

Abstract
Although Parkinson’s disease was first diagnosed nearly 200 years ago, its effective treatment still remains elusive for most of those diagnosed. The gold standard of treatment for most patients is 3,4-dihydroxy-l-phenylalanine. This drug works for most individuals early in the disease; however, resistant symptoms start to emerge after several years of treatment. There has been increased interest in finding novel therapies to help Parkinson’s disease patients. Such strategies may have the benefit of not only treating the symptomatic issues of the disorder, but might also offer promise in protecting dopaminergic neurons from further degeneration. One such target that is now receiving much attention from the scientific community is the metabotropic glutamate receptor mGluR4. In this article, we briefly review Parkinson’s disease and then recent work in the mGluR area, with a focus on the efforts being made toward finding and optimizing novel mGluR4 positive allosteric modulators (PAMs). Preclinically in rodent models, mGluR4 activation has offered much promise as a novel treatment of Parkinson’s disease. Additionally, the specific use of PAMs, rather than direct-acting agonists at the orthosteric glutamate site, continues to be validated as a viable treatment option for this target. It is anticipated that continued progress in this area will further our understanding of the potential of mGluR4 modulation as a novel symptomatic and potentially disease-modifying treatment for Parkinson’s disease.
Parkinson’s disease
Parkinson’s disease (PD) is a debilitating movement disorder with a lifetime risk of 2% (>1 million people in the USA are afflicted), making PD the second most common neurode-generative disease after Alzheimer’s disease [1]. Typical disease onset occurs in those aged 45 years or above, but early-onset PD can occur in those aged as young as 35 years [1–4]. PD was first described in 1817 by an English physician, James Parkinson, as a clinical condition he coined ‘shaking palsy’ or ‘paralysis agitans’. His initial observations, “involuntary tremulous motion, with lessened muscular power, in part not in action and even when supported; with a propensity to bend the trunk forwards and pass from a walking to a running pace; the senses and intellect being uninjured” are still the basis for modern clinical diagnosis [5]. PD is further defined by “the unilateral or asymmetric onset of bradykinetic rigid syndrome with resting tremor and a subsequent good response to an adequate dose of a dopaminergic agent given for a sufficient period” [1–4]. The three hallmark symptoms of PD are bradykinesia (slowness of movement), rigidity and tremor. We now know that, in addition to the pronounced motor symptoms (dopaminergic), PD patients suffer from significant cognitive impairment and emotional disturbances (some of which are nondopaminergic, i.e., cholinergic, adrenergic and serotonergic in nature) such as fatigue, depression, slowness in thinking (brady-phrenia) and sleep disorders [1–4,6,7]. Some of these are disease mediated, while others are the result of the dopamine (DA)-replacement therapy. Definitive diagnosis of PD occurs at postmortem analysis, where two distinctive features, the selective loss of dopaminergic neurons in the substantia nigra (typically 20% of the normal population) and the occurrence of Lewy bodies (intracellular inclusions composed of the proteinαare present [1–4,6,7].
The loss of mesencephalic dopaminergic neurons as the major preceptor of the motor disturbances in PD is strongly supported by animal studies [2–4,6–8]. When injected bilaterally into the substantia nigra, toxins such as 6-hydroxydopamine selectively destroy dopaminergic neurons and afford animals PD phenotypes [2–4,6–9]. Another toxin, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), rapidly passes the blood–brain barrier (BBB) and is oxidized, via monoamine oxidase (MAO), to 1-methyl-4-phenylpyridinium (MPP+) [10]. MPP+ selectively accumulates in dopaminergic neurons via the DA transporter, leading to dopaminergic cell death by interfering with mitochondrial function. MPTP-treated primates display a PD phenotype that mirrors the human condition [10]. Thus, these findings on the selective loss of dopaminergic neurons in the substantia nigra have led to the development of DA-replacement therapies for the treatment of PD.
Dopamine itself as a therapy for PD proved ineffective, as it does not cross the BBB. Pioneering work in DA-replacement therapy then focused on 3,4-dihydroxy-l-phenylalanine (1), a brain penetrant precursor of DA (Figure 1). However, due to a rapid decarboxylation prior to crossing the BBB, the librated vasoactive DA elicited severe adverse cardiovascular events. Groups at Merck and Roche independently solved this issue by combination therapy of L-DOPA with peripheral inhibitors of aromatic amino acid decarboxylase, such as (2), which provided increased DA levels in the CNS without cardiovascular side effects [3,4]. After the success of L-DOPA, direct-acting DA agonists were introduced to PD patients, including 3 (D2 agonist), 4 (D1/2 agonist), 5 and 6 (both D2/3 agonists). Another complimentary approach to elevate central DA levels focused on manipulations of the release, re-uptake and degradation of DA. This effort has resulted in the use of inhibitors of catabolic enzymes, such as MAO and catechol-O-methyl transferase (COMT). For example, 7 and 8 inhibit MAO and COMT, respectively, and are now used for the treatment of PD, often in conjunction with L-DOPA [3,4]. Despite these major advances in symptomatic therapy for the treatment of PD, DA-replacement strategies have significant problems [11,12]. First, there are ‘wearing off ’ and ‘on-off’ phenomena where DA-replacement therapy loses efficacy over time. Second, dsykinesias (grimacing, head bobbing and oscillatory rocking movements of the extremities) are observed in many patients. Third, long-term DA-replacement therapy causes psychotic symptoms and behavioral disturbances (hallucinations, paranoia, mania and anxiety). During the course of treatment, PD patients begin with low-dose L-DOPA or a DA agonist, doses of the drugs are gradually increased and then combinations of these agents with MAO and COMT inhibitors are often employed [2–4,6–9]. From onset of PD to death, typically a 20-year span, resistant symptoms begin to emerge at approximately 8–15 years, with cognitive decline settling in for the final 5 years of the disease [11,12]. In fact, there is discussion that DA-replacement therapy may actually accelerate disease progression through increased oxidative stress and damage, although it has not been definitively demonstrated that LDOPA is toxic to patients [9,13]. Owing to these limitations of L-DOPA therapy, particularly late in the disease and because L-DOPA alone does not correct the progressive neurodegeneration, alternative mechanisms are actively being sought that avoid direct modulation of the DA system.
Figure 1.

Commonly used agents for dopamine-replacement therapy: 3,4-dihydroxy-L-phenylalanine (1), benserazide (2), bromocriptine (3), pergolide (4), ropinole (5), pramipexole (7), selegeline (8) and entacapone (9).
Understanding basal ganglia circuitry provides new insights into potential pharmacological therapies for PD
The basal ganglia (BG) are a set of interconnected nuclei that play a key role in the control of movement. The primary input nucleus of the BG is the striatum (caudate, putamen and nucleus accumbens), which is innervated by the cortex and subcortical structures, such as the thalamus. These inputs are predominantly excitatory glutamatergic projections. The primary output nuclei of the BG in primates are the internal globus pallidus (GPi) and the related substantia nigra pars reticulata (SNr). In a simplified model, inhibitory GABAergic projections from the striatum project to the output nuclei through two major routes, a direct pathway and an indirect pathway [14–18]. The direct pathway projects directly to the GPi/SNr and inhibits activity in these output nuclei. The indirect pathway originates from a different population of GABAergic striatal neurons and projects to the external DAGPi (GPe) in primates. The GPe (referred to as DAGPi or GP in rodents and other nonprimates) sends GABAergic projections to the subthalamic nucleus (STN), which then projects to the GPi/SNr via excitatory glutamatergic projections. The opposing inhibitory and excitatory signals from the direct and indirect pathways, respectively, provide a delicate balance to the overall activity of the output nuclei (Figure 2). While this is a highly oversimplified model of BG circuitry [18–20], it provides a useful framework within which to study BG function.
Figure 2. Simplified model of basal ganglia circuitry in normal physiology and Parkinson’s disease.

Excitatory (glutamatergic) projections are shown by light arrows and inhibitory (GABAergic) projections are shown by dark arrows. (A) In the normal BG circuit, direct (striatum to GPi/SNr) and indirect (striatum to GPe) projections to the output nuclei are balanced by striatal dopaminergic tone. D1 receptor-containing neurons stimulate transmission through the direct pathway and D2 receptor-containing neurons inhibit transmission through the indirect pathway. These circuits converge at the GPi/SNr with a balance of inhibitory (via direct pathway) and excitatory (via indirect pathway) inputs to properly regulate inhibitory output to the thalamus. mGluR4 (circle) is localized at the synapse between the striatum and GPe (striatopallidal synapse) as well as synapses between the STN and SNc. (B) In the Parkinson’s BG circuit, the loss of striatal dopamine produces an imbalance in the direct and indirect pathways leading to too much inhibitory tone from the output nuclei. mGluR4-mediated decreases in GABA (striatopallidal synapse) and glutamate (STN–SNc synapse) release are hypothesized to restore balance to the output nuclei and possibly prevent further degeneration of SNc neurons via excitotoxic mechanisms. GPe: External segment of the globus pallidus; GPi: Internal segment of the globus pallidus; SNc: substantia nigra pars compacta; SNr: substantia nigra pars reticulata; STN: subthalamic nucleus.
The intricate balance of activity between the direct and indirect pathways is maintained, in part, by the modulation of striatal neurons by dopaminergic projections from the substantia nigra pars compacta (SNc). Release of DA from terminals of the nigrostriatal projections facilitates transmission through the direct pathway (via activation of D1 DA receptors) and inhibits transmission through the indirect pathway (via activation of D2 receptors) [23]. In this simplified model, the loss of dopaminergic input to the striatum leads to increased activation of the indirect pathway and decreased activity in the direct pathway, eventually resulting in increased and inappropriate inhibition of thalamocortical neurons (Figure 2B) [14,15].
Increased understanding of BG function has led to new strategies for treating PD. Recent advances in surgical interventions in PD, such as pallidotomy and deep-brain stimulation of the STN or GPi, have provided dramatic palliative benefit to PD patients and a refinement of the model of BG dysfunction in PD [2–8]. Excitingly, modulation of STN activity via deep-brain stimulation also as the potential to be disease modifying and this has been supported by preclinal primate studies [24], although it remains to be seen if these results will translate to PD patients [25,26]. Nevertheless, the success of these surgical manipulations suggests that pharmacological agents that correct dysfunctions in the indirect pathway circuit may provide substantial relief of motor symptoms in PD patients and could provide much needed disease-modifying treatments.
Metabotropic glutamate receptors: the role of mGluR4 in inhibiting striatopallidal neurotransmission
The metabotropic glutamate receptors (mGluRs) are members of class C of the G-protein-coupled receptors and possess an extracellular domain, called the Venus flytrap domain, which contains the glutamate binding site in addition to a seven transmembrane-spanning domain [27]. There have been eight mGluR subtypes identified in mammalian brain and these are classified into three groups based on sequence homology, G-protein-coupling profile and ligand specificity [28]. Group I mGluRs include mGluR1 and mGluR5, which couple via Gq to phospholipase C, Group II includes mGluR2 and mGluR3, and Group III includes mGluRs 4, 6, 7 and 8; both group II and group III mGluRs couple to inhibition of adenylyl cyclase when expressed in cell lines but also exert modulatory control of ion channels.
Due to the ability of mGluRs to exert a neuromodulatory role in the control of both glutamatergic and GABAergic neurotransmission, there has been much interest in developing novel mGluR ligands that can be used to treat a variety of neurological and psychiatric disorders. However, most ligands developed thus far bind to the highly conserved orthosteric glutamate binding site in the Venus flytrap domain [29]. Due to the high conservation of this binding site, development of compounds that are selective for individual mGluRs has proven to be a difficult endeavor. Despite this limitation, mGluR4 has received much attention as a therapeutic target for multiple indications [30–33].
Expression studies have demonstrated that mGluR4 is highly expressed at the striatopallidal synapse within the indirect pathway of the BG (Figure 2) [34,35]. In 2003, Valenti and coworkers described group III mGluR-mediated modulation of the striatopallidal synapse and demonstrated that reduction of GABA release at the striatopallidal synapse by a group III receptor agonist could be a potentially novel approach for the treatment of PD [36]. In this study, electrical stimulation in rat brain slices evoked GABAA-mediated inhibitory postsynaptic currents (IPSCs) in all three types of GP neurons. Importantly, the group III mGluR-selective agonist L-(+)-2-amino-4-phosphonobutyric acid (L-AP4)(10) inhibited these IPSCs by decreasing GABA release via a presynaptic mechanism of action. Moreover, L-AP4 exhibited a pharmacological profile consistent with activation at mGluR4. Significantly, this effect of L-AP4 on striatalpallidal transmission was absent in mGluR4 knock out mice, affording strong evidence that mGluR4 mediates this effect. Overall, these data indicated that activation of mGluR4 could decrease the excessive inhibition of the GPe that has been suggested to occur in PD. Valenti et al. also found that ICV injections of L-AP4 produced robust efficacy in both acute (haloperidol-induced catalepsy and resperine-induced akinesia) and chronic (forelimb asymmetry in unilateral 6-hydroxydopamine [6-OHDA] rats) PD models [36]. Additionally, further studies demonstrated that mGluR4 activation could affect excitatory neurotransmission from the STN to the SNc, suggesting that mGluR4 activation could reduce glutamate release onto dying dopaminergic (DA) neurons [37]. This work is further supported by other neuroprotection studies in rodents, demonstrating that L-AP4 can protect the nigrostriatal system against lesions induced by 6-hydroxydopamine [38]. Collectively, these data have suggested that selective activation of mGluR4 should be further explored as a potential new approach for the treatment of PD.
Many of the initial studies evaluating the function of group III mGluRs, including mGluR4, used the orthosteric agonist glutamate, 9, as the basis for ligand design. Prototypical group III agonists include 10 [39–42], 11 [43,44], 12 [45] or 13 [46–48] (Figure 3). Compound 13, aminocyclopentane, has been reported in the literature numerous times as (1S,3R,4S); however, this designation is incorrect as this molecule is meso and therefore the designation of carbon 1 as S cannot be assigned (the compound is superimposable with its mirror image). We have assigned the 3R and 4S for the sake of identifying whether the carboxylic acid groups are trans to the amino group (or cis). We would like to make sure all further use of this molecule adopts the correct terminology for an achiral (meso) compound (Figure 4). Although these compounds contain moieties that limit their development as true drug leads (multiple carboxylic acids, phospho-nate groups), these orthosteric agonists have provided some insight into the possible beneficial effects of modulating this group of mGluRs and, importantly, mGluR4. L-AP4 is a close analogue of L-glutamate and has agonist activity against all four of the group III mGluRs. [49]. L-AP4 has been demonstrated to be active in haloperidol-induced catalepsy and reserpine-induced akinesia models [36], two pharmacological models of PD, suggesting that that activation of group III mGluRs produces significant antiparkinsonian action. L-AP4 has also been examined in a chronic striatal DA-depletion model (unilateral 6-OHDA-lesioned rats) [36]. In this study, L-AP4 was as efficacious as L-DOPA, the prototypical anti-PD drug. Recently, L-AP4 has been reported to recover motor behaviors in various PD models, again substantiating potential neuroprotective effects induced by group III mGluR activation [50].
Figure 3.

Glutamate (9), L-AP4 (10), L-SOP (11), APCD (12), APCT (13) and other phosphonate-containing group III mGluR agonists.
Figure 4.

APCT-I (13).
In addition to L-AP4, other compounds have been extensively studied in antiparkinsonian rodent models (11–13). Compound 11 was demonstrated by MacInnes et al. in 2004 to reverse reserpine-induced akinesia via icv injection [43]. In addition to (12) was shown to exert a strong suppressive effect on amphetamine-induced rotational behavior of 6-OHDA-lesioned rats in a dose- and time-dependent manner following intrapallidal injection [45]. Lopez et al. demonstrated that intrapallidal injection of (13), had antiparkinsonian actions in both acute and chronic rat models of PD [46]. These pioneering studies suggest that activation of group III mGluRs should have a beneficial effect on PD. A number of analogues of (12) and APCD have been synthesized and tested (14–17); however, many of these compounds still suffer from issues with selectivity and CNS penetration (Figure 3) [51,52].
Discovery of the first positive allosteric modulator of mGluR4, (-)-PHCCC (18)
Due to the lack of selectivity of orthosteric ligands for the mGluRs, much effort has now been directed at identifying and examining the utility of compounds that act via allosteric sites on the receptor. This strategy has been successfully employed for other G-protein-coupled receptors, including mGluRs, such as mGluR 1, 2 and 5 [53]. These allosteric compounds can regulate receptor function in both positive and negative directions. Positive allosteric modulators (PAMs) have little to no effect on the receptor alone but can dramatically potentiate the effects of the endogenous ligand. Negative allosteric modulators antagonize the activity of orthosteric agonists; in contrast to antagonists that compete for binding of the orthosteric agonists, negative allosteric modulators block agonist activity in a noncompetitive fashion. In addition to greater receptor subtype selectivity, there are several additional advantages to developing PAMs over classical agonists as therapeutic agents. First, the effects of an allosteric modulator are saturable, such that no additional allosteric effect is possible once the allosteric sites are occupied, even in the presence of excessive dosage; in contrast, classical orthosteric agonists have no such ‘ceiling’ to their effects and overdose is an issue. Second, a true positive allosteric modulator will only exert its effects, that is, enhancing the agonist response, when the endogenous agonist (e.g., glutamate) is present, maintaining temporal and spatial activity of the endogenous ligand. Indeed, these two attributes suggest that PAMs may induce less receptor desensitization compared with classical agonists [29,53].
A breakthrough in the study of mGluR4 came with the discovery that (-)-N-phenyl-7-(hydroxyimino)cyclopropa[b]chromen-1acarboxamide (18), could potentiate mGluR4 function (Figure 5) [22,54]. (±)-PHCCC was first reported in the literature in 1996 by Annoura et al., along with CPCCOEt, a novel antagonist of mGluR1 and mGluR5 [55]. This group demonstrated that both (18) and (19) were potent antagonists of mGluR1 with IC50s of 23 and 10 μM, respectively. These compounds were also determined not to be partial agonists since there were no observed effects on basal Ca2+ mobilization at concentrations of 1–100 μM. In addition to mGluR1 antagonism, 100 μM CPCCOEt, completely inhibited glutamate-induced Ca2+ mobilization in mGluR5-CHO cells, leading the authors to surmise that these compounds were potent and selective group I mGluRs sub-type antagonists. In this study the authors also offered a glimpse into the structure–activity relationship (SAR) of 18 by noting that compounds lacking the cyclopropyl group or the oxime were inactive in these assays.
Figure 5. PHCCC potentiates the effects of glutamate in cells expressing mGluR4.
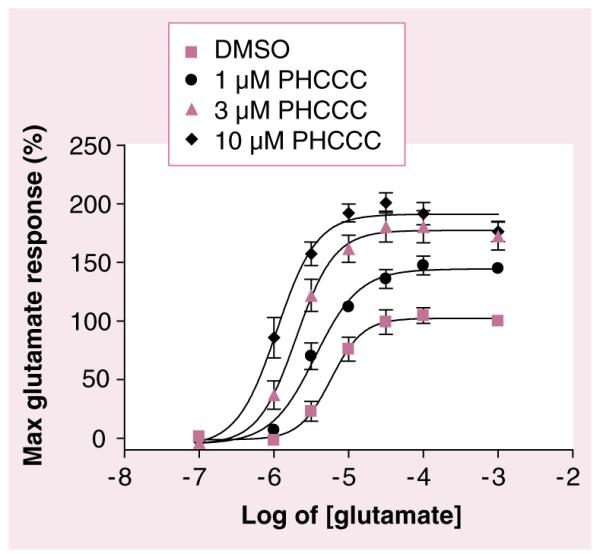
Cells were incubated with indicated concentrations of PHCCC prior to the addition of increasing concentrations of glutamate. PHCCC progressively shifts the potency of glutamate (EC50) to the left by 1.7-fold (at 1 μM), 3.1-fold (at 3 μM), and 5.8-fold (at 10 μM). Data were independently generated for this review by CM Niswender using mGluR4-CHO cells as described in Niswender et al. [65].
DMSO: Dimethyl sulfoxide.
Following these studies, Litschig et al. [55] demonstrated that CPCCOEt inhibited receptor signaling without affecting glutamate binding [56]. CPCCOEt selectively inhibited gluta-mate-induced increases in intracellular calcium at human mGluR1b with an IC50 of 6.5 μM. In addition, CPCCOEt demonstrated no agonist or antagonist activity at human mGluR2, 4a, 5a, 7b or 8a (up to 100 μM). The authors also demonstrated that CPCCOEt acted in a noncompetitive manner by decreasing the efficacy of glutamate-stimulated phosphoinositide hydrolysis without affecting glutamate potency. Lastly, by systematically exchanging segments and single amino acids, it was demonstrated that two amino acids, Thr 815 and Thr 818, were responsible for mediating the noncompetitive antagonism of CPCCOEt at mGluR1b [22].
In 2003, Maj et al. and Marino et al. reported the first receptor subtype-selective positive modulator of mGluR4, PHCCC, (18) [22,53]. As PHCCC is a racemic mixture of two enantiomers, Maj et al. synthesized and tested (±)-PHCCC along with the isolated enantiomers (+)-PHCCC and (18) and determined that the activity is solely due to its (–)-enantiomer. Using cell-based assays, PHCCC was shown to increase agonist potency and enhance maximum efficacy and, at high concentrations, was shown to directly activate mGluR4 with low efficacy [54]. The half maximal effective concentration (EC50) of PHCCC at mGluR4 was 4.1 μM and PHCCC progressively shifted the glutamate concentration–response curve to the left (Figure 6) [22]. In these studies, PHCCC was shown to be a relatively selective PAM (inactive against mGluR2, 3, 5a, 6, 7b and 8a); however, it did show partial antagonist activity at mGluR1b. In addition, PHCCC was shown to be neuroprotective against β-amyloid peptide and NMDA toxicity in mixed cultures of mouse cortical neurons [54].
Figure 6.
The mGluR4-positive allosteric modulator (–)-PHCCC (18) and the mGluR1 antagonist (–)-CPCCOEt (19).
As discussed previously, studies had suggested that group III orthosteric agonists mediated inhibition of striatopallidal transmission and antiparkinsonian effects via mGluR4. In further support of this hypothesis, Marino et al. demonstrated that PHCCC could potentiate the effects of L-AP4 at the striatopallidal synapse, reducing GABA release [22]. PHCCC was further shown to reverse haloperidol-induced catalepsy and reserpine-induced akinesia in rats when administered intracerebroventricularly and also reversed haloperidol-induced catalepsy in mice when given systemically [57]. These studies provided further proof of concept indicating that mGluR4 was a relevant therapeutic target for symptomatic treatment of PD. Excitingly, additional studies with PHCCC demonstrated that the compound could prevent dopaminergic neuron degeneration induced by MPTP in mice [57]. These promising results indicated that mGluR4 activation could provide a novel mechanism for not only treating the symptoms of PD but also slowing disease progression.
While PHCCC has been a breakthrough compound for exploring the therapeutic potential of mGluR4 activation in a variety of disease states, such as anxiety [58], neuroprotection [59,60] and oncology [60], in addition to PD, the SARs for analogues of this molecule have been reported to be ‘flat’ (as noted by Annoura [54]) with very little modification being tolerated, which is common with other mGluR PAMs [61,62]. Additionally, PHCCC is a mGluR1 antagonist, has recently been shown to be a direct agonist of mGluR6 [63] and possesses poor physicochemical properties and limited brain penetration. Additionally, other potential and possibly unknown off-target effects of PHCCC could formally contribute to its antiparkinsonian action, suggesting that identification and examination of other chemical scaffolds with mGluR4 PAM activity would provide further evidence for the promise of mGluR4 activation in PD therapy.
Discovery of additional, novel allosteric modulators of mGluR4
Owing to the current deficiencies of the PHCCC scaffold, other approaches have been taken to identify new, more selective mGluR4 PAMs with improved physiochemical properties. Recently, several distinct, novel series of mGluR4 PAMs have been described (Figure 7). The cyclohexylcarboxylic acid amide 20 is a compound discovered from a high-throughput screening (HTS) campaign initiated at Vanderbilt University, TN, USA [64]. In this screening program, the primary cell line used for screening was a Chinese hamster ovary cell line expressing human mGluR4 (hmGluR4) and a chimeric G-protein, Gqi5, to couple mGluR4 to calcium mobilization. Compounds were confirmed using a secondary assay in which the rat mGluR4 receptor was expressed in HEK cells along with subunits of G-protein-regulated inwardly rectifying potassium channels (GIRKs) [65]. This strategy was designed to confirm activity at both the rat and human receptors while monitoring different signaling pathways to ensure compounds exhibited activity in multiple signal transduction cascades. A number of analogues of 20 were discovered through HTS; however, the 3,5-dichloroamide derivative proved to have the best activity at both hmGluR4 and rat mGluR4 (rmGluR4) (750 and 560 nM, respectively) [64]. This compound demonstrated superior potency and similar efficacy in shifting the glutamate concentration–response curve when compared to PHCCC in both the human and rat mGluR4 assays. In addition, 20 did not potentiate or antagonize responses of any of the other mGluR4 subtypes, indicating that this compound was selective among these mGluRs as a mGluR4 PAM.
Figure 7.

Recently disclosed mGluR4-positive allosteric modulators from Vanderbilt University, TN, USA.
To further profile this ligand, both the cis-and trans-regioisomers were synthesized and evaluated for potency and efficacy at human and rat mGluR4. These studies revealed that the cis-regioisomer (VU0155041) was similar in potency to 20 (798 nM, hmGluR4; 693 nM, rmGluR4); and that the trans-isomer (not shown) was much less active (potency > 10 μM). Additionally, 21 was separated via chiral liquid chromatography into the two enantiomers, (1R,2S) and (1S,2R), each being equipotent. Additional in vitro experiments revealed that, in contrast to PHCCC’s effects in the same assay, 21 demonstrated relatively strong partial agonist activity when examined in the rmGluR4 assay in the absence of glutamate [64]. It is also interesting to note that PHCCC was unable to potentiate or antagonize the agonist activity of 21, suggesting that these two compounds may potentiate mGluR4 via different sites; this possibility will require verification by mutagenesis studies. Encouragingly, 21 was evaluated at MDS-Pharma Services for activity on radio-ligand binding at 68 different targets, including G-protein-coupled receptors, ion channels and transporters and demonstrated no effect on binding at any target examined.
An additional advantage of 21 versus PHCCC for in vivo studies was the solubility of the compound in aqueous vehicles versus the dimethyl sulfoxide or other toxic vehicles required for PHCCC solubility. Encouragingly, 21 was found to exhibit antiparkinsonian effects in haloper-idol-induced catalepsy and reserpine-induced akinesia in vivo antiparkinsonian rodent models [64]. At the time of writing, this compound represented only the second unique chemical scaffold with mGluR4 PAM activity to show efficacy in rodent models of PD, further validating the role of mGluR4 as a therapeutic target for PD.
Three other novel chemical scaffolds, identified via HTS, have been reported by Vanderbilt in 2008 as mGluR4 PAMs 22, 23 and 24 (Figure 7). The pyrazolo[3,4-d]pyrimidine scaffold (22) was disclosed as a novel chemical scaffold with micromolar activity [66]. Pyrazole 22 was equi-potent with PHCCC (4.6 vs 4.1 μM); however, pyrazole (22) displayed a superior shift of the glutamate agonist response curve (27.2-fold at 30 μM compared with 5.5-fold shift for PHCCC). Selectivity experiments demonstrated that 22 was a full antagonist of mGluR1 (IC50: 2.6 μM). Although PHCCC was shown to be a partial antagonist of mGluR1, which was not surprising since it was derived from a series of mGluR1 antagonists (CPCCOEt), it was somewhat unexpected that a structurally unrelated series would also show mGluR1 antagonism. This could suggest that 18 and 22 share a common allosteric binding site on mGluR4 that is also conserved in mGluR1. Future mutagenesis experiments will be required to address this possibility.
Structure–activity relationship modifications were undertaken to further explore the pyrazolo[3,4-d]pyrimidine scaffold. A number of compounds were synthesized to determine the effects of changes to the upper amino substituent; additional modifications were made to evaluate the bottom aryl group. The study commenced with the resynthesis of 22 and reconfirmation of activity, which resulted in a compound that demonstrated equivalent potency but with a less robust fold shift (EC50: ~5 μM, fold shift: 12) [66]. Knowing that allosteric modulator modifications can yield ‘flat’ SAR results, an iterative design employing multidimensional diversity libraries was undertaken in an effort to expand the scope and breadth of the SAR. Unfortunately, very few synthesized compounds demonstrated any mGluR4 PAM activity (~4%). This could be explained by an allosteric binding site for 22 and PHCCC being very shallow. Given the fact that this series demonstrated a nonresponsive SAR and the additional finding that 22 was found to be unstable in fortified liver microsome preparations (9% of parent remaining after 90 min), this series was abandoned as an active discovery scaffold. Although this series did not yield a viable drug candidate, 22 still represents a significant advance due to the large shift (12–27-fold) and the novel chemical scaffold that does not contain the oxime or NH moieties that are speculated to contribute to the poor pharmacokinetic properties of PHCCC.
We have now published two other alternative mGluR4 PAM scaffolds, 23 and 24, based on a functionalized benzylidene hydrazinyl-3-methylquinazoline core or a bis-2,3-dihydroquinazolin-4(1H)-one core, respectively [67]. Although ‘flat’ SAR is a known issue with allosteric ligands, these two scaffolds proved extremely difficult to modify. HTS identified three novel 3-methylquinazolines that afforded a concentration-dependent potentiation of an EC20 of glutamate in the hmGluR4 assay. When examined using concentration–response curves, 23 was the best compound, with an EC50 of 1.7 μM against mGluR4 while having no effect in the absence of glutamate. Resynthesis of 23 demonstrated significant improvements over PHCCC both in terms of potency and efficacy with an EC50 value of 650 M and an extremely robust 36-fold shift of the glutamate concentration–response curvewhen examined at 30 μM. In addition, when screened for selectivity against other mGluRs, 23 was selective for mGluR4 in both activation (EC20 glutamate potentiation) and inhibition (EC80 depression) assays. The absence of any activation or inhibition of the other mGluRs is a marked improvement over PHCCC and the previous mGluR4 PAM (22) due to their activities against mGluR1. Due to the differences in selectivity of 23 versus PHCCC and 22, it is possible that 23 may modulate mGluR4 via a second allosteric site. Multi-dimensional diversity synthesis was undertaken in an attempt to better evaluate the SAR of this series. Compounds were made to explore the aromatic group at the hydrazino imine terminus, as well as the effect of replacement of the labile hydrazino imine moiety. Unfortunately, all of these modifications failed to produce any other compounds from this series that demonstrated improved potency and since the hydrazino imine moiety could not be replaced, this series was not advanced further.
The lead from the last series evaluated, 24, has some structural features similar to PHCCC. 24 exhibited a potency against hmGluR4 of 1.8 μM, comparable to PHCCC (4.1 μM); however, this compound only modestly shifted the glutamate response curve (2.7-fold) [67]. Microwave-assisted chemical synthesis was employed for this series since it was highly amenable to rapid SAR analysis. Two separate 24-membered libraries were synthesized, keeping either the 4-methylaniline or the 2-phenylpropanal constant. From this synthetic endeavor, 48 compounds were synthesized and only three compounds were found to have any activity against hmGluR4. The resynthesis of 24 demonstrtated similar potency but reduced efficacy; two other compounds exhibited activity only above 10 μM. Owing to the structural similarities to PHCCC, 24 may bind in a similar site that is not amenable to functionalization for SAR. Although these last two scaffolds did not yield any leads that were advanced further, in a field that lacks selective ligands, they still represent two more novel mGluR4 PAMs.
Lastly, both Merck and Amgen have presented data at meetings in 2008 revealing compounds with mGluR4 PAM activity. Researchers at Merck Research Labs presented data at the Sixth International Metabotropic Glutamate Receptors Meeting in Taormina, Sicily [67]. Two compounds, 25 (in vitro EC50: 45 nM) and 26 (EC50: 160 nM), were presented and demonstrated to have good activity against haloperidol-induced catalepsy when given intracerebroventricularly (Figure 8). These compounds were high enough affinity to radiolabel and it was demonstrated that PHCCC could displace their binding to the receptor. Additionally, Amgen presented data at the Society for Neuroscience 2008 meeting describing a potent and selective positive allosteric modulator of mGluR4 [68]. This compound was shown to modulate striatopallidal synapse and restore balance in the BG motor circuit. The structure of the lead compound was not disclosed; however, it was reported to have a potency in a hmGluR4 assay of 20 nM and shift the glutamate concentration–response curve 5.2-fold to the left. Moreover, intracerebroventricular administration of this compound reversed motor impairment in reserpinized rats. Both of these results provide further evidence that mGluR4 PAMs will be beneficial for the treatment of PD.
Figure 8.

Recently disclosed mGluR4-positive allosteric modulators from Merck research laboratories.
Future perspective
Almost 200 years have passed since James Parkinson first described the symptoms of the motor disorder that now bears his name. The past 10 years have witnessed amazing progress in our understanding of the etiology and pathology of PD, leading to a significant improvement in the quality of life of PD patients. However, disease-modifying approaches, strategies to address the non-motor symptoms of the disorder and treatments exploiting nondopaminergic approaches represent a major unmet medical need. Advances in genetics have identified key gene mutations (PARK1–11, PINK1, LRRK2) that suggest new molecular targets for therapeutic intervention. Other nondopaminergic approaches show promise in preclinical models, such as A2A antagonists, NR2B antagonists, selective M4 antagonists and the mGluR4 PAMs/selective mGluR4 agonists [70–74]. It remains to be seen if any of these new molecular targets will represent standalone therapies for PD, or if combinations of drugs targeting multiple molecular targets will prove most effective [75–77], as in the polypharmacology approach for the treatment of other CNS disorders, such as schizophrenia. It is anticipated that the next decade holds exciting promise for the both symptomatic and disease-modifying treatment of PD.
Executive summary.
Introduction
Parkinson’s disease (PD) is a debilitating movement disorder.
Current treatments include dopamine-replacement therapies.
Resistance to dopamine-replacement therapies begins to emerge during treatment and may actually accelerate disease progression through increased oxidative stress and damage.
Understanding basal ganglia circuitry provides new insights into potential pharmacological therapies for PD.
Basal ganglia are a set of interconnected nuclei that play a key role in the control of movement.
The opposing inhibitory and excitatory signals from the direct and indirect pathways provide a delicate balance to the overall activity of the output nuclei.
Increased understanding of basal ganglia function has led to new strategies for treatment of PD.
Metabotropic glutamate receptors (mGluRs): the role of mGluR4 in inhibiting striatopallidal neurotransmission
mGluRs are members of class C G-protein-coupled receptors.
mGluRs have the ability to exert a neuromodulatory role in the control of both glutamatergic and GABAergic neurotransmission.
GABA release from the striatum to the external segment of the globus pallidus (GPe) within the indirect pathway is increased in PD due to dopamine neuron degeneration in the substantia nigra pars compacta.
mGluR4 is highly expressed at the striatopallidal synapse within the indirect pathway; activation of mGluR4 reduces GABA release, which is predicted to normalize basal ganglia output. Work conducted with orthosteric ligands of mGluR4 has shown efficacy in a number of anti-parkinsonian rodent models when administered via intracerebroventricular injection.
Discovery of the first positive allosteric modulator of mGluR4, (–)-PHCCC
PHCCC, a chemical compound derived from the mGluR1/5 antagonist CPCCOEt, was the first known mGluR4-positive allosteric modulator.
PHCCC was shown to reverse haloperidol-induced catalepsy and reserpine-induced akinesia in rats when administered intracerebroventricularly.
These studies provided proof-of-concept studies indicating that allosteric modulation of mGluR4 is a potential therapeutic strategy for symptomatic treatment of PD.
In addition, PHCCC was shown to prevent dopaminergic neuron degeneration induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; indicating that mGluR4 activation/potentiation could provide a novel mechanism for slowing disease progression.
Discovery of additional, novel allosteric modulators of mGluR4
Research efforts at Vanderbilt University, TN, USA, have led to the discovery of a novel ago-potentiator of mGluR4 (VU0155041), a cyclohexylcarboxylic acid amide.
VU0155041 is a selective mGluR4 potentiator and has been shown to reverse haloperidol-induced catalepsy and reserpine-induced akinesia in rats when administered intracerebroventricularly.
Four additional novel chemical scaffolds were introduced in 2008 as selective mGluR4-positive allosteric modulators and a number of these compounds are active in both the haloperidol-induced catalepsy and reserpine-induced akinesia models.
Future perspective
mGluR4 receptor-positive allosteric modulation remains an active area of research to bring about symptomatic as well as potentially disease-modifying potential without invasive surgical procedures.
Recent advances in genetics suggest new molecular targets for therapeutic intervention, as well as other nondopaminergic approaches that show promise in preclinical models (A2A antagonism and NR2B antagonist, for example) have brought much needed excitement to the field of Parkinson’s research.
Glossary
- Parkinson’s disease
A neurodegenerative disease resulting primarily from the degeneration of dopaminergic neurons in the substantia nigra pars compacta in the brain that impairs motor skills, speech and can result in behavioral disturbances
- Metabotropic glutamate receptors
Family C G-protein-coupled-receptors that transduce G protein-regulated intracellular signaling cascades in response to glutamate. These receptors complement the ionotropic glutamate receptors by providing a mechanism whereby glutamate can modulate synaptic transmission
- Mglur4
Subtype 4 receptor of the metabotropic receptors
- L-aP4
An orthosteric agonist of the group III metabotropic glutamate receptors
- allosteric Modulator
A ligand (or compound) that binds to a target at a site other than the endogenous agonist binding site
- Phccc
Prototype mGluR4-positive allosteric modulator
- vu0155041
A novel, recently identified modulator of mGluR4 that differs from PHCCC by exhibiting both allosteric agonist and positive allosteric modulator activity in vitro
Footnotes
Financial & competing interests disclosure Corey Hopkins, Craig Lindsley and Colleen Niswender all report that salaries are paid partly by the Michael J Fox Foundation for Parkinson’s Research. This research was supported in part by grants from the NIH. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Bibliography
Papers of special note have been highlighted as:
■ of interest
■■ of considerable interest
- 1.Dauer W, Predborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39(6):889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 2.Schapira AHV. Neurobiology and treatment of Parkinson’s disease. Trends Pharmacol. Sci. 2009;30(1):41–47. doi: 10.1016/j.tips.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 3.Hefti FF. Parkinson’s disease. In: Hefti FF, editor. Drug Discovery for Nervous System Diseases. Wiley-Interscience; NJ, USA: 2005. pp. 183–204. [Google Scholar]
- 4.Fahn S. Description of Parkinson’s disease as a clinical syndrome. Ann. NY Acad. Sci. 2003;991:1–14. doi: 10.1111/j.1749-6632.2003.tb07458.x. [DOI] [PubMed] [Google Scholar]
- 5.Parkinson J. An Essay on the Shaking Palsy. Sherwood, Neely and Jones; London, UK: 1817. ■ First report of what is now known as Parkinson’s disease.
- 6.Chaudhuri KR, Healy DG, Schapira AHV. Non-motor symptoms of Parkinson’s disease: diagnosis and management. Lancet Neurol. 2006;5(3):235–245. doi: 10.1016/S1474-4422(06)70373-8. [DOI] [PubMed] [Google Scholar]
- 7.Warner TT, Schapira AHV. Genetic and environmental factors in the cause of Parkinson’s disease. Ann. Neurol. 2003;53(Suppl 3):S16–S25. doi: 10.1002/ana.10487. [DOI] [PubMed] [Google Scholar]
- 8.Jellinger KA. Recent developments in the pathology of Parkinson’s disease. J. Neural Transm. Suppl. 2002;62:347–376. doi: 10.1007/978-3-7091-6139-5_33. [DOI] [PubMed] [Google Scholar]
- 9.Jankovic J. Parkinson’s disease therapy: treatment of early and late disease. Chin. Med. J. 2001;114(3):227–234. [PubMed] [Google Scholar]
- 10.Langston JW. The etiology of Parkinson’s disease with emphasis on the MPTP story. Neurology. 1996;47(6):S153–S160. doi: 10.1212/wnl.47.6_suppl_3.153s. [DOI] [PubMed] [Google Scholar]
- 11.Baas H, Beiske AG, Ghika J, et al. Catechol-O-methyltransferase inhibition with tolcapone reduces the “wearing off” phenomenom and levodopa requirements in fluctuating Parkinsonian patients. Neurology. 1998;50(5 Suppl 5):S46–S53. doi: 10.1212/wnl.50.5_suppl_5.s46. [DOI] [PubMed] [Google Scholar]
- 12.Stocchi F. The levodopa wearing-off phenomenon in Parkinson’s disease: pharmacokinetic considerations. Expert Opin. Pharmacother. 2006;7(10):1399–1407. doi: 10.1517/14656566.7.10.1399. [DOI] [PubMed] [Google Scholar]
- 13.Schapira AHV. The clinical relevance of levodopa toxicity in the treatment of Parkinson’s disease. Mov. Disord. 2008;23(Suppl 3):S515–S520. doi: 10.1002/mds.22146. [DOI] [PubMed] [Google Scholar]
- 14.Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12(10):366–375. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- 15.DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 1990;13(7):281–285. doi: 10.1016/0166-2236(90)90110-v. [DOI] [PubMed] [Google Scholar]
- 16.Ciliax B, Greenamyre J, Levey A. Functional biochemistry and molecular neuropharmacology of the basal ganglia and motor systems. In: Watts RL, editor. Movement Disorders: Neurological principles and practice. McGraw-Hill Professional; 1997. pp. 99–118. [Google Scholar]
- 17.Bergman H, Wichmann T, DeLong MR. Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science. 1990;249:1436–1438. doi: 10.1126/science.2402638. [DOI] [PubMed] [Google Scholar]
- 18.Wichmann T, DeLong MR. Functional neuroanatomy of the basal ganglia in Parkinson’s disease. Adv. Neurol. 2003;91:9–18. [PubMed] [Google Scholar]
- 19.Bevan MD, Magill PJ, Terman D, Bolam JP, Wilson CJ. Move to the rhythm: oscillations in the subthalamic nucleus-external globus pallidus network. Trends Neurosci. 2002;25(10):525–531. doi: 10.1016/s0166-2236(02)02235-x. [DOI] [PubMed] [Google Scholar]
- 20.Boraud T, Bezard E, Bioulac B, Gross CE. From single extracellular unit recording in experimental and human parkinsonism to the development of a functional concept of the role played by the basal ganglia in motor control. Prog. Neurobiol. 2002;66(4):265–283. doi: 10.1016/s0301-0082(01)00033-8. [DOI] [PubMed] [Google Scholar]
- 21.Wichmann T, DeLong MR. Functional and pathopysiological models of the basal ganglia. Curr. Opin. Neurobiol. 1996;6(6):751–758. doi: 10.1016/s0959-4388(96)80024-9. [DOI] [PubMed] [Google Scholar]
- 22.Marino MJ, Williams DL, Jr, O’Brien JA, et al. Allosteric modulation of group III metabotropic glutamate receptor 4: a potential approach to Parkinson’s disease treatment. Proc. Natl Acad. Sci. USA. 2003;100(23):13668–13673. doi: 10.1073/pnas.1835724100.■■ Important study showing that the novel metabolic glutamine receptor 4 positive allosteric modulator N-phenyl-7-(hydroxyimino)cyclopropa[B]chromen-1A-carboxamide (PHCCC) corrects motor deficits in antiparkinsonian rodent models.
- 23.Gerfen CR, Engber TM, Mahan LC, et al. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science. 1990;250:1429–1432. doi: 10.1126/science.2147780. [DOI] [PubMed] [Google Scholar]
- 24.Wallace BA, Ashkan K, Heise CE, et al. Survival of midbrain dopaminergic cells after lesion or deep brain stimulation of the subthalmic nucleus in MPTP-treated monkeys. Brain. 2007;130(8):2129–2145. doi: 10.1093/brain/awm137. [DOI] [PubMed] [Google Scholar]
- 25.Breit S, Schulz JB, Benabid AL. Deep brain stimulation. Cell Tissue Res. 2004;318(1):275–288. doi: 10.1007/s00441-004-0936-0. [DOI] [PubMed] [Google Scholar]
- 26.Warnke PC. STN stimulation and neuroprotection in Parkinson’s disease – when beautiful theories meet ugly facts. J. Neurol. Neurosurg. Psychiatr. 2005;76(9):1186–1187. doi: 10.1136/jnnp.2004.061481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Ann. Rev. Pharmacol. Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205.■■ Seminal work describing the functional utility of the metabolic glutamine receptors.
- 28.Schoepp DD, Jane DE, Monn JA. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology. 1999;38(10):1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- 29.Marino MJ, Conn PJ. Glutamate-based therapeutic approaches: allosteric modulators of metabotropic glutamate receptors. Curr. Opin. Pharmacol. 2006;6(1):98–102. doi: 10.1016/j.coph.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 30.Marino MJ, Hess JF, Liverton N. Targeting the metabotropic glutamate receptor mGluR4 for the treatment of diseases of the central nervous system. Curr. Top. Med. Chem. 2005;5(9):885–895. doi: 10.2174/1568026054750263. [DOI] [PubMed] [Google Scholar]
- 31.Niswender CM, Jones CK, Conn PJ. New therapeutic frontiers for metabotropic glutamate receptors. Curr. Top. Med. Chem. 2005;5(9):847–857. doi: 10.2174/1568026054750254. [DOI] [PubMed] [Google Scholar]
- 32.Yang ZQ. Agonists and antagonists for group III metabotropic glutamate receptors 6, 7, and 8. Curr. Top. Med. Chem. 2005;5(9):913–918. doi: 10.2174/1568026054750272. [DOI] [PubMed] [Google Scholar]
- 33.Lavreysen H, Dautzenberg FM. Therapeutic potential of group III metabotropic glutamate receptors. Curr. Med. Chem. 2008;15(7):671–684. doi: 10.2174/092986708783885246. [DOI] [PubMed] [Google Scholar]
- 34.Bradley SR, Standaert DG, Levey AI, Conn PJ. Distribution of group III mGluRs in rat basal ganglia with subtype-specific antibodies. Ann. NY Acad. Sci. 1999;868:531–534. doi: 10.1111/j.1749-6632.1999.tb11322.x. [DOI] [PubMed] [Google Scholar]
- 35.Corti C, Aldegheri L, Somogyi P, Ferraguti F. Distribution and synaptic localisation of the metabotropic glutamate receptor 4 (mGluR4) in the rodent CNS. Neuroscience. 2002;110(3):403–420. doi: 10.1016/s0306-4522(01)00591-7. [DOI] [PubMed] [Google Scholar]
- 36.Valenti O, Marino MJ, Wittmann M, et al. Group III metabotropic glutamate receptor-mediated modulation of the striatopallidal synapse. J. Neurosci. 2003;23(18):7218–7226. doi: 10.1523/JNEUROSCI.23-18-07218.2003.■■ Important paper demonstrating that reduction of GABA release at the striatopallidal synapse by a group III mGluR agonist could be a potentially novel approach for the treatment of PD.
- 37.Valenti O, Mannaioni G, Seabrook GR, Conn PJ, Marino MJ. Group III metabotropic glutamate-receptor-mediated modulation of excitatory transmission in rodent substantia nigra pars compacta dopamine neurons. J. Pharmacol. Exp. Ther. 2005;313(3):1296–1304. doi: 10.1124/jpet.104.080481. [DOI] [PubMed] [Google Scholar]
- 38.Vernon AC, Zbarsky V, Datla KP, Dexter DT, Croucher MJ. Selective activation of group III metabotropic glutamate reeptors by L-(+)-2-amino-4-phosphonobutyric acid protects the nigrostriatal system against 6-hydroxydopamine toxicity in vivo. J. Pharmacol. Exp. Ther. 2007;320(1):397–409. doi: 10.1124/jpet.106.108159. [DOI] [PubMed] [Google Scholar]
- 39.Johansen PA, Chase LA, Sinor AD, Koerner JF, Johnson RL, Robinson MB. Type 4a metabotropic glutamate receptor: identification of new potent agonists and differentiation from the L-(+)-2-amino-4-phosphonobutanoic acid-sensitive receptor in the lateral perforant pathway in rats. Mol. Pharmacol. 1995;48(1):140–149. [PubMed] [Google Scholar]
- 40.Trombley PQ, Westbrook GL. L-AP4 inhibits calcium currents and synaptic transmission via a G-protein-coupled glutamate receptor. J. Neurosci. 1992;12(6):2043–2050. doi: 10.1523/JNEUROSCI.12-06-02043.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomsen C, Hampson DR. Contribution of metabotropic glutamate receptor mGluR4 to l-2-[3H]amino-4-phosphonobutyrate binding in mouse brain. J. Neurochem. 1999;72(2):835–840. doi: 10.1046/j.1471-4159.1999.0720835.x. [DOI] [PubMed] [Google Scholar]
- 42.Naples MA, Hampson DR. Pharmacological profiles of the metabotropic glutamate receptor ligands [3H]L-AP4 and [3H]CPPG. Neuropharmacology. 2001;40(2):170–177. doi: 10.1016/s0028-3908(00)00128-3. [DOI] [PubMed] [Google Scholar]
- 43.MacInnes N, Messenger MJ, Duty S. Activation of group III metabotropic glutamate receptors in selected regions of the basal ganglia alleviates akinesia in the reserpine-treated rats. Br. J. Pharmacol. 2004;141(1):15–22. doi: 10.1038/sj.bjp.0705566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wan H, Cahusac PMB. The effects of L-AP4 and L-serine-O-phosphate on inhibition in primary somatosensory cortex of the adult rat in vivo. Neuropharmacology. 1995;34(8):1053–1062. doi: 10.1016/0028-3908(95)00091-j. [DOI] [PubMed] [Google Scholar]
- 45.Agari T, Yasuhara T, Matsui T, et al. Intrapallidal metabotropic glutamate receptor activation in a rat model of Parkinson’s disease: behavioral and histological analyses. Brain Res. 2008;1203:189–196. doi: 10.1016/j.brainres.2008.01.051. [DOI] [PubMed] [Google Scholar]
- 46.Lopez S, Turle-Lorenzo N, Acher F, De Leonibus E, Mele A, Amalric M. Targeting group III metabotropic glutamate receptors produces complex behavioral effects in rodent models of Parkinson’s disease. J. Neurosci. 2007;27(25):6701–6711. doi: 10.1523/JNEUROSCI.0299-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Konieczny J, Wardas J, Kuter K, Pilc A, Ossowska K. The influence of group III metabotropic glutamate receptor stimulation by (1S,3R,4S)-1-aminocyclopentane-1,3,4-tricarboxylic acid on the parkinsonian-like akinesia and striatal proenkephalin and prodynorphin mRNA expression in rats. Neuroscience. 2007;145(2):611–620. doi: 10.1016/j.neuroscience.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 48.Acher FC, Tellier FJ, Azerad R, Brabet IN, Fagni L, Pin JPR. Synthesis and pharmacological characterization of aminocyclopentanetricarboxylic acids: new tools to discriminate between metabotropic glutamate receptor subtypes. J. Med. Chem. 1997;40(19):3119–3129. doi: 10.1021/jm970207b. [DOI] [PubMed] [Google Scholar]
- 49.Sibille P, Lopez S, Brabet I, et al. Synthesis and biological evaluation of 1-amino-2-phosphono methylcyclopropanecarboxylic acids, new group III metabotropic glutamate receptor agonists. J. Med. Chem. 2007;50(15):3585–3595. doi: 10.1021/jm070262c. [DOI] [PubMed] [Google Scholar]
- 50.Betts MTJ, O’Neill MJ, Mitchell SN, Duty S. The group III mGlu receptor agonist L-AP4 affords both neurochemical and functional neuroprotection against a 6-hydroxydopamine lesion of the substantia nigra in rats. Presented at: 6th International Meeting on Metabotropic Glutamate Receptors; Taoromino, Sicily, Italy. 14–19 September.2008. [Google Scholar]
- 51.Selvam C, Goudet C, Oueslati N, Pin JP, Acher C. L-(+)-2-amino-4-thiophosphonobutyric acid (L-thioAP4), a new potent agonist of group III metabotropic glutamate receptors: increased distal acidity affords enhanced potency. J. Med. Chem. 2007;50(19):4656–4664. doi: 10.1021/jm070400y. [DOI] [PubMed] [Google Scholar]
- 52.Filosa R, Marinozzi M, Costantino G, Hermit MB, Thomsen C, Pellicciari R. Synthesis and biological evaluation of (2S)-and (2R)-2-(3’-phosphonobicyclo[1.1.1] pentyl)glycines as novel group III metabotropic glutamate receptor ligands. Bioorg. Med. Chem. 2006;14(11):3811–3817. doi: 10.1016/j.bmc.2006.01.027. [DOI] [PubMed] [Google Scholar]
- 53.Conn PJ, Christopoulous A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat. Rev. Drug Discov. 2009;8(1):41–54. doi: 10.1038/nrd2760.■ Describes allosteric modulation and the ongoing efforts to translate these modulators in to clinical candidates.
- 54.Maj M, Bruno V, Dragic Z, et al. (-)-PHCCC, a positive allosteric modulator of mGluR4: characterization, mechanism of action, and neuroprotection. Neuropharmacology. 2003;45(7):895–906. doi: 10.1016/s0028-3908(03)00271-5.■■ Independent study describing the activity of PHCCC, showing that compound function was mediated via the mGluR4 transmembrane domain and neuroprotective activity of the compound.
- 55.Annoura H, Fukunaga A, Uesugi M. A novel class of antagonists for metabotropic glutamate receptors, 7-(hydroxyimino) cyclopropa[b]chromen-1a-carboxylates. Bioorg. Med. Chem. Lett. 1996;6(7):763–766.■ First report of PHCCC as a mGluR1 antagonist.
- 56.Litschig S, Gasparini F, Ruegg D, et al. CPCCOEt, a noncompetitive metabotropic glutamate receptor 1 antagonist, inhibits receptor signaling without affecting glutamate binding. Mol. Pharmacol. 1999;55(3):453–461. [PubMed] [Google Scholar]
- 57.Battaglia G, Busceti CL, Molinaro G, et al. Pharmacological activation of mGluR4 metabotropic glutamate receptors reduces nigrostriatal degeneration in mice treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J. Neurosci. 2006;26(27):7222–7229. doi: 10.1523/JNEUROSCI.1595-06.2006.■■ Study showing that the novel mGluR4 PAM PHCCC is neuroprotective.
- 58.Stachowicz K, Klak K, Klodzinska A, Chojnacka-Wojcik E, Pilc A. Anxiolytic-like effects of PHCCC, an allosteric modulator of mGluR4 receptors, in rats. Eur. J. Pharmacol. 2004;498(1–3):153–156. doi: 10.1016/j.ejphar.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 59.Canudas AM, Di Giorgi-Gerevini V, Iacovelli L, et al. PHCCC, a specific enhancer of type 4 metabotropic glutamate receptors, reduces proliferation and promotes differentiation of cerebellular granule cell neuroprecursors. J. Neurosci. 2004;24(46):10343–10352. doi: 10.1523/JNEUROSCI.3229-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Iacovelli L, Arcella A, Battaglia G, et al. Pharmacological activation of mGluR4 metabotropic glutamate receptors inhibits the growth of medulloblastomas. J. Neurosci. 2006;26(32):8388–8397. doi: 10.1523/JNEUROSCI.2285-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao Z, Wisnoski DD, O’Brien JA, et al. Challenges in the development of mGluR5 positive allosteric modulators: the discovery of CPPHA. Bioorg. Med. Chem. Lett. 2007;17(24):1386–1391. doi: 10.1016/j.bmcl.2006.11.081. [DOI] [PubMed] [Google Scholar]
- 62.Lindsley CW, Wisnoski DD, Leister WH, et al. Discovery of positive allosteric modulators for the metabotropic glutamate subtype 5 from a series of N-(1,3-diphenyl-1H-pyrazol-5-yl) benzamides that potentiate function in vivo. J. Med. Chem. 2004;47(24):5825–5828. doi: 10.1021/jm049400d. [DOI] [PubMed] [Google Scholar]
- 63.Beqollari D, Kammermeier PJ. The mGlu4 receptor allosteric modulator N-phenyl-7-(hydroxyimino)cyclopropa[b]chromen-1acarboxamide acts as a direct agonist at mGlu6 receptors. Eur. J. Pharmacol. 2008;589(1–3):49–52. doi: 10.1016/j.ejphar.2008.06.054. [DOI] [PubMed] [Google Scholar]
- 64.Niswender CM, Johnson KA, Weaver CD, et al. Discovery, characterization, and antiparkinsonian effect of novel positive allosteric modulators of metabotropic glutamate receptor 4. Mol. Pharmacol. 2008;74(5):1345–1358. doi: 10.1124/mol.108.049551.■■ Important study utilizing a novel, aqueous soluble, novel and selective mGluR4 PAM showing antiparkinsonian effects in in vivo rodent models.
- 65.Niswender CM, Johnson KA, Luo Q, et al. A novel assay of Gi/o-linked G protein-coupled receptor coupling to potassium channels provides new insights into the pharmacology of the group III metabotropic glutamate receptors. Mol. Pharmacol. 2008;73(4):1213–1224. doi: 10.1124/mol.107.041053. [DOI] [PubMed] [Google Scholar]
- 66.Niswender CM, Lebois EP, Luo Q, et al. Positive allosteric modulators of the metabotropic glutamate receptor subtype 4 (mGluR4): part I: discovery of pyrazolo[3,4-d]pyrimidines as novel mGluR4 positive allosteric modulators. Bioorg. Med. Chem. Lett. 2008;18(20):5626–5630. doi: 10.1016/j.bmcl.2008.08.087.■ Describes a novel mGluR4 PAM from an alternative chemical scaffold.
- 67.Williams R, Niswender CM, Luo Q, Le U, Conn PJ, Lindsley CW. Positive allosteric modulators of the metabotropic glutamate receptor subtype 4 (mGluR4). Part II: challenges in hit-to-lead. Bioorg. Med. Chem. Lett. 2009;19(3):962–966. doi: 10.1016/j.bmcl.2008.11.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reynolds IJ. Metabotropic glutamate receptors as therapeutic targets in Parkinson’s disease. Presented at: 6th International Meeting on Metabotropic Glutamate Receptors; Taoromino, Sicily, Italy. 14–19 September.2008. ■ Novel chemical scaffolds that show activity via intracerebroventricularly injection in in vivo antiparkinsonian rodent models.
- 69.Ortuno D, Cheng C, Weiss M, Bergeron M, Shanker Y. Identification and characterization of a potent and selective positive allosteric modulator of mGluR4. Presented at: Society for Neuroscience 2008; Washington, DC, USA. 15–19 November, 2008. [Google Scholar]
- 70.Gillepsie RJ, Bamford SJ, Botting R, et al. Antagonists of the human A2A adenosine receptor. 4. Design, synthesis, and preclinical evaluation of 7-aryltriazolo[4,5-d] pyrimidines. J. Med. Chem. 2009;52(1):33–47. doi: 10.1021/jm800961g. [DOI] [PubMed] [Google Scholar]
- 71.Slee DH, Zhang X, Moorjani M, et al. Identification of novel, water-soluble, 2-amino-N-pyrimidin-4-yl acetamides as A2A receptor antagonists with in vivo efficacy. J. Med. Chem. 2008;51(3):400–406. doi: 10.1021/jm070623o. [DOI] [PubMed] [Google Scholar]
- 72.Liverton NJ, Bednar RA, Bednar B, et al. Identification and characterization of 4-methylbenzyl 4-[(pyrimidin-2-ylamino) methyl]piperidine-1-carboxylate, an orally bioavailable, brain penetrant NR2B selective N-methyl-D-aspartate receptor antagonist. J. Med. Chem. 2007;50(4):807–819. doi: 10.1021/jm060983w. [DOI] [PubMed] [Google Scholar]
- 73.Schwarz RD, Nelson BC, Augelli-Szafran CE, et al. Pharmacological characterization of PD102807: an m4 subtype selective muscarinic antagonist. Life Sci. 1997;60(13):1167. [Google Scholar]
- 74.Augelli-Szafran CE, Jaen JC, Moreland DW, Nelson CB, Penvose-Yi JR, Schwarz RD. Identification and characterization of m4 selective muscarinic antagonists. Bioorg. Med. Chem. Lett. 1998;8(15):1991–1996. doi: 10.1016/s0960-894x(98)00351-5. [DOI] [PubMed] [Google Scholar]
- 75.Van der Schyf CJ, Youdim MBH. Multifunctional drugs as neurotherapeutics. Neurotherapeutics. 2009;6(1):1–3. doi: 10.1016/j.nurt.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Petzer JP, Castagnoli N, Jr, Schwarzschild MA, Chen JF, Van der Schyf CJ. Dual-target-directed drugs that block monoamine oxidase B and adenosine A2A receptors for Parkinson’s disease. Neurotherapeutics. 2009;6(1):141–151. doi: 10.1016/j.nurt.2008.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cavalli A, Bolognesi ML, Minarini A, et al. Multi-target-directed ligands to combat neurodegenerative diseases. J. Med. Chem. 2008;51(3):347–372. doi: 10.1021/jm7009364. [DOI] [PubMed] [Google Scholar]