, Schmeer N, Karl W. Appl Environ Microbiol. 1997;63:4272–4281. doi: 10.1128/aem.63.11.4272-4281.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]