

Abstract
Inhibitor of growth 2 (ING2) is associated with chromatin remodeling and regulation of gene expression by binding to a methylated histone H3K4 residue and recruiting HDAC complexes to the region. The aim of our study is to investigate the regulation of ING2 expression and the clinical significance of upregulated ING2 in colon cancer. Here, we show that the ING2 mRNA level in colon cancer tissue increased to more than twice than that in normal mucosa in the 45% of colorectal cancer cases that we examined. A putative NF-κB binding site was found in the ING2 promoter region. We confirmed that NF-κB could bind to the ING2 promoter by EMSA and luciferase assays. Subsequent microarray analyses revealed that ING2 upregulates expression of matrix metalloproteinase 13 (MMP13), which enhances cancer invasion and metastasis. ING2 regulation of MMP13 expression was confirmed in both ING2 overexpression and knock down experiments. MMP13 expression was further induced by coexpression of ING2 with HDAC1 or with mSin3A, suggesting that the ING2-HDAC1-mSin3A complex members regulates expression of MMP13. In vitro invasion assay was performed to determine functional significance of ING2 upregulation. ING2 overexpressed cells exhibited greater invasive potential. Taken together, upregulation of ING2 was associated with colon cancer and MMP13-dependent cellular invasion, indicating that ING2 expression might be involved with cancer invasion and metastasis.
Keywords: ING2, MMP13, chromatin remodeling, colon cancer, metastasis, NF-κB
Colorectal cancer is the third most common type of cancer in the United States, with an estimated 148,810 new cases and the second leading cause of cancer-related deaths in the United States, with an estimated 49,960 deaths in 2008.1
Inhibitor of growth 2 (ING2), which is a member of ING gene family, was identified by searching EST databases for any similarity to the ING1 gene. A previous report has shown that the ING2 mRNA is expressed in testis at the highest level among normal tissues, and also emphasizes that ING2 mRNA expression is remarkably increased in colon cancer tissue compared to nonmalignant tissue.2 ING2 as well as ING1b are considered to be candidate tumor suppressor genes, which cooperate with p53 and share many biological functions, such as apoptosis, cell cycle regulation and senescence.3-7 Our recent report has shown that ING2 expression is suppressed by nutlin-3a-induced p53, resulting in the induction of senescence in normal human fibroblasts and a cancer cell line.8 ING family proteins functionally form complexes with several factors possessing intrinsic histone acetyltransferase (HAT) and histone deacetylase complex (HDAC) activities, thus, linking the function of ING genes to chromatin remodeling and transcriptional regulation through histone modifications.9-15 Among ING proteins, ING2 especially has a potential ability to bind to the trimethylated histone H3 at lysine 4 (H3K4me3). H3K4me3 correlates with transcriptional activation, while methylations occurred at many other histone residues are associated with transcriptional suppression.16-18 Previous reports suggest that ING2 only induces gene expression and does not repress expression of any known genes in normal fibroblasts.19 ING2 also interacts with phosphatidylinositol 5-phosphate (PtdIns5P).20,21 PtdIns5P, which may function in a nuclear signaling pathway, has been implicated as a critical regulator of nuclear signaling events during cell-cycle progression.22,23 Cellular stress, such as UV irradiation or oxidative stress, causes the accumulation of nuclear PtdIns5P, resulting in the activation of ING2.24,25 Oxidative and nitrosative stress are linked with inflammation-associated cancer including colon carcinoma.26
We report here evidence that pathophysiological expression of ING2 may enhance invasion and metastasis of colon cancer by increasing the expression of MMP13.
Material and methods
Clinical samples and RNA extraction
Five colorectal cancer samples were obtained from BD Bioscience (San Jose, CA). Additional surgical specimens were obtained with IRB approval from 34 patients with colorectal cancer undergoing surgical operation from 1996 to 1997 at Maryland University hospital. The median age of the patients was 59.8 years. The carcinomas were staged according to TNM classification. These cases included stage I, 10 cases; stage II, 9 cases; stage III, 8 cases and stage IV, 7 cases. Malignant and adjacent nonmalignant portions of each specimen were used for RNA extraction. Total cellular RNA was extracted using TRIzol (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions.
RNA probe for ING2 and in situ hybridization of ING2 transcripts
Colon cancer specimens were fixed in 20% phosphate-buffered formalin (pH 7.4) at room temperature, embedded in paraffin and cut into 4 μm sections. In situ hybridization was carried out using Digoxigenin (DIG)-labeled cRNA probes and DIG RNA Labeling Mix (Roche Applied Science, Indianapolis, IN) according to the manufacturer’s instructions. An ING2-specific probe was generated using T7 polymerase (Ambion, Austin, TX) and human ING2 cDNA (780 bp) inserted into pTRI-amp-18 (Ambion) that was linearized by digesting with Xba I, followed by alkaline hydrolysis. After in vitro transcription by T7 RNA polymerase, the product was digested with treatment of 40 mM NaHCO3 and 60 mM Na2CO3 (pH 10.2), making each 150 bp length probe of whole ING2 cDNA.
Immunohistochemical staining
Immunohistochemical staining was performed by using serial sections of paraffin-embedded colon cancer specimens. Antigen retrieval and immunochemical staining were performed as previously described.27 Sections were incubated with a 1:100 dilution of the rabbit polyclonal anti-ING2, which was generated against C-terminal 14mer amino acid of ING2.
Cell culture and reagents
Cultured human colon cancer cell lines including DLD-1, LS174T, RKO, SW620, SW837 and WiDr as well as a human kidney cell line, 293, were originally obtained from the American Type Culture Collection (Rockville, MD). The HCT116 human colon cancer cell lines (p531+/+ and p53-/-) were kindly provided by Dr. Bert Vogelstein (Johns Hopkins University School of Medicine, Baltimore, MD). These cells were grown at 37°C in the presence of 5% CO2 in the recommended media. In all experiments, cells were plated for 48 hr before treatment. Wedelolactone (Calbiochem, San Diego, CA), MG132 (Sigma), doxorubicin (Sigma, St. Louis, MO) and a MMP13 inhibitor (Calbiochem) were used at the indicated concentrations. These compounds were dissolved in dimethyl sulfoxide (DMSO).
Realtime reverse-transcription PCR (RT-PCR) analysis of mRNA expression
Total RNA from each cultured cell line was extracted using TRIzol (Invitrogen) according to the manufacturer’s protocol. Five micrograms of total RNA were used for the synthesis of first-strand cDNA using the SuperScript III First Strand cDNA Synthesis Kit (Invitrogen) following the manufacturer’s instructions. In the conventional PCR, ING2 and β-actin mRNA transcripts were amplified, respectively, using the following primer sets: forward 5′-GCGAGAGCTGGACAACAAAT-3′; reverse 5′-GACACTTG GTTGCATAAGCAG-3′; forward 5′-GCTCGTCGTCGACAACGGCTC-3′ and reverse 5′-CAAACATGATCTGGGTCATCTTCTC-3′. Realtime RT-PCR analysis was performed using ABI prism 7500 (Applied Biosystems, Foster City, CA) with a TaqMan probe provided by the manufacturer. The TaqMan probes used for the real-time PCR analysis were: ING2 (Id Hs00357543_m1), MMP13 (Id Hs00233992_m1), β-actin (Id Hs99999903_m1) and GAPDH (Id Hs99999905_m1). The relative amount of targeted gene’s transcripts was normalized by the amount of β-actin or GAPDH transcript in the same cDNA.
Nuclear extract preparation, western blot analysis and immunoprecipitation
The nuclear extraction was performed using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce, Rockford, IL) according to the manufacturer’s protocol. Protein lysates were prepared as described previously.28 For immunoprecipitation, 293 cells transfected with FLAG-ING2 expression plasmids were harvested at 48 hr posttransfection. The cellular extracts (400 μg) were immunoprecipitated with FLAG M2 resin (Sigma) for 4 hr at 4°C, centrifuged and washed. Protein aliquots were separated on SDS-polyacrylamide gels and transferred onto nitrocellulose membranes. Blots were probed with a primary antibody for 1 hr at room temperature, and then incubated with a horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA) for 30 min at room temperature. Bound antibodies were detected by the enhanced chemiluminescence (ECL) detection reagents (Amersham Pharmacia Biotech, Piscataway, NJ) and visualized by autoradiography. The primary antibodies used for western blot analysis were: mouse monoclonal anti-phospho-NF-κB p65 (Cell signaling technology, Danvers, MA), mouse monoclonal anti-p65, rabbit polyclonal anti-mSin3A, rabbit polyclonal anti-HDAC1 (Santa Cruz Biotechnology), mouse monoclonal anti-FLAG M2, mouse monoclonal anti-β-actin (Sigma), rabbit polyclonal anti-α-lamin A/C (Santa Cruz Biotechnology) and rabbit polyclonal anti-ING2.
Electrophoretic mobility shift analysis for NF-κB
Electrophoretic mobility shift assays (EMSAs) were performed using the LightShift Chemiluminescent EMSA Kit (Pierce). Recombinant NF-κB p50 protein (Promega, Madison, WI) and nuclear extracts of cells were added to the reaction. Biotin end-labeled, double-stranded oligonucleotides, which contain a putative NF-κB consensus binding site (5′-CGCCGAGGGGATCCCCACTGCT-3′) on the ING2 regulatory region, were then added to the reaction mixtures and incubated for 20 min at room temperature. In competition experiments, unlabeled oligonucleotides (100-fold excess) were incubated with the extracts for 5 min before adding the labeled probe. For super-shift experiments, each anti-p50 and anti-p65 antibody (Rockland Immunochemicals, PA) was added to the reaction mixture prior to adding the labeled probe, and the incubation was continued for 20 min at room temperature. The oligonucleotides used in our study were as follows: NF-κB consensus sequence (consensus) 5′-AGTT GAGGGGACTTTCCCAGGC-3′ and cAMP responsive element binding protein consensus sequence (CREB) 5′-AGAGATTGCCT GACGTCAGAGAGCTAG-3′. Aliquots of these mixtures were loaded onto a 6% polyacrylamide gel (Invitrogen) with 0.5× tris-glycine as the running buffer and transferred to a nylon membrane. Biotin end-labeled DNA and DNA—protein bands were visualized by autoradiography.
Detection of the ING2 promoter activity
The reporter constructs of the ING2 promoter (A) containing a −1,251 to +140 region, (B) containing a −162 to +140 region and (C) containing a +64 to +140 region from the putative transcription start site were prepared by PCR amplification from the human genomic DNA of Beas2B, which is the aneuploid Ad12-SV40 immortalized cell line derived from a normal human lung epithelial cell,29 and cloned into the pGL-3 basic vector (Promega). The luciferase reporter assay was performed using a dual luciferase reporter assay system (Promega). A pGL3 firefly luciferase-reporter plasmid (1.6 μg) of each construct (A—C) were cotransfected with a 0.16 μg of the pRL Renilla luciferase plasmid into cells cultured in 12-well plates using the Lipofectamine 2000 reagent (Invitrogen). Cell lysates were obtained by adding 250 μl of cell lysis buffer per well. Luciferase activity was measured by using 20 μl of cell lysate per assay tube in a single-photon channel of a scintillation counter (Beckman, Fullerton, CA). The level of firefly luciferase activity was normalized by that of the Renilla luciferase activity in each experiment.
Plasmids and adenovirus constructs
The cells (1 × 106) were seeded in 6-well plates, 1 day before the transfection. The cells were transfected with the pcDNA3.1-ING2 vector using the Lipofectamine 2000 (Invitrogen) as previously described.3 FLAG tagged ING2 expression vector using FLAG-CMV-6 vector (Sigma) was generated and transfected into cells for IP western assays. Each HDAC1 and mSin3A cDNA was generated by PCR amplification from the human genomic DNA of Beas2B, and inserted into pcDNA3.1 vector (Invitrogen). Control cells were transfected with the same amount of pcDNA3.1 vectors. The ING2 complete cDNA expression vector with a cytomegalo-virus promoter was transferred into an adenovirus-packing cell line following the manufacturer’s instructions. The recombinant plasmids were linearized and propagated in HEK293 cells, and high-titer purified preparations (~1010 plaque-forming units/ml) were generated by the Gene Expression Laboratory of NCI-Frederick (Frederick, MD). The transfection efficiency of HCT116 cell lines was determined by counting GFP-positive cells at 24 and 48 hr postinfection. GFP gene expression in HCT116 cells treated with the GFP inserted adenovirus (adeno-GFP) at a multiplicity of infection (MOI) of 10 was observed in approximately 90% of the cells. The optimization of transfection in LS174T and SW620 was determined in the same way. LS174T and SW620 were infected with 50 and 20 MOI, respectively. Viral infection was allowed to proceed for a further 24–48 hr.
Microarray analysis
Microarray experiments were performed as previously described.30 Samples for the microarray experiments were prepared as follows: both HCT116 p53+/+ andHCT116 p53-/- cells were infected with 10 MOI adeno-ING2 or same amount of adeno-GFP. Cells were harvested at 48 hr after the transfection. The data analysis was performed using the BRB Array Tools (http://linus.nci.nih.gov/BRB-ArrayTools.html). The basic raw data and derived ratio measurements were then uploaded to the National Cancer Institute Micro-Array Database system, which provides the bioinformatics and analysis tools necessary for the interpretation of gene expression data.
ELISA
To analyze NF-κB binding activity, NF-κB, p65 ELISA kit was purchased from Assay designs (Ann Arbor, MI) and performed according to the manufacturer’s protocol. The assay uses streptavidin-coated plates with bound NF-κB biotinylated-consensus sequence to capture only the active form of NF-κB. The nuclear extracts of all cell lines (each 1 μg) were applied for this experiment.
MMP13 in the culture supernatants was measured by a sandwich enzyme-linked immunoassay using the MMP-13 ELISA Kit (Calbiochem) following the manufacturer’s instructions.
Knock down of p65 and ING2 expression using siRNA
siRNA oligonucleotides for NF-κB p65 (sc-29410) was purchased from Santa Cruz Biotechnology. Three kinds of siRNAs were prepared for knocking down of ING2. The ING2 siRNA sequences were designed as the following: siRNA ING2-1, 5′-ACAUGCAGAGGAACGUGUCUGUGCU-3′; ING2-2, 5′-ACA CAAAUGCUCGAAUUGGUGGAAA-3′; and ING2-3, 5′-ACAUACUGCUUAUGCAACCAAGUGU-3′. The scrambled siRNA sequences were 5′-ACAAAGUUCGCAAUUGGGUGACAAA-3′. For the siRNA transfection, 1.5 × 105 cells were plated in 6-well plates 1 day before the transfection. Cells were transfected with 40 nM of siRNA using Lipofectamine RNAiMAX (Invitrogen) following the manufacturer’s instructions and harvested at 72 hr.
Invasion assay
The cell invasion assay was conducted using a 24-well BD Bio-Coat Tumor Invasion System (BD Bioscience). Briefly, HCT116 cells were infected with either adeno-GFP or adeno-ING2 for 24 hr. Cells were harvested, seeded (5 × 103) into the top chamber and incubated at 37°C for 22 hr. The cells were post-stained with 4 μg/ml of Calcein-AM (Molecular Probes, Carlsbad, CA) in Hank’s buffered salt solution at 37°C, 5% CO2 for 1 hr. The labeled cells that invaded the BD Matrigel Matrix and passed through the pores of the BD FluoroBlok membrane were detected by fluorescence spectrometry (Victor II, Perkin—Elmer Life and Analytical Sciences, Boston, MA).
Results
ING2 mRNA transcripts are increased in human colon cancer tissue
Because previous report showed that ING2 mRNA transcripts were increased in colon cancer,2 we first examined the expression of ING2 mRNA in human colorectal cancers by the conventional RT-PCR. The ING2 mRNA level was increased in 3 out of 5 colon cancer samples, compared to matched noncancer colon tissue (Fig. 1a, upper panel). We further performed the real-time RT-PCR using the additional 34 colorectal cancers. β-actin was used as an internal control. The ING2 mRNA level in cancer tissue was upregulated when compared to nonmalignant mucosa, and the difference was statistically significant at p < 0.00001. In 18 of the 34 cases (45%), ING2 mRNA expression in cancer tissue was more than twice as high as the expression in corresponding noncancerous tissue (Fig. 1a, lower panel). This association was observed more prominently in patients with early stages of cancers (I and II) than in the advanced stages (III and IV) (Fig. 1a, lower panel). To investigate the distribution of ING2 mRNA expression in colon cancer tissue, in situ hybridization was performed using a RNA probe specific for ING2 transcripts. Figure 1b-a showed that ING2 mRNA expression, which was detected by the antisense probe, was enhanced in cancer tissue compared to noncancerous tissue at low magnification. More specifically, ING2 mRNA expression was found to be upregulated in cancer cells (Fig. 1b-b and Supporting Information Fig. S1), while it was undetectable in noncancerous mucosa and stroma (Fig. 1b-c and Supporting Information Fig. S1). No staining was observed in cancer cells using the sense probe as a negative control (Fig. 1b-d).
Figure 1.

ING2 mRNA is upregulated in colon cancer. (a) Upper panel, RT-PCR analysis of ING2 mRNA in 5 colon cancer samples, which were purchased from BD Bioscience. β-actin was used as the internal control. Lower panel, Results of real-time RT-PCR analyses of ING2 mRNA levels of in 34 human colon cancer tissues and corresponding nonmalignant mucosa. Cancerous tissues (Ca) and nonmalignant mucosa (N) were pre-pared from the same patient. Paired t test was performed to ascertain statistical significance between the amount in cancer tissue and in nonmalignant mucosa. The ING2 and β-actin expression vector were used for calculating the copy number. Y-axis shows the copy number of ING2 per 10,000 copies of β-actin. (b) Representative staining of ING2 mRNA by in situ hybridization in human colon cancer tissues (a, b and d) and nonmalignant mucosa (c). (a) included both cancerous tissues (Ca) and nonmalignant mucosa (N). The anti-sense probe (a, b and c) or sense probe (d) were used for detecting the ING2 mRNA transcripts. Bar, 200 μm (a), 100 μm (b,c and d).(c) Typical examples of immunohistochemical localization of ING2 in colon cancer cells and surrounding nonmalignant colonic epithelia. ×40; H&E staining. The cases with upregulated ING2 mRNA in cancer tissues (a and b) and the case with no alteration of ING2 mRNA level was seen between cancer tissues and normal tissues (c). Ca, cancer cells; N, nonmalignant colonic epithelial cells. (d) Upper panel, RT-PCR analysis of ING2 mRNA in 8 colon cancer cell lines. The ING2 expression was normalized by β-actin. Lower panel, ING2 protein expression was determined by western blotting using total cell lysates prepared from 8 colon cancer cell lines. Thirty-five μg were applied for the gel. (e) The distribution of ING2 protein in colon cancer cells was observed by western blotting. α-lamin A/C and β-actin were used as control of nuclear fractions and cytoplasmic cell fractions, respectively. C, cytoplasm; N, nuclear extracts.
We further performed immunohistochemical staining to examine ING2 protein expression in colon cancer. In the case with highly upregulated ING2 mRNA, strong expression of ING2 was detected in both nuclear and cytoplasm of cancer cells compared to normal mucosa (Fig. 1c-a and b). No staining was observed in the case that ING2 mRNA level was not altered in cancer tissues and normal mucosa (Fig. 1c-c).
The ING2 mRNA level correlates with protein expression in cultured human colon cancer cell lines
The ING2 mRNA level in 8 colon cancer cell lines was determined by the real-time RT-PCR and normalized with β-actin mRNA transcripts. The ING2 mRNA level was varied depending on the cell lines (Fig. 1d, upper panel). In 3 of those cell lines (DLD-1, RKO and SW837), ING2 mRNA expression was relatively higher than other cell lines. The ING2 mRNA level in WiDr cells was the lowest among those cell lines. We further investigated whether ING2 mRNA expression correlated with ING2 protein expression. ING2 protein expression was correlated with the mRNA level in these cell lines (Fig. 1d, lower panel, and Supporting Information Fig. S2). The localization of endogenous ING2 protein in these colon cancer cell lines was predominantly in the nucleus (Fig. 1e).
NF-κB activation is associated with ING2 expression in colon cancer
To explore the mechanism of the ING2 regulation in colon cancer, we analyzed the ING2 promoter regulatory region. A putative NF-κB consensus binding sequence was found around 130 bp upstream from the translational start site of the ING2 regulatory region (Supporting Information Table S1). We first investigated the association with ING2 and NF-κB binding activity using colon cancer cell lines by ELISA assay. The level in DLD-1, RKO and SW837 cells was much stronger than in other cell lines (Fig. 2a). Next, phospho-p65 expression was examined by western blotting since phospho-p65 was used as the marker of activated NF-κB.31,32 Phospho-p65 expression was detected in almost all of the colon cancer cell lines (Supporting Information Fig. S3). The expression level was correlated with the result of ELISA assay in the cell lines. The level of ING2 mRNA was similar to levels of activated NF-κB as measured by binding assays.
Figure 2.

NF-κB activation positively regulates ING2 expression. (a) NF-κB binding activity of colon cancer cell lines was measured using NF-κB, p65 ELISA kit. Chemiluminescent results were detected using a luminometer. Columns, average of 3 independent experiments; Bars, SD. (b) The ING2 mRNA expression in RKO and SW837 cells, which were treated with either 2.5 μM MG132 or 100 μM wedelolactone or DMSO for 3 and 6 hr, was analyzed by realtime RT-PCR. β-actin mRNA transcripts were used as a internal control. (c) Upper panel, Phospho-p65 and p65 expression was determined by western blotting in HCT116 and WiDr cells, which were treated with 3 μM doxorubicin for 0.5, 1 and 2 hr. Lower panel, Realtime RT-PCR was performed for detecting the ING2 mRNA expression in HCT116 and WiDr cells, which were treated with 3 μM doxorubicin for 6 and 12 hr. The ING2 mRNA level was normalized using β-actin mRNA transcripts. Columns, average of 3 independent experiments; Bars, SD. (d) Upper panel, Knockdown of p65 expression was performed by siRNA technology. RKO and SW837 cells were treated with the oligonucleotides for siRNA experiments (p65 siRNA). Cells were transfected with 40 nM of siRNA and incubated for 72 hr. A random sequence control (control siRNA) was used as a control. p65 expression was determined by western blotting. β-actin was probed as an internal control. Lower panel, Realtime RT-PCR was performed for detecting ING2 mRNA transcripts using the same samples as used for the upper panel experiment. ING2 expression was normalized by β-actin mRNA transcripts. Columns, average of 3 independent experiments; Bars, SD.
When RKO and SW837 cells were treated with either MG132 or wedelolactone, which were potent NF-κB inhibitors,33,34 the ING2 mRNA level in both cell lines was remarkably decreased in a time-dependent manner (Fig. 2b). It is well-known that a high concentration of doxorubicin activates NF-κB.35 Therefore, HCT116 and WiDr cells, which showed the relatively low expression of ING2, were treated with 3 μM doxorubicin. Phospho-p65 expression was increased quickly under this condition (Fig. 2c, upper panel), and ING2 mRNA expression was upregulated in a time-dependent manner (Fig. 2c, lower panel).
We further examined a knock down effect of p65 on ING2 expression using p65 siRNA oligonucleotides. p65 expression was reduced by treatment with p65 siRNA oligonucleotides in both RKO and SW837 cells (Fig. 2d, upper panel). ING2 mRNA level was suppressed by knock down of p65 in these cell lines (Fig. 2d, lower panel).
NF-κB is bound to the ING2 promoter region
To analyze the ability of NF-κB to bind the ING2 promoter regulatory region, EMSAs were preformed. Biotin-labeled, double-stranded oligonucleotides including the putative NF-κB site in the ING2 promoter regulatory region were generated to examine the binding with NF-κB. The putative NF-κB site in ING2 interacted with p50, as well as typical NF-κB consensus oligonucleotides (Fig. 3a). This binding was completely inhibited by the 100-fold excess unlabeled oligonucleotides as a competitor and not inhibited by another transcriptional sequence of CREB protein. Next, we prepared the nuclear extracts from HCT116 and RKO cells to confirm the NF-κB binding to the ING2 regulatory region. The shifted-bands were detected in the putative NF-κB site in the ING2 regulatory region (Fig. 3b). Each antibody to p50 and p65 generated a super-shifted complex, further confirming that the binding was specific. Moreover, when the unlabelled double-stranded oligonucleotides corresponding to the ING2 regulatory region were used as competitors, the formation of complexes was inhibited, supporting the specificity of the DNA-NF-κB complexes for the putative site.
Figure 3.

NF-κB bounds to ING2 promoter and regulates ING2 expression. (a) Electrophoretic mobility-shift assay (EMSA) was carried out using biotinated oligonucleotides generated from NF-κB consensus DNA-binding sequence (consensus) and the candidate of NF-κB consensus DNA-binding sequence on the ING2 regulatory region (ING2). Recombinant p50 were used for detecting a shifted band. Each unlabeled oligonucleotides (100-fold excess), which were described in the Material and Methods section, was used as a competitor. (b) The biotinated oligonucleotides of the consensus NF-κB sequence in the ING2 regulatory region was analyzed for the ability to form a super-shifted complex with DNA binding proteins in nuclear extracts from HCT116 and RKO cells. Anti-p50 and anti-p65 antibodies were incubated with the nuclear extracts. Unlabeled oligonucleotides (100-fold excess) of the consensus NF-κB sequence in the ING2 regulatory region was used as a competitor. (c) Three kinds of luciferase constructs using pGL-3 basic vector were designed as A, B and C. Each 1.6 μg of a pGL-3 luciferase reporter and 0.16 μg of Renilla luciferase assay vector pRL were cotransfected into RKO and WiDr cells. The firefly luciferase activity was measured and normalized by Renilla luciferase activity, 24 hr later. Columns, Average of relative luciferase activity in 3 independent experiments; bars, SD.
Next, regulation of ING2 expression though binding to the putative NF-κB responsive element (NRE) in ING2 promoter was examined by luciferase assay using 3 constructs including different length of ING2 promoter region (Fig. 3c). The longest construct named “A” (−1,251 to +140) included the putative NRE (NRE: −138 to −128) with extra 1,113 bp upstream sequence of ING2 promoter. The construct “B” (−162 to +140) included the putative NRE, but not the extra upstream sequence, and the construct “C” (+64 to +140) did not include the putative NRE. In RKO cells, which have high expression of phospho-p65 (Fig. 2a), promoter activity of construct A and construct B, which have the putative NRE sequence, was almost the same and ~4 times stronger than that of construct C, which did not include the putative NRE sequence (Fig. 3c). The promoter activity of all constructs was very low in WiDr cells, which have low NF-κB expression (Fig. 2a), indicating that NF-κB binds to the putative NRE in ING2 promoter and transactivate its expression.
ING2 regulates MMP13 expression
To investigate genes that are regulated by ING2 in colorectal cancer, expression profiles generated by microarray analysis in control virus infected cells and adeno-ING2 infected cells were compared. ING2 protein was highly expressed in the adeno-ING2-infected cells (Fig. 4a, left panel) and the overexpression of ING2 did not affect cell growth and morphology in HCT116 p53+/+ and p53-/- cells (Fig. 4a, right panel). As the result of microarray analysis, matrix metalloproteinase 13 (MMP13) was upregulated in the ING2-overexpressed cells (Supporting Information Table S2). The microarray result was verified by real-time RT-PCR. The MMP13 mRNA level was increased at similar level in both adeno-ING2-infected HCT116 p53+/+ and p53-/- cells compared to adeno-GFP expressed cells, though p53+/+ cells exhibited modest changes in gene expression in response to ING2 but the p53-/- cells exhibited much greater effects in the microarray data (Fig. 4b). To confirm this finding in other cell lines, we further examined 2 other colon cancer cell lines, SW620 and LS174T, in which the ING2 expression was relatively low. MMP13 mRNA expression was increased by adeno-ING2 infection in both SW620 and LS174T. Next, we investigated the production of MMP13 protein using ELISA to evaluate the correlation with the mRNA induction and its protein level. The amount of MMP13 protein was increased in the ING2-overexpressed cells (Fig. 4c).
Figure 4.

The ING2 expression is positively associated with the MMP13 expression. (a) Left, ING2 protein expression in adeno-GFP or adeno-ING2 infected HCT116 cells was determined by western blotting. β-actin was probed as an internal control. Right, Microscopic photographs of HCT116 p53+/+ (a, b and c) and p53-/- (d, e and f) infected with adeno-ING2 (10MOI) for 24 hr (b and e) and 48 hr (c and f). a and d showed the cells before the infection. (b) The MMP13 mRNA transcripts in ING2 overexpressing cells were analyzed by realtime RT-PCR. Adeno-ING2 or adeno-GFP were exposed to the colon cancer cell lines, HCT116 p53+/+ (10MOI), HCT116 p53-/- (10MOI), SW620 (20MOI) and LS174T (50MOI) for 48 hr. The expression was normalized by β-actin mRNA transcripts. Columns, average of 3 independent experiments; Bars, SD. (c) MMP13 protein level was determined by ELISA. The supernatants were collected from the cell treated with adeno-ING2 or adeno-GFP. Columns, average of 3 independent experiments; Bars, SD. (d) Upper panel, Knockdown of ING2 expression was performed by siRNA technology. HCT116 and SW837 cells were treated with 3 different oligonucleotides for siRNA experiments (ING2 siRNA 1, 2 and 3). Cells were transfected with 40 nM of siRNA and incubated for 72 hr. A random sequence control (ING2 siRNA—control) was used as a control. ING2 expression was determined by western blotting. β-actin was probed as an internal control. Lower panel, Realtime RT-PCR was performed for detecting MMP13 mRNA transcripts using the same samples as earlier. MMP13 expression was normalized by β-actin mRNA transcripts. Columns, average of 3 independent experiments; Bars, SD. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
To further investigate the regulation of MMP13 expression by ING2, we performed the knocking down of ING2 expression in HCT116 and SW837 cells using the 3 different ING2 siRNA oligonucleotides. ING2 protein expression was remarkably decreased by the ING2 siRNA treatments at 72 hr after the transfection (Fig. 4d, upper panel). Under this condition, the MMP13 mRNA transcripts were also consistently downregulated in all of the ING2 siRNA-treated cells (Fig. 4d, lower panel).
MMP13 expression was enhanced by the combination with ING2 and HDAC1
Previous reports have shown that ING2 interacted with mSin3A and HDAC1, suggesting that ING2 was associated with chromatin remodeling.9-11 To investigate whether the association between ING2 and these molecules affects transcriptional regulation of MMP13, we first confirmed the interaction of these molecules by immunoprecipitation method. The expression of mSin3A and HDAC1 was associated as estimated by IP-western assays in ING2-overexpressed 293 cells, while no band was detected in control cells (Fig. 5a). We then examined MMP13 expression when ING2, HDAC1 and/or mSin3A were overexpressed. ING2 or HDAC1 overexpression could induce the MMP13 mRNA expression, while MMP13 expression was not changed in mSin3A-overexpressed cells (Fig. 5b). Furthermore, the MMP13 mRNA level was synergistically increased by the combination of ING2 and HDAC1 overexpression. Although mSin3A overexpression was not able to induce the MMP13 expression, the combination of ING2 and mSin3A could induce the MMP13 expression synergistically (Fig. 5b).
Figure 5.

MMP13 expression is enhanced by the combination of ING2 with either HDAC1 or mSin3A. (a) Immunoprecipitation (IP) was performed using anti-FLAG, anti-HDAC1, or anti-mSin3A antibody for confirming a previously reported physical association between ING2, HDAC1 and mSin3A. Cell lysate was prepared from 293 cells, to which FLAG-ING2 vector was transfected and incubated for 48 hr. (b) MMP13 mRNA expression was analyzed by realtime RT-PCR. The indicated amount of each vector was transfected to 293 cells using the Lipofectamine 2000 following manufacturer’s protocol. Cells were harvested at 48 hr posttransfection. MMP13 expression was normalized by β-actin mRNA transcripts. Columns, average of 3 independent experiments; Bars, SD.
MMP13 mRNA expression correlates with ING2 mRNA in colon cancer
On the basis of the finding that ING2 induces MMP13 expression, we investigated the correlation between ING2 and MMP13 mRNA levels in colon cancer. MMP13 mRNA expression was positively correlated with the ING2 mRNA in the 34 colorectal cancers, which were used for Figure 1b (Fig. 6; p = 0.0002).
Figure 6.
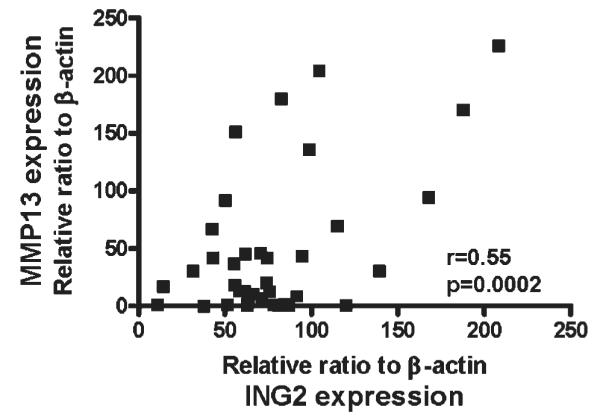
MMP13 expression is correlated with ING2 expression. Both ING2 and MMP13 mRNA levels in colon cancer tissue were determined by realtime RT-PCR. The colon cancer samples were same with Figure 1b. Both ING2 and MMP13 expression were normalized by β-actin mRNA transcripts. Pearson’s correlation analysis was performed.
ING2-induced MMP13 increases in vitro cancer cell invasion
As MMP13 is implicated in cancer invasion and metastasis,36 we performed the invasion assay using the ING2-overexpressing cells. The ability of invasion was significantly increased in the ING2-overexpressing cells compared to the control cells. When cells were treated with the MMP13 inhibitor, the ability was attenuated in the ING2-overexpressing cells (Fig. 7).
Figure 7.
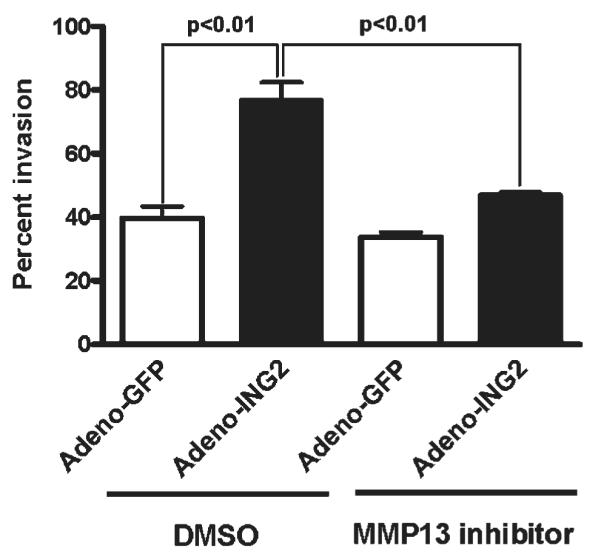
MMP13 expression is associated with tumor invasion. Using the HCT116 cells infected with adenoviral constructs expression either GFP or ING2 described in Figure 3, a cell invasion assay was performed using a 24-well BD BioCoat Tumor Invasion System as described in Material and Methods. Statistical analysis was performed by Student’s t-test. Columns, average of 3 independent experiments; Bars, SD.
Discussion
We first investigated the ING2 expression using colon cancer samples. In approximately half of all the cases, the ING2 mRNA level was remarkably increased in the cancer tissue compared to the adjacent noncancerous tissue. These results were consistent with a previous report.2 Moreover, we showed that ING2 mRNA was highly expressed in carcinoma cells, when compared to stroma cells. In these cases that ING2 mRNA level was increased in cancer tissues, ING2 protein expression in cancer tissues was also higher than in normal tissues. When we examined the correlation between the ING2 protein level and its mRNA level using colon cancer cell lines, ING2 protein expression was observed predominantly in nuclear and positively correlated with its mRNA expression level. It has been reported that ING2 expression was reduced in hepatocellular carcinoma and melanoma.27,37 However, it remains unclear why ING2 expression is downregulated in hepatocellular carcinoma and melanoma. The deletion of ING2 and the epigenetic mechanism may be related with the alteration in some of organs. The gene regulation and functions may be different depending on the interacting proteins and each organ. ING2 may have multiple functions like TGF-beta in cancer.
To understand why the ING2 mRNA transcripts were upregulated in cancer cells, we analyzed the ING2 promoter region to know whether there was a consensus DNA binding site of cancer-associated transcriptional factors. A putative NF-κB binding site was detected on the promoter. Indeed, this sequence was a complete match with a basic NF-κB consensus sequence.38 It is well-known that NF-κB expression is activated in several cancers including colon cancer and is antiapoptotic.39-43 We investigated the association between ING2 mRNA expression and NF-κB binding activity by ELISA assay and phospho-p65 expression, indicating that the level of ING2 mRNA was correlated with NF-κB binding activity. We further demonstrated that ING2 expression was altered by various stimulations that affected NF-κB activation or suppression. The ING2 mRNA expression was increased by doxorubicin treatment in HCT116 and WiDr cells, and was remarkably decreased by MG132 and wedelolactone, potent NF-κB inhibitors, in RKO and SW837 cells. Knock down of p65 induced downregulation of ING2 expression. Moreover, we confirmed the direct association between ING2 expression and the NF-κB transcription factor using the 2 methods, EMSAs and luciferase assay. NF-κB specifically binds to the ING2 promoter region. Therefore, NF-κB activation may be one of the mechanisms that induce ING2 expression in colon cancer. We recently reported that ING2 expression was suppressed by p53 activation through the inhibition of Sp1 binding to the promoter region.8 Although luciferase activity in WiDr cells was independent with NF-κB binding, basal ING2 expression might be regulated by Sp1. ING2 is regulated by NF-κB or p53 through the ING2 promoter regions depending on the condition. There are similar genes that are suppressed by p53 and upregulated by NF-κB. Survivin and focal adhesion kinase (FAK), which are well known as cancer-related genes, also have the binding sites of both p53 and NF-κB on its promoter regions.44-47 The expression of these genes is suppressed by p53 and upregulated by either NF-κB, as well as ING2.
We hypothesized that ING2 might regulate genes involved with cell proliferation, cell adhesion and cell motility, leading us to focus on MMP13. Matrix metalloproteinases play a crucial role in tumor cell invasion and metastasis due to their ability to digest basement membrane and extracellular matrix components, thereby facilitating cell movement through connective tissues.48,49 MMP13 expression was upregulated in many types of cancers, including colon cancer.48,50-59 We demonstrated that ING2 overexpression induces MMP13 mRNA transcripts and protein expression, and the knocked-down ING2 attenuated MMP13 expression.
Previous reports have shown that ING2 interacts with HDAC1 and mSin3A, and has a potential ability to bind to the H3K4me3, indicating that ING2 plays an essential role for chromatin remodeling.9-15 Hence, ING2 may have a dynamic role in regulating a subset of genes. Based on this concept and our microarray analysis of cells overexpressing ING2, we investigated whether these ING2 complexes could affect MMP13 expression. The combination with ING2 and HDAC1 or mSin3A overexpression remarkably enhances MMP13 expression, when compared to only ING2 overexpression. Although HDAC1 and mSin3A are generally corepressors,60 our novel results indicated that when complexed with ING2, HDAC1 or mSin3A can enhance MMP13 expression. It has been reported that HDAC1 serves as a coactivator for the gluco-corticoid receptor, and this activator function is dynamically modulated by acetylation.61 ING proteins including ING2 have function as critical regulators of chromatin acetylation.11 Moreover, SIRT1 is recruited by ING proteins and inhibits R2-associated mSin3A/HDAC1 transcriptional repression activity.62 These findings suggest that mSin3A/HDAC1 complexes function not only as a corepressor but also as a coactivator with interacting molecules such as ING2, which may modify HDAC1 complexes through the acetylation and the recruitment of SIRT1. Therefore, our novel finding that MMP13 expression was induced by the complexes of ING2 and mSin3A/HDAC1 could be reasonable.
The MMP13 mRNA level is upregulated in the colon carcinoma cells.52,53 We found a significant positive correlation with ING2 and MMP13 mRNA expression. MMP13 is regulated by multiple transcription factors.49,63 Our results are consistent with the hypothesis that ING2 increases MMP13 expression by chromatin remodeling, resulting in MMP13-dependent cancer invasion.
Supplementary Material
Acknowledgements
The authors thank Mrs. Dorothea Dudek-Creaven and Dr. Tom Holroyd for editorial and Mrs. Karen MacPherson for bibliographic assistance.
Grant sponsors: NIH, NCI, CCR.
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer. J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Shimada Y, Saito A, Suzuki M, Takahashi E, Horie M. Cloning of a novel gene (ING1L) homologous to ING1, a candidate tumor suppressor. Cytogenet Cell Genet. 1998;83:232–5. doi: 10.1159/000015188. [DOI] [PubMed] [Google Scholar]
- 3.Garkavtsev I, Kazarov A, Gudkov A, Riabowol K. Suppression of the novel growth inhibitor p33ING1 promotes neoplastic transformation. Nat Genet. 1996;14:415–20. doi: 10.1038/ng1296-415. [DOI] [PubMed] [Google Scholar]
- 4.Garkavtsev I, Grigorian IA, Ossovskaya VS, Chernov MV, Chumakov PM, Gudkov AV. The candidate tumour suppressor p33ING1 cooperates with p53 in cell growth control. Nature. 1998;391:295–8. doi: 10.1038/34675. [DOI] [PubMed] [Google Scholar]
- 5.Feng X, Hara Y, Riabowol K. Different HATS of the ING1 gene family. Trends Cell Biol. 2002;12:532–8. doi: 10.1016/s0962-8924(02)02391-7. [DOI] [PubMed] [Google Scholar]
- 6.Nagashima M, Shiseki M, Miura K, Hagiwara K, Linke SP, Pedeux R, Wang XW, Yokota J, Riabowol K, Harris CC. DNA damage-inducible gene p33ING2 negatively regulates cell proliferation through acetylation of p53. Proc Natl Acad Sci USA. 2001;98:9671–6. doi: 10.1073/pnas.161151798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pedeux R, Sengupta S, Shen JC, Demidov ON, Saito S, Onogi H, Kumamoto K, Wincovitch S, Garfield SH, McMenamin M, Nagashima M, Grossman SR, et al. ING2 regulates the onset of replicative senescence by induction of p300-dependent p53 acetylation. Mol Cell Biol. 2005;25:6639–48. doi: 10.1128/MCB.25.15.6639-6648.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumamoto K, Spillare EA, Fujita K, Horikawa I, Yamashita T, Appella E, Nagashima M, Takenoshita S, Yokota J, Harris CC. Nutlin-3a activates p53 to both down-regulate inhibitor of growth 2 and up-regulate mir-34a, mir-34b, and mir-34c expression, and induce senescence. Cancer Res. 2008;68:3193–203. doi: 10.1158/0008-5472.CAN-07-2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pena PV, Davrazou F, Shi X, Walter KL, Verkhusha VV, Gozani O, Zhao R, Kutateladze TG. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature. 2006;442:100–3. doi: 10.1038/nature04814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi X, Hong T, Walter KL, Ewalt M, Michishita E, Hung T, Carney D, Peña P, Lan F, Kaadige MR, Lacoste N, Cayrou C, et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature. 2006;442:96–9. doi: 10.1038/nature04835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doyon Y, Cayrou C, Ullah M, Landry AJ, Côté V, Selleck W, Lane WS, Tan S, Yang XJ, Côté J. ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Mol Cell. 2006;21:51–64. doi: 10.1016/j.molcel.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 12.Loewith R, Meijer M, Lees-Miller SP, Riabowol K, Young D. Three yeast proteins related to the human candidate tumor suppressor p33 (ING1) are associated with histone acetyltransferase activities. Mol Cell Biol. 2000;20:3807–16. doi: 10.1128/mcb.20.11.3807-3816.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choy JS, Tobe BT, Huh JH, Kron SJ. Yng2p-dependent NuA4 histone H4 acetylation activity is required for mitotic and meiotic progression. J Biol Chem. 2001;276:43653–62. doi: 10.1074/jbc.M102531200. [DOI] [PubMed] [Google Scholar]
- 14.Vieyra D, Loewith R, Scott M, Bonnefin P, Boisvert FM, Cheema P, Pastyryeva S, Meijer M, Johnston RN, Bazett-Jones DP, McMahon S, Cole MD, Young D, Riabowol K. Human ING1 proteins differentially regulate histone acetylation. J Biol Chem. 2002;277:29832–9. doi: 10.1074/jbc.M200197200. [DOI] [PubMed] [Google Scholar]
- 15.Kuzmichev A, Zhang Y, Erdjument-Bromage H, Tempst P, Reinberg D. Role of the Sin3-histone deacetylase complex in growth regulation by the candidate tumor suppressor p33(ING1) Mol Cell Biol. 2002;22:835–48. doi: 10.1128/MCB.22.3.835-848.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T. Active genes are trimethylated at K4 of histone H3. Nature. 2002;419:407–11. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- 17.Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ, III, Gingeras TR, Schreiber SL, Lander ES. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–81. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 18.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–12. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 19.Feng X, Bonni S, Riabowol K. HSP70 induction by ING proteins sensitizes cells to tumor necrosis factor alpha receptor-mediated apoptosis. Mol Cell Biol. 2006;26:9244–55. doi: 10.1128/MCB.01538-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gozani O, Karuman P, Jones DR, Ivanov D, Cha J, Lugovskoy AA, Baird CL, Zhu H, Field SJ, Lessnick SL, Villasenor J, Mehrotra B, et al. The PHD finger of the chromatin-associated protein ING2 functions as a nuclear phosphoinositide receptor. Cell. 2003;114:99–111. doi: 10.1016/s0092-8674(03)00480-x. [DOI] [PubMed] [Google Scholar]
- 21.Kaadige MR, Ayer DE. The polybasic region that follows the plant homeodomain zinc finger 1 of Pf1 is necessary and sufficient for specific phosphoinositide binding. J Biol Chem. 2006;281:28831–6. doi: 10.1074/jbc.M605624200. [DOI] [PubMed] [Google Scholar]
- 22.Clarke JH, Letcher AJ, D’santos CS, Halstead JR, Irvine RF, Divecha N. Inositol lipids are regulated during cell cycle progression in the nuclei of murine erythroleukaemia cells. Biochem J. 2001;357:905–10. doi: 10.1042/0264-6021:3570905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roberts HF, Clarke JH, Letcher AJ, Irvine RF, Hinchliffe KA. Effects of lipid kinase expression and cellular stimuli on phosphatidylinositol 5-phosphate levels in mammalian cell lines. FEBS Lett. 2005;579:2868–72. doi: 10.1016/j.febslet.2005.04.027. [DOI] [PubMed] [Google Scholar]
- 24.Bunce MW, Gonzales ML, Anderson RA. Stress-ING out: phosphoinositides mediate the cellular stress response. Sci STKE. 2006;360:46. doi: 10.1126/stke.3602006pe46. [DOI] [PubMed] [Google Scholar]
- 25.Jones DR, Bultsma Y, Keune WJ, Halstead JR, Elouarrat D, Mohammed S, Heck AJ, D’santos CS, Divecha N. Nuclear PtdIns5P as a transducer of stress signaling: an in vivo role for PIP4Kbeta. Mol Cell. 2006;23:685–95. doi: 10.1016/j.molcel.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 26.Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nat Rev Cancer. 2003;3:276–85. doi: 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]
- 27.Zhang HK, Pan K, Wang H, Weng DS, Song HF, Zhou J, Huang W, Li JJ, Chen MS, Xia JC. Decreased expression of ING2 gene and its clinicopathological significance in hepatocellular carcinoma. Cancer Lett. 2008;261:183–92. doi: 10.1016/j.canlet.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 28.Hofseth LJ, Khan MA, Ambrose M, Nikolayeva O, Xu-Welliver M, Kartalou M, Hussain SP, Roth RB, Zhou X, Mechanic LE, Zurer I, Rotter V, et al. The adaptive imbalance in base excision-repair enzymes generates microsatellite instability in chronic inflammation. J Clin Invest. 2003;112:1887–94. doi: 10.1172/JCI19757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reddel RR, Ke Y, Kaighn ME, Malan-Shibley L, Lechner JF, Rhim JS, Harris CC. Human bronchial epithelial cells neoplastically transformed by v-Ki-ras: altered response to inducers of terminal squamous differentiation. Oncogene Res. 1988;3:401–8. [PubMed] [Google Scholar]
- 30.Staib F, Robles AI, Varticovski L, Wang XW, Zeeberg BR, Sirotin M, Zhurkin VB, Hofseth LJ, Hussain SP, Weinstein JN, Galle PR, Harris CC. The p53 tumor suppressor network is a key responder to microenvironmental components of chronic inflammatory stress. Cancer Res. 2005;65:10255–64. doi: 10.1158/0008-5472.CAN-05-1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu J, Nakano H, Sakurai H, Colburn NH. Insufficient p65 phosphorylation at S536 specifically contributes to the lack of NF-kappaB activation and transformation in resistant JB6 cells. Carcinogenesis. 2004;25:1991–2003. doi: 10.1093/carcin/bgh198. [DOI] [PubMed] [Google Scholar]
- 32.Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005;30:43–52. doi: 10.1016/j.tibs.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 33.Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell. 1994;78:773–85. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 34.Kobori M, Yang Z, Gong D, Heissmeyer V, Zhu H, Jung YK, Gakidis MA, Rao A, Sekine T, Ikegami F, Yuan C, Yuan J. Wedelolactone suppresses LPS-induced caspase-11 expression by directly inhibiting the IKK complex. Cell Death Differ. 2004;11:123–30. doi: 10.1038/sj.cdd.4401325. [DOI] [PubMed] [Google Scholar]
- 35.Das KC, White CW. Activation of NF-kappaB by antineoplastic agents. Role of protein kinase C. J Biol Chem. 1997;272:14914–20. doi: 10.1074/jbc.272.23.14914. [DOI] [PubMed] [Google Scholar]
- 36.Ala-Aho R, Johansson N, Baker AH, Kahari VM. Expression of collagenase-3 (MMP-13) enhances invasion of human fibrosarcoma HT-1080 cells. Int J Cancer. 2002;97:283–9. doi: 10.1002/ijc.1619. [DOI] [PubMed] [Google Scholar]
- 37.Lu F, Dai DL, Martinka M, Ho V, Li G. Nuclear ING2 expression is reduced in human cutaneous melanomas. Br J Cancer. 2006;95:80–6. doi: 10.1038/sj.bjc.6603205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baeuerle PA. The inducible transcription activator NF-kappa B: regulation by distinct protein subunits. Biochim Biophys Acta. 1991;1072:63–80. doi: 10.1016/0304-419x(91)90007-8. [DOI] [PubMed] [Google Scholar]
- 39.Rayet B, Gelinas C. Aberrant rel/nfkb genes and activity in human cancer. Oncogene. 1999;18:6938–47. doi: 10.1038/sj.onc.1203221. [DOI] [PubMed] [Google Scholar]
- 40.Yu HG, Yu LL, Yang Y, Luo HS, Yu JP, Meier JJ, Schrader H, Bastian A, Schmidt WE, Schmitz F. Increased expression of RelA/nuclear factor-kappa B protein correlates with colorectal tumorigenesis. Oncology. 2003;65:37–45. doi: 10.1159/000071203. [DOI] [PubMed] [Google Scholar]
- 41.Evertsson S, Sun XF. Protein expression of NF-kappaB in human colorectal adenocarcinoma. Int J Mol Med. 2002;10:547–50. [PubMed] [Google Scholar]
- 42.Lind DS, Hochwald SN, Malaty J, Rekkas S, Hebig P, Mishra G, Moldawer LL, Copeland EM, 3rd, Mackay S. Nuclear factor-kappa B is upregulated in colorectal cancer. Surgery. 2001;130:363–9. doi: 10.1067/msy.2001.116672. [DOI] [PubMed] [Google Scholar]
- 43.Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–10. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 44.Kawakami H, Tomita M, Matsuda T, Ohta T, Tanaka Y, Fujii M, Hatano M, Tokuhisa T, Mori N. Transcriptional activation of survivin through the NF-kappaB pathway by human T-cell leukemia virus type I tax. Int J Cancer. 2005;115:967–74. doi: 10.1002/ijc.20954. [DOI] [PubMed] [Google Scholar]
- 45.Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J Biol Chem. 2002;277:3247–57. doi: 10.1074/jbc.M106643200. [DOI] [PubMed] [Google Scholar]
- 46.Golubovskaya V, Kaur A, Cance W. Cloning and characterization of the promoter region of human focal adhesion kinase gene: nuclear factor kappa B and p53 binding sites. Biochim Biophys Acta. 2004;1678:111–25. doi: 10.1016/j.bbaexp.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 47.Golubovskaya VM, Finch R, Kweh F, Massoll NA, Campbell-Thompson M, Wallace MR, Cance WG. p53 regulates FAK expression in human tumor cells. Mol Carcinog. 2008;47:373–82. doi: 10.1002/mc.20395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Freije JM, Diez-Itza I, Balbin M, Sánchez LM, Blasco R, Tolivia J, López-Otín C. Molecular cloning and expression of collagenase-3, a novel human matrix metalloproteinase produced by breast carcinomas. J Biol Chem. 1994;269:16766–73. [PubMed] [Google Scholar]
- 49.Giambernardi TA, Grant GM, Taylor GP, Hay RJ, Maher VM, McCormick JJ, Klebe RJ. Overview of matrix metalloproteinase expression in cultured human cells. Matrix Biol. 1998;16:483–96. doi: 10.1016/s0945-053x(98)90019-1. [DOI] [PubMed] [Google Scholar]
- 50.Balbin M, Pendas AM, Uria JA, Jimenez MG, Freije JP, Lopez-Otin C. Expression and regulation of collagenase-3 (MMP-13) in human malignant tumors. APMIS. 1999;107:45–53. doi: 10.1111/j.1699-0463.1999.tb01525.x. [DOI] [PubMed] [Google Scholar]
- 51.Hilska M, Roberts PJ, Collan YU, Laine VJ, Kössi J, Hirsimäki P, Rahkonen O, Laato M. Prognostic significance of matrix metalloproteinases-1, -2, -7 and -13 and tissue inhibitors of metalloproteinases-1, -2, -3 and -4 in colorectal cancer. Int J Cancer. 2007;121:714–23. doi: 10.1002/ijc.22747. [DOI] [PubMed] [Google Scholar]
- 52.Leeman MF, McKay JA, Murray GI. Matrix metalloproteinase 13 activity is associated with poor prognosis in colorectal cancer. J Clin Pathol. 2002;55:758–62. doi: 10.1136/jcp.55.10.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roeb E, Arndt M, Jansen B, Schumpelick V, Matern S. Simultaneous determination of matrix metalloproteinase (MMP)-7. MMP-1, -3, and -13 gene expression by multiplex. PCR in colorectal carcinomas. Int J Colorectal Dis. 2004;19:518–24. doi: 10.1007/s00384-004-0592-6. [DOI] [PubMed] [Google Scholar]
- 54.Mori D, Nakafusa Y, Miyazaki K, Tokunaga O. Differential expression of Janus kinase 3 (JAK3), matrix metalloproteinase 13 (MMP13), heat shock protein 60 (HSP60), and mouse double minute 2 (MDM2) in human colorectal cancer progression using human cancer cDNA microarrays. Pathol Res Pract. 2005;201:777–89. doi: 10.1016/j.prp.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 55.Bostrom PJ, Ravanti L, Reunanen N, Aaltonen V, Söderström KO, Kähäri VM, Laato M. Expression of collagenase-3 (matrix metalloproteinase-13) in transitional-cell carcinoma of the urinary bladder. Int J Cancer. 2000;88:417–23. [PubMed] [Google Scholar]
- 56.Elnemr A, Yonemura Y, Bandou E, Kinoshita K, Kawamura T, Takahashi S, Tochiori S, Endou Y, Sasaki T. Expression of collagenase-3 (matrix metalloproteinase-13) in human gastric cancer. Gastric Cancer. 2003;6:30–8. doi: 10.1007/s101200300004. [DOI] [PubMed] [Google Scholar]
- 57.Etoh T, Inoue H, Yoshikawa Y, Barnard GF, Kitano S, Mori M. Increased expression of collagenase-3 (MMP-13) and MT1-MMP in oesophageal cancer is related to cancer aggressiveness. Gut. 2000;47:50–6. doi: 10.1136/gut.47.1.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johansson N, Airola K, Grenman R, Kariniemi AL, Saarialho-Kere U, Kahari VM. Expression of collagenase-3 (matrix metalloproteinase-13) in squamous cell carcinomas of the head and neck. Am J Pathol. 1997;151:499–508. [PMC free article] [PubMed] [Google Scholar]
- 59.Airola K, Johansson N, Kariniemi AL, Kahari VM, Saarialho-Kere UK. Human collagenase-3 is expressed in malignant squamous epithelium of the skin. J Invest Dermatol. 1997;109:225–31. doi: 10.1111/1523-1747.ep12319441. [DOI] [PubMed] [Google Scholar]
- 60.Xu L, Glass CK, Rosenfeld MG. Coactivator and corepressor complexes in nuclear receptor function. Curr Opin Genet Dev. 1999;9:140–7. doi: 10.1016/S0959-437X(99)80021-5. [DOI] [PubMed] [Google Scholar]
- 61.Qiu Y, Zhao Y, Becker M, John S, Parekh BS, Huang S, Hendarwanto A, Martinez ED, Chen Y, Lu H, Adkins NL, Stavreva DA, Wiench M, Georgel PT, Schiltz RL, Hager GL. HDAC1 acetylation is linked to progressive modulation of steroid receptor-induced gene transcription. Mol Cell. 2006;22:669–79. doi: 10.1016/j.molcel.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 62.Binda O, Nassif C, Branton PE. SIRT1 negatively regulates HDAC1-dependent transcriptional repression by the RBP1 family of proteins. Oncogene. 2008;27:3384–92. doi: 10.1038/sj.onc.1211014. [DOI] [PubMed] [Google Scholar]
- 63.Leeman MF, Curran S, Murray GI. The structure, regulation, and function of human matrix metalloproteinase-13. Crit Rev Biochem Mol Biol. 2002;37:149–66. doi: 10.1080/10409230290771483. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.