

Abstract
Technology based on surface plasmon resonance (SPR) has allowed rapid, label-free characterization of protein-protein and protein-small molecule interactions, from quantitative measurements of binding kinetics and thermodynamics and concentrations in complex samples to epitope analysis. SPR has become the gold standard in industrial and academic settings, in which typically the interaction between a pair of soluble binding partners is characterized in detail or a library of molecules is screened for binding against a single soluble protein. In spite of these successes, the technology is only beginning to be adapted to the needs of membrane-bound proteins. Including G protein-coupled receptors (GPCR), ion channels and other growth, immune and cellular receptors, these proteins are difficult to study in situ but represent promising targets for drug and biomarker development. Existing technologies, such as BIAcore™, have been adapted for membrane protein analysis by building supported lipid layers or vesicle capture on existing chips. Newer technologies, still in development, will allow membrane proteins to be presented in native or near-native formats. These include SPR nanopore arrays, in which lipid bilayers containing membrane proteins stably span small pores that are addressable from both sides of the bilayer. Here, we discuss successes with current SPR instrumentation and the potential for SPR nanopore arrays to enable quantitative, high-throughput screening of GPCR ligands, biomarker discovery involving membrane bound proteins and basic cellular biology.
Keywords: SPR, GPCR, membrane protein, autoantibody, protein array, supported lipid bilayer
Introduction
Technological advances facilitate scientific breakthroughs by providing previously inaccessible data and accelerating the pace of scientific discovery. In particular, modern biology has been transformed by the ability to describe biological phenomena in quantitative physical terms, a development provided by innovations such as surface plasmon resonance (SPR) to measure protein-ligand binding kinetics. Similarly, assay miniaturization has allowed development of high throughput screening (HTS) programs, from developments as simple as increasing the density of assay plates from 96 to 1536 wells to integrated lab-on-a-chip devices. Combined into a single device, SPR and HTS could allow rapid quantitative analysis of, for instance, thousands of small molecule ligands binding a cell surface receptor in order to identify agonists meeting specific criteria.
While many ligand screening programs rely on equilibrium binding as a first level of analysis, subsequent characterization of “hits” includes detailed characterization of binding kinetics and selectivity for the receptor of interest. The current standard for characterization of binding partners is quantitation of association and dissociation rate constants by SPR, most successfully characterized by BIAcore™. This technology typically works by tethering one binding partner to a microfluidic chip constructed from a thin gold film atop a glass support. To measure equilibrium and binding kinetics, a solution containing ligand flows across the surface. As ligand binds the immobilized partner, the mass of material bound to the surface increases. This change is detected as a change in the angle of polarized light reflected from the bottom surface of the chip. (see Figure 1). SPR has been extensively used in industry and academia for antibody engineering [1] and drug screening programs as well as to understand basic mechanisms of molecular recognition [2]. Binding kinetics are important to quantify as small differences can provide a rationale for selecting lead molecules during development and binding kinetics will impact both the dosing and potency of a molecule in vivo. Mechanistically, kinetic analysis of site-directed variants provides insight into the mechanism and dynamics of binding [3].
Figure 1. Comparison of SPR technologies.
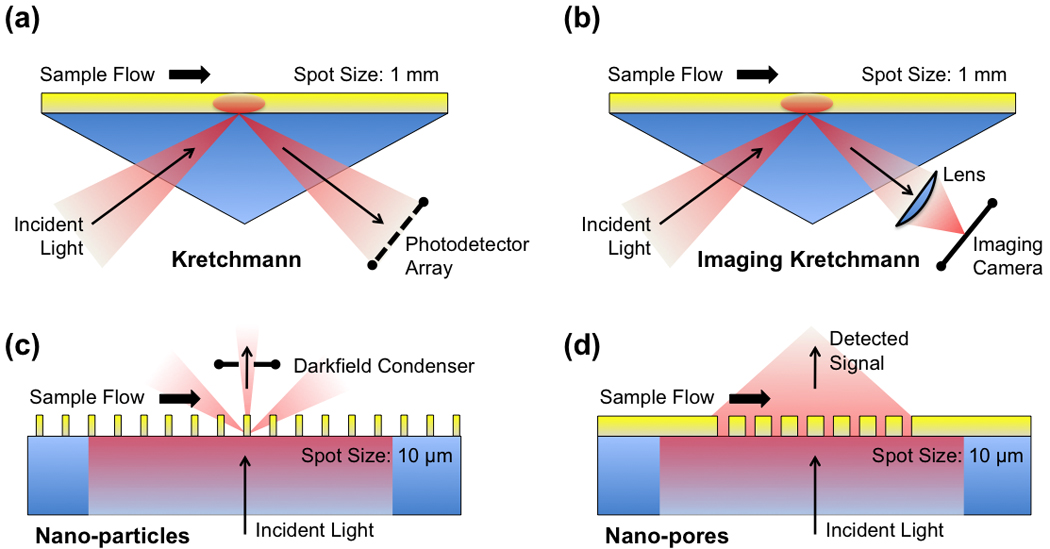
a) The standard BIAcore™ measurements with a prism-based Kretschmann setup have a large sensing spot size. (b) SPR imaging uses a similar setup, but with imaging optics for the detectors. (c) Nanoparticle arrays use a dark-field condenser for collecting the signal. (d) Nanopore arrays have a high spatial resolution and can easily be made highly multiplexed.
However, membrane-bound proteins, which require a lipid bilayer for native function, present a series of challenges for currently available SPR technologies. Membrane-bound proteins are an important class of molecules for several reasons – almost half of the 100 best-selling drugs on the market are targeted to membrane-bound proteins [4]. These proteins represent the interface between a cell and its surroundings, mediating responses to growth factors and immune cells and representing potential diagnostic and therapeutic targets. While 30% of genes in the human genome are predicted to encode for membrane proteins, these molecules remain poorly characterized, largely due to difficulties in purifying protein for analysis. As an example, the structure of only the second G protein-coupled receptor (GPCR) was solved in 2007 after enormous effort [5–8].
To enable rapid, quantitative screening of ligands binding GPCRs and identification of membrane-bound immune and tumor-associated biomarkers, these ligand-receptor interactions must be probed in lipid bilayers that resemble their native membrane environment. To interface with existing SPR instrumentation, membrane proteins can be immobilized as detergent “solubilized” protein, deposited in supported lipid bilayers or trapped in vesicles which are subsequently captured. Newer SPR-based technologies offer the potential to analyze membrane proteins in completely native environments. One option is a periodic metallic nanopore array supporting free-standing lipid bilayers on a gold film (see 1d). In this format, membrane proteins would be presented in a lipid bilayer that mimics the natural biological membrane to allow functional studies and label-free kinetic measurements. This review will focus on the applications of existing and emerging SPR technologies for ligand screening programs, biomarker discovery for cancer and basic cellular biology. Alternative options do exist for label-free kinetic biosensing, such as a quartz crystal microbalance, nanomechanical resonators [9], nanowire sensors [10] and high-Q optical microcavities [11], but these are beyond the scope of this review.
Membrane proteins and lipid bilayers
Lipid membranes are responsible for compartmentalizing the many functions and components of a cell, including the cell itself (see Figure 2). However, in order to replicate and interact with its surroundings, cells need to transport molecules across membranes, detect and respond to external molecules and to interact with other cells. These processes are particularly complex in the eukaryotic cell, and are mediated by a host of peripheral, integral and transmembrane proteins. The lipid bilayers themselves are a complex mosaic of different lipids, with cholesterol, sphingolipids and lipo-proteins and membrane proteins forming “lipid rafts,” membrane microdomains serving to transiently compartmentalize membrane functions such as formation of the immune synapse [12].
Figure 2. Membrane protein topology.

A, Type I integral membrane protein with an alpha helical transmembrane domain and a cytoplasmic c-terminus; B, Type II integral membrane protein with an extracellular c-terminus; C, Type III and IV multi-pass transmembrane proteins (including GPCRs); D, a beta-barrel protein, such as the eight stranded, anti-parallel bacterial outer membrane protein OmpA; E, a lipid- or GPI-linked peripheral membrane protein; and F, a peripheral membrane protein with an alpha helix lying in the plane of the lipid bilayer.
A major challenge to the biochemical study of membrane proteins in general, and seven-transmembrane GPCRs in particular, has been the lack of robust recombinant expression systems resulting in purification of large (milligram) quantities of pure, functional material (for review, see [13]). In fact, despite intense efforts, only two GPCR crystal structures have been solved, that of the highly expressed native bovine rhodopsin [14] and the recombinant human beta-2 adrenoceptor [8]. Challenges include low-level endogenous expression, poorly understood folding and stability pathways, host cell toxicity and the need to solubilize these integral membrane proteins with detergents or lipids. However, advances are being made using various expression hosts and fusion proteins, with bacterial systems able to produce 0.5–2 mg/L canabinoid and bradykinin receptors [15], respectively, and the yeast S. cerevisiae producing ~4 mg/L adenosine A2A receptor [16].
Experimental approaches for analysis of membrane proteins
Soluble membrane proteins
Membrane proteins are frequently studied using a variety of “soluble” formats because of the ease of experimentation. In the simplest case, proteins tethered to the membrane via a single pass alpha helix or lipid-linked anchor are simply produced as truncated extracellular variants. Because the functional domain folds independently of the anchor, truncation usually results in a properly folded soluble variant of the original membrane protein which faithfully reproduces many protein functions. Truncation has been widely used, especially for analysis of immune recognition proteins with low expression levels and weak binding affinities, such as the T cell receptor and major histocompatibility complex proteins [17, 18], which limits analysis on the cell membrane. For multi-pass transmembrane proteins such as GPCRs, which have significant hydrophobic domains and altered tertiary structures and binding affinities in the absence of a lipid bilayer, two options are available. Surfactant screening can identify a detergent whose presence allows the protein to be purified from the cell membrane while retaining function [19]. Alternatively, the hydrophobic surface residues usually in contact with the lipid tails of the membrane can be altered to hydrophobic residues to generate a completely solubilized variant, an approach which has resulted in crystallization of the pentameric transmembrane protein phospholamban [20]. While successful, there is a valid concern is that the amino acid necessary for solubility may modify the protein’s function and compromise interactions with accessory proteins.
Cell capture technologies
When recombinant soluble expression is not an option, or when membrane proteins need to be studied in situ, binding of soluble ligands can be used to measure the binding affinity and approximate the number of receptors on the cell surface. Typically labeled with fluorescent or radioactive probes, the soluble ligand is incubated with cells prior to analysis by low throughput methods such as flow cytometry or ELISA. This approach is widely used for its ease but is unable to deconvolute complexity – for instance, if co-receptors are involved in binding and influence the binding kinetics, this information is lumped into a single equilibrium binding constant. In an effort to access the same information with high-throughput and multiplexing capabilities, ligand capture has been extended to array formats, in which the soluble binding partner is immobilized in a feature on the array and cells specifically binding the ligand are quantified under equilibrium binding conditions.
This approach has been used most extensively with antibody arrays, in which a panel of monoclonal or recombinant antibodies specific for different membrane proteins are immobilized in discrete features on the array surface. A report by Borrebaeck et al used 20 recombinant single-chain antibodies recognizing different cell-surface receptors to detect corresponding cells in mixed cell populations, representing a semi-quantitative technology for rapid profiling of the plasma membrane [21]. Similar immobilized antibody arrays have been used to phenotype characterization of leukemic, stem and blood cells and have also been combined with planar wave-guide detection systems [22]. Immobilized pMHC complexes have created arrays for T cell capture to characterize cellular immune responses to cancer and vaccination [23–25]. While these arrays are readily adapted to high-throughput analysis, their reliance on equilibrium-based measurements limits the quality of the information. For instance, two anti-HIV antibodies binding the same protein with similar Kd, ~35 nM achieved equilibrium behavior with very different binding mechanisms, as the on-rates differed by five-fold while the off-rates differed by six-fold [26].
Supported lipid bilayers
A compromise between the completely native environment of the cell membrane and the ready analysis of a soluble protein is a supported lipid bilayer (SLB), in which membrane proteins and lipids are immobilized on a solid support (see Figure 3). In this format, membrane proteins are analyzed in native or near-native environments with the practical appeal of easy preparation, stability, patterning and availability of compatible surface characterization techniques. First exploited to study the requirements for T cell activation [27] and the interaction of cholera toxin with the cell surface ganglioside GM1, SLBs can be formed by vesicle fusion, microcontact printing or direct deposition of lipids onto a solid surface to achieve protein-lipid ratios between 1:500 to 1:5000 for large transmembrane proteins (see supporting material for more information). The key advantages are that the solid support confers excellent mechanical stability while the lipids retain their fluid nature and the system is compatible with many surface characterization techniques. Supported lipid membranes on silicon or SiO2-based substrates have been successfully used as a model systems for investigating natural cell membranes in pioneering work by several groups [28–31]. Detection can be achieved by a number of optical techniques, including fluorescence, SPR and plasmon waveguide analysis.
Figure 3. Membrane protein immobilization for in vitro analysis.

A, capture of detergent-solubilized membrane proteins by a c-terminal peptide tag and an immobilized antibody; or formation of lipid layers, including, B, lipid monolayers self-assembled on hydrophobic surfaces, including the BIAcore HPA chip; C, lipid bilayers formed on hydrophilic surfaces; D, tethered or polymer-cushioned lipid bilayers reduce frictional drag of membrane proteins along the solid surface; E, capture of vesicles by single-stranded DNA tethers, anti-LPS antibodies or the L1 BIAcore chip; F, localized SPR signal from nanocrystals; G, suspended lipid bilayers over nanopores to allow access to both sides of the lipid bilayer; and H, dropley interface bilayers [42].
A thin layer of water (1–2 nm) beneath the lipid layer acts as a lubricant and allows lateral and rotational mobility. However, there is evidence of friction between the lipids and the solid support, as lipid diffusion coefficients in a supported bilayer are more than two times slower than in a free-floating bilayer under identical conditions [32]. Mobility of transmembrane proteins is ever further reduced, due to drag of the external loops against the surface and incorporation of native membrane proteins with large intracellular domains is impossible [33]. The common solution is to lift the bilayer away from the solid support by some type of spacer molecule, such as a polymer cushion [34], a hydrogel [30] or a DNA tether [35]. However, it has been challenging to form stable lipid bilayers on planar noble metal films (gold or silver) without extensive surface modifications [4]. The problem is compounded by the intrinsic roughness of as-deposited metal films, which interferes with lipid membrane formation and reduces transmembrane protein lateral mobility and function. While the use of a polymer cushion or a hydrogel layer [36] (see Figure 3) can heal these surface defects, the addition of a passivation layer can sharply degrade the SPR detection sensitivity. Tethered lipid bilayers can partially overcome this challenge by lifting the membrane a few nanometers above the substrate [37], it requires difficult chemistry, and the membrane is only accessible from above, making this technique not readily applicable to natural cell membranes.
Suspended lipid bilayers
An even more physiologic environment for analysis would be a free-standing or suspended lipid bilayer, allowing the membrane to be addressable from both sides of the bilayer. Early efforts to create suspended lipid bilayers over micron-sized pores (so called “black lipids” because of their black appearance) were limited by the poor stability of the suspension. Recent developments in nano-fabrication have allowed the metal substrate to be machined to include nanometer-sized pores (~30 nm to microns). Lipid bilayers deposited by vesicle fusion, Langmuir-Blodgett or detergent dialysis techniques (similar to the methods described for SLBs above, see [38] for review) span these pores which can be characterized by AFM indentation force as a measure of elasticity [39, 40]) or electrochemical impedance spectroscopy. These experiments have revealed that the bilayers respond to stress by local bending rather than lateral tension. Danelon et al [41] were able to spread native membranes across silicon nitride films containing apertures of 50–600 nm in diameter and total surface areas of coverage of 100 µm2. Remarkably, not only did this approach allow access to both sides of the membrane, but it preserved the native orientation of the membrane proteins.
SPR instrumentation
For a variety of applications, including membrane protein ligand screening, biomarker discovery and cellular signaling, it is critical to measure and quantify binding rates and affinities, and not only the mere presence of binding events easily obtainable with basic fluorescence imaging. Surface Plasmon Resonance (SPR) techniques enable such real-time, label-free quantification of molecular binding kinetics and affinities [43–47] and are currently the gold standard for quantifying the binding kinetics of molecules. In these techniques, capture molecules immobilized on a thin gold film are immersed in a liquid solution containing analytes, and surface plasmon waves probe the molecular activity on the surface (see Figure 1).
A surface plasmon (SP) wave is a rippling motion of the conduction electrons of a metal (typically gold), right at the interface between the metal and a sample solution. As an SP wave propagates along a gold-liquid interface, its wavelength changes when it encounters a thin layer of biomolecules bound to the gold film. By monitoring the changing behavior of the SP waves in real time, affinity and binding kinetics between capture molecules immobilized on the gold surface and target molecules in the liquid can be obtained. Since SPs are coupled to free electrons, for a given energy they have a larger momentum than free-space electromagnetic waves, necessitating various geometries to increase the momentum of the exciting light, such as the use of an optical prism or grating. In BIAcore™, a convergent light cone illuminates the detection spot (~1.6 mm spot size) on a gold film via prism coupling in total internal reflection mode. The angular distribution of the reflected light is measured by a photodiode array in real-time, scanning for a steep drop in intensity that indicates the resonant excitation of SPs. As molecules bind to the surface of the gold, the resonance angle changes. This gives a local refractive index sensitivity of Δn/n ~10−6. In various formats, this technique has found wide application in pharmaceutical development (small molecules and proteins) and in basic research and has also been successfully commercialized [48].
In contrast to radioactive or fluorescent labeling methods, label-free SPR kinetic assays provide several unique advantages: 1) ligand-analyte binding kinetics can be probed without the costly and time-consuming labeling process that can also interfere with the binding interactions; 2) binding kinetics and affinities can be measured directly, as opposed to only the mere presence of binding events; and 3) a wide range of molecular interactions – especially low affinity interactions that require a large amount of antibodies for saturation – can be characterized with less reagent consumption than other equilibrium measurement techniques.
SPR technology for membrane proteins: state of the art and challenges
While the SPR technique has been successfully commercialized by several companies, most notably BIAcore™ (GE Healthcare), its main function has been measuring the average affinity between known pairs of purified proteins immobilized over a large area (~1 mm2) of the gold surface. For many membrane protein applications, a new class of SPR technology is needed that is capable of directly measuring antibody-antigen interactions occurring on a cell membrane at a much higher spatial resolution than BIAcore™ can offer, and with a consequent increase in multiplexing capabilities. Furthermore, a solid gold film as the sensing surface does not provide a natural environment to study cell-surface antigens that are positioned within a lipid bilayer, as the procedures of isolating and immobilizing membrane proteins often adversely affects their function.
For high-throughput, functional studies of transmembrane protein binding kinetics using real-time label-free SPR techniques, the following key challenges must be addressed: throughput, imaging resolution, and maintenance of biological function of transmembrane proteins on a gold film. In its current implementation, BIAcore™ is a low throughput instrument that can measure binding kinetics from only four channels with an associated cost of $300K. For membrane protein microarray applications, it is necessary to simultaneously measure kinetics from thousands of samples spots. SPR microscopy (sometimes called SPR imaging) based on a similar setup is one such technique. Another type of high-throughput SPR instrument utilizes a diffraction grating instead of a prism, to convert incident light into SP waves. The FlexCHIP system uses this mechanism to measure binding kinetics from 400 sample spots, but at a reduced sensitivity compared to BIAcore™. Both approaches, however, suffer from low imaging resolution and limited field-of-view, because the image plane is tilted at a sizable angle to the sample surface, creating significant optical aberrations and prohibits the use of high-resolution imaging optics. Finally, while BIAcore™ is good at measuring kinetics between capture ligands immobilized on gold surface and target molecules in solution, it cannot easily be applied to functional studies of membrane proteins because the gold surface of the sensor ship may perturb the biological activity of these proteins, as illustrated in Figure 2.
Conventional prism-based SPR platforms
Two specialized chips, the HPA and L1, have been developed by to facilitate membrane protein analysis on BIAcore™ systems. The hydrophobic association analysis, or HPA chip includes a covalently attached monolayer of long-chain alkanethiol groups attached to the gold surface. When injected over the surface, small unilamellar vesicles containing membrane proteins rupture and fuse to form a supported lipid monolayer on the surface of the chip. This chip has been used, for instance, to analyze recombinant antibodies binding LPS molecules and to demonstrate bacterial species selectivity [49]. In contrast, the L1 chip presents a surface coated with carboxymethyl dextran with terminal alkane groups to capture liposomes containing integral membrane proteins in a lipid bilayer. The exact form of the captured lipid membranes is not precisely known but appears to be dominated by captured liposomes rather than a lipid bilayer [50]. This chip was developed specifically to allow identification of orphan GPCR ligands by coupled SPR-mass spectrometry analysis, in which a library of potential ligands is injected, molecules binding non-specifically washed away while binding ligands are eluted and recovered for identification by mass spec. A direct comparison of the two chips analyzed coagulation factor VIII binding to synthetic membranes containing phosphatidyl choline and varying amounts of phosphatidyl serine (4% to 25%). In this study, the L1 chip provided superior sensitivity, most likely due to the presence of more binding sites due to the capture of vesicles versus planar bilayers. Apart from the different immobilization techniques, the chips are used in a similar manner as the standard CM5 chip, with cycles of binding and regeneration.
Early reports demonstrated that functional light-mediated activation of rhodopsin and the subsequent dissociation of G proteins could be monitored by plasmon-waveguide resonance systems in tethered lipid bilayers [51, 52] and used to measure GPCR-G protein affinities in the presence of agonists and antagonists [53]. Standard CM5 chips have been used to study binding of detergent-solubilized neurotensin receptor-1 GPCR to immobilized peptide ligand [54], while the L1 chip has been used to capture GPCR-containing micelles in a proof-of-concept experiment with transducin [55]. Myszka’s group has used two complementary approaches to study GPCRs on BIAcore chips. In the first incarnation, the CXCR4 GPCR (a co-receptor for the gp120 protein during HIV invasion of T cells) was expressed with a c-terminal peptide tag and purified in the presence of detergent. This solubilized CXCR4 was then immobilized via an antibody specific for the peptide tag. Second, to more closely mimic the receptor’s native environment, CXCR4 was immobilized from crude supernatant via the 1D4 tag-specific antibody on a chip with alkanes, followed by reconstitution of the lipid bilayer around the receptor [56].
Next generation SPR instrumentation based on nano-structured materials
Despite the success of BIAcore™ for ligand screening and pharmaceutical research, it is clear from the previous sections that there is a critical need for a new generation of SPR technology capable of high-throughput microarray sensing, detecting small ligands with higher sensitivity, and integrating transmembrane proteins. Emerging SPR technologies based on patterned nanostructures, such as noble metal nanoparticles and nanostructured metal films, provide new design freedoms, enhanced detection sensitivity, and the unique geometry to address some of these challenges. These nanostructured SPR sensors can be divided into two categories: 1) nanoparticle-based sensors utilizing localized surface plasmon resonance (LSPR) [57] and 2) the inverse structure utilizing nanopore arrays in a thin metal film [58]. While both systems harness collective oscillation of conduction electrons, the LSPR in nanoparticles and the propagating SPR wave in a metal film perforated with nanopores exhibit very different characters.
A particularly desirable feature of these patterned metal nanostructures is their ability to directly convert incident light into SPR, obviating the need for a bulky coupling prism used in the BIAcore™ system. On the curved surface of a metal nanoparticle, light can directly couple into a LSPR that has the symmetry of a time-varying dipole. For more detail, see “Online Supporting Material: Nanoparticle-based LSPR biosensors.” Similarly, a subwavelength hole patterned in a metal film can also efficiently couple incident light into SP waves. The elimination of a prism can considerably simplify the optical design, assembly and alignment of an SPR imaging system that is required for high-throughput imaging.
Furthermore, compared with an unpatterned gold or silver film, metal nanoparticles or nanopores can resonantly amplify the intensity of incident light by up to 10,000 times. Such strong field enhancement was shown to significantly increase the Raman scattering cross-section of surface-adsorbed molecules by as much as 1012, thereby facilitating the identification of the bound molecules via surface-enhanced Raman scattering (SERS) [59, 60]. While label-free SPR technology can measure the affinity of binding partners, it does not reveal a chemical signature for the bound molecules. By coupling SPR and SERS measurements in nano-structured metals, it will be possible to identify “hits” in a high-throughput SPR binding screen and then capture the vibrational signature of the bound target molecule using SERS, which will be an important step forward for biomarker discovery.
Nanopore SPR sensors
While the LSPR of metal nanoparticles give them strong plasmon resonance effects, the inverse structure, i.e. nanopores in a metal film, also exhibits unique SPR characteristics because of the extraordinary optical transmission (EOT) effect [61]. An obvious distinction between a nanopore array patterned in a continuous metal film and an array of disconnected nanoparticles is that the former can support propagating SPR (used in BIAcore™) whereas the latter can only sustain the short-range LSPR.
A single subwavelength nanopore milled through a thin gold film will transmit very little incident light. It can, however, convert the incident light into an SP wave, acting as a local source for SP waves, like a stone tossed into a pond will generate surface waves from a single point. When many of these nanopores, or SP sources, are arranged in a periodic array (Figure 4), at certain resonance wavelengths (Figure 4b), the SP waves constructively interfere and intensify, efficiently “funneling” their energy through the tiny nanopores. Molecules on the gold surface sharply modulate the resonance wavelength (Figure 4b) and this funneling process. On the other side of the gold film, these funneled SP waves are then re-converted into light, which freely propagates away. Overall, the optical transmission is far more efficient than one would expect considering only the openings created by the tiny nanopores. By continuously measuring the transmitted light, molecular binding events, binding rates and affinities can be monitored as a function of time. Thus each nanopore array behaves as a single SPR biosensor.
Figure 4.
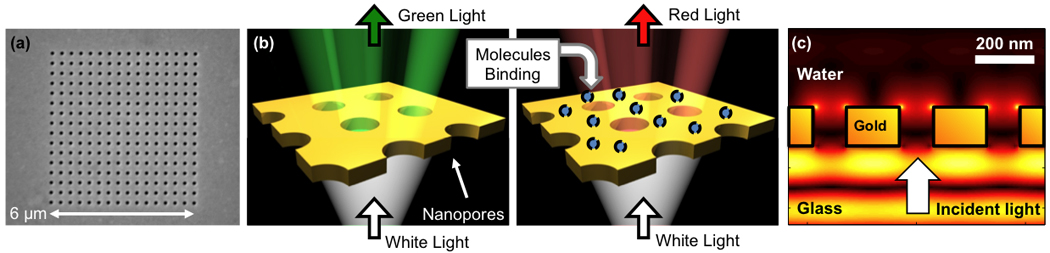
(a) A periodic array of nanopores milled through a thin gold film. (b) At resonant wavelengths, the incident light is efficiently transmitted, giving a sharp transmission peak that is easily monitored. As molecules bind, the peak shifts, modulating the transmission. (c) Side view: Computer simulation of light transmission (“funneling”) through a nanopore array. Intense optical energy is observed, confined within ~100 nm from the gold surface. Molecular binding on or near the gold surface sharply modulates this field distribution, and the optical transmission process, providing the basis for measuring binding events.
Several groups have demonstrated the potential of nanopore arrays for label-free SPR biosensing. Following the discovery of the EOT effect, Brolo et al. first demonstrated a proof-of-concept using periodic nanopore arrays in a gold film for biochemical sensing [62]. There, using a broadband light source and a spectrometer, they reported a 4nm shift of the EOT transmission peaks after the immobilization of a molecular monolayer on the gold surface. Using a tunable IR laser source (1520–1570 nm), Tetz et al. demonstrated refractive index sensing and estimated the sensitivity of periodic nanopore arrays to be close to 10−6, comparable to BIAcore™ [63]. Larson and coworkers demonstrated the potential of the nanopore platform for highly multiplexed analysis of ligand interactions [64–67]. Furthermore, recent advances in the fabrication of large-area nanopore arrays in a metal film [68, 69] show promise to push this technology further toward next generation SPR biosensing that is more sensitive, miniaturized, with the ability to multiplex and use very small amounts of sample. In our group, we recently reported using shape-enhanced periodic nanopore arrays in a microfluidic flow cell for real-time measurements of molecular binding with a 50% improvement in sensitivity [70].The shape-enhancement came from producing sharp apexes by overlapping two circular nanopores. For multiplex, microarray applications, Lesuffleur et al. used periodic nanopore arrays with laser illumination and an imaging camera, which was also incorporated with multiple microfluidic channels as shown in Figure 5 [71, 72].
Figure 5.

Real-time SPR sensing platform based on periodic nanopore arrays: (a) With a standard microscope, CCD camera, and laser, a (b) microfluidic chip with (c) multiple parallel channels is (d) illuminated from below and imaged. Each bright spot is a single nanopore array, whose brightness changes with molecular binding events [72].
Later, Lindquist, et al. demonstrated sub-micron-resolution nanopore-based SPR imaging with enhanced sensitivity and sensor-to-sensor isolation (Figure S1) [73]. Recent work by Ferreira et al. has even shown that each nanopore on a glass substrate can detect attomolar concentrations of proteins using in-hole SPR effects [74]. Detailed reviews of the physics and applications of nanopore arrays in metal films can be found in a review article by Gordon et al. [75]. Using a random array of nanopores, it is possible to perform LSPR detection, since there is no longer any long-range order to support travelling SP and pore-to-pore interference effects, as in EOT. Dahlin et al. utilized this technique for membrane sensing [76]. However, the ability to harness propagating SPs in a continuous gold film can increase the possible probing range (beyond the 10~30 nm mentioned previously) as well as the tunability of the structure. The periodic nanopore structure, therefore, is uniquely suited for SPR biosensing, achieving high sensitivities, seamless integration with inexpensive optics in a transmission-mode setup, and high-resolution, highly multiplexed detection. The nanopore array is also distinctive in its geometry, which may be suitable for investigating trans-membrane proteins.
Novel SPR sensing scheme based on suspended lipid membrane over nanopores
Toward membrane protein sensing applications, we note that nanopore arrays provide a unique geometry, since a thin lipid bilayer can be suspended over the nanopores while maintaining mechanical stability and being surrounded by a buffer on both sides (Figure 6). Membrane proteins can thereby be seamlessly integrated with the SPR sensing capability of periodic nanopore arrays, maintaining their functionality in an environment that more closely mimics their natural state. Furthermore, membrane proteins integrated in the free-standing lipid bilayer can be easily accessed from both sides, making this approach more attractive for studying membrane protein interactions than planar lipid bilayers supported on a flat substrate. Each lipid monolayer will join at the nanopore area to form bilayers spanning the nanopore. While the formation of pore-spanning lipid bilayers was previously studied by several groups using atomic force microscopy (AFM) or impedance measurements [38, 39, 77, 78], the unique ability of metallic nanopore arrays that can concurrently act as a mechanical support for lipid membrane as well as an ultra-sensitive SPR biosensor has not been realized.
Figure 6.
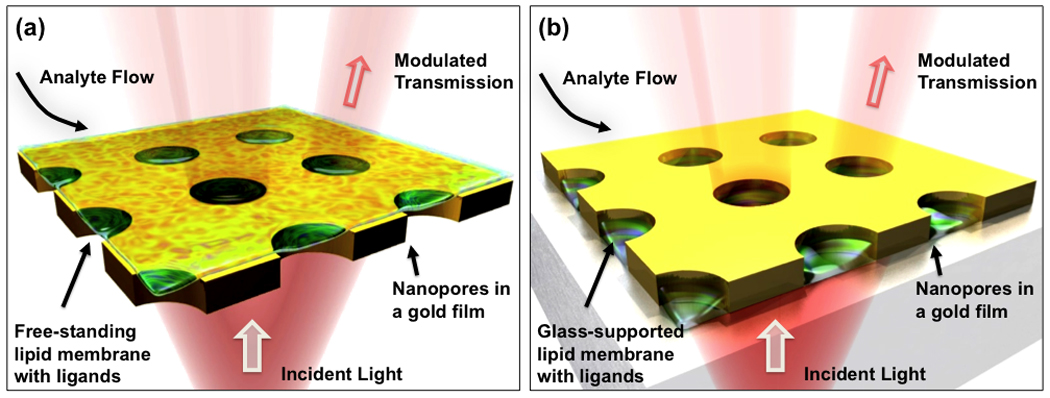
Proposed nanopore sensing schemes: (a) A cell membrane is reconstituted on a glass substrate and is surrounded by a thin gold film sidewall inside each nanopore. Ligands binding to transmembrane proteins can drastically modulate light transmission through the nanopores, enabling label-free SPR measurements. (b) A cell-membrane that is freestanding and surrounded with a buffer solution on both sides.
Most existing work for making periodic nanopore arrays relied on milling metallic holes through a metal film deposited on a glass substrate. This process results in dead-ended nanopores, suitable for substrate-supported lipid membranes (Figure 6a). A few groups have demonstrated processing schemes for making free-standing nanopores, suitable for flow-through SPR sensing [79] or for lipid membrane sensing [80]. (Figure 7). The process typically begins with backside etching of a silicon wafer covered with a thin nitride film. A thin gold film is then deposited on the free-standing nitride membrane, through which the nanopores are then milled. Microfluidic channels can then access both the top and the bottom openings of the nanopores, allowing both sides of a suspended lipid membrane to be in contact with a buffer solution (or with two different buffers, as is the case for the inside and outside of a cell).
Figure 7.
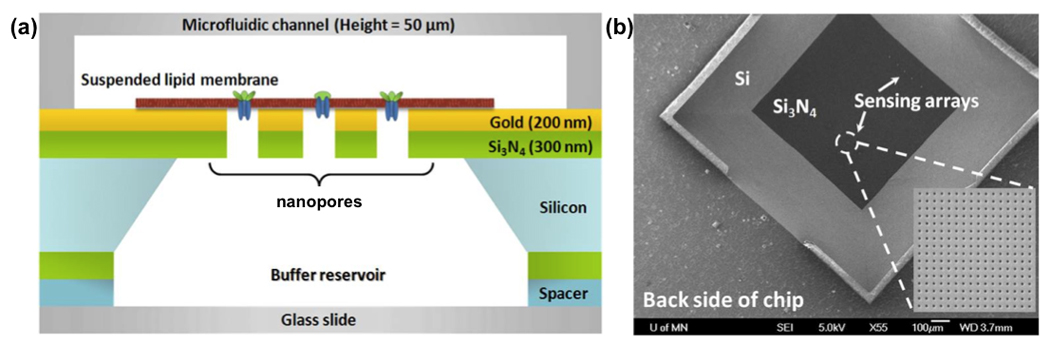
Schematic of the nanopore platform made on a suspended nitride membrane. (A) The ability to fill both sides of the membrane with a buffer and access them makes this geometry a unique platform to study transmembrane proteins. (B) SEM image of a free-standing silicon nitride membrane on a silicon wafer (flipped for imaging). Adapted from [80].
We believe the scheme proposed in Figure 6 and Figure 7 provides a new platform for studying transmembrane proteins such as GPCRs and ion channels. Small nanopores perforated through a thin gold layer are ideally suited to provide mechanical support, since smaller diameter free-standing membranes are more stable than larger diameter membranes, and for detection of molecular binding events, since the gold film sustains SPR effects. Importantly, such a set-up would allow transmembrane proteins to be presented in native or near-native environments and allow interrogation from both sides of the membrane. This will be particularly important for fundamental studies of signal transduction, in which ligand binding on the extracellular side of the membrane triggers association and dissociation of multiple membrane proteins on the internal or cytoplasmic side of the membrane, such as occurs during GPCR activation.
APPLICATIONS OF SPR TO MEMBRANE PROTEINS
G-protein coupled receptors (GPCR) ligand screening
GPCRs are the largest family of membrane proteins in human genome and while sequence homology across the family is low, all exhibit a seven-transmembrane α-helical topology. The majority of hormones and neurotransmitters communicate extracellular information to cells via GPCRs, and drugs acting on GPCRs can impact a broad spectrum of diseases. While endogenous ligands have been proposed for several hundred GPCRs, there remain over 100 “orphan” GPCRs for which ligands have yet to be identified and likely represent opportunities for new drug development. Moreover, even for those GPCRs for which suitable pharmacologically active drugs have been identified, modified ligands with greater binding specificity, affinity and selectivity for a given GPCR could represent an improved drug. Importantly, it has been observed that the ligand equilibrium dissociation constant (Kd) scales with biological responses, such that a partial agonist is less avid than a full agonist. These dissociation constants range from 0.2 – 3000 nM for the small molecule and peptide ligands (0.5 – 8 kDa) [81], falling precisely within the normal detection window of an SPR instrument. Sensitive, high throughput activity screens are currently used to identify novel and more efficient molecules from large chemical libraries, although with the recent advances in recombinant GPCR expression and structural characterization [82], structure-based drug design is becoming an increasingly attractive approach.
Currently available assays to assess GPCR ligand binding affinity and specificity fall into two main categories: those that use radio-labeled ligands to measure binding to cells over-expressing a specific GPCR [81] and those that use complex cell-based assays to indirectly measure downstream events of the signal transduction cascade (e.g., intracellular cAMP or intracellular Ca2+ concentrations, see Figure 8 [83]). A major limitation of the simple cellular receptor binding assay is the uncertainty in the GPCR concentration [84] and that over-expressed GPCRs may exhibit constitutive activation. Molecular assays, such as those employing SPR imaging, that directly report ligand binding kinetics and G-protein activation could bridge the gap between simple binding assays and the complexity of cell-based systems. An ideal GPCR screen should be simple, non-radioactive, with a high signal to noise ratio, contain minimal reagent additions and be amenable to automation [85].
Figure 8. GPCR-G protein coupled activation.

In step (1), the agonist-GPCR interaction promotes a series of conformational changes favoring GPCR interactions with G proteins. In step (2), formation of an agonist-GPCR-G protein tri-molecular complex induces G protein conformational changes resulting in (3) the exchange of the α subunit bound GDP for GTP. Step (4), the activated G protein dissociates to form a GTP-bound α subunit and a βγ complex. The dissociated G proteins then regulate the activity of a number of intracellular effector proteins, resulting in changes in cAMP or calcium levels and regulation of signal transduction pathways. These activities stop when the GTP is hydrolyzed to GDP and the αβγ G protein complex reforms. [Adapted from [83].]
SPR imaging has the potential to address the limitations of current GPCR ligand screening methods, although the sensitivity and throughput remain inadequate for screening of large chemical libraries. Three distinct SPR approaches include (for review, see [86]. (1) Immobilization of GPCR in lipid bi-layers, with ligand-binding dose-response curves monitored by G-protein alpha unit dissociation and the consequent decrease in SPR signal [52] Because the Gα sub-unit is relatively large (~45 kDa), the SPR signal is more sensitive than that resulting from direct monitoring of small molecular weight ligand binding (~0.5–8 kDa). (2) Antibody-capture or immobilization of detergent-solubilized GPCR, followed by SPR imaging of ligand binding [56, 87, 88]. (3) Immobilization of biotinylated ligand, followed by capture of the high molecular weight, detergent-solubilized GPCR [54]. The most appealing approach for development of high-throughput screens is immobilization of the GPCR in a suspended lipid bilayer across a nanopore. In this way, ligand can be added to one side of the bilayer with the G protein attached to the other side. GPCR activation could then be sensitively and directly monitored by α subunit dissociation, without the complications associated with monitoring downstream functional effects, such as changes in cAMP levels.
Biomarker discovery with membrane-bound antigens
Both cancer and autoimmune diseases induce auto-antibodies – cancer due to expression of protein variants or disregulation of key proteins which can be recognized by the humoral immune system, aberrant recognition of self-antigens in autoimmune disease. When considered alone as diagnostic tools, most auto-antibodies show poor sensitivity and/or specificity for their associated diseases. While it is usually difficult to identify a single biomarker for which the presence of specific antibodies is diagnostic of disease presence and severity, there is evidence that auto-antibody binding patterns can indicate disease pathology and severity years before the onset of clinical symptoms [89]. For instance, there is some evidence that the occurrence of auto-antigens to specific antigens in lung cancer may have prognostic relevance and tumor regression has been demonstrated in some patients with small cell lung carcinoma and auto-antibodies to onconeural antigens [90]. Similarly, a set of 18 signaling proteins has been identified that can distinguish Alzheimer’s from control patients with 90% accuracy [91].
Sensitive, high-throughput, equilibrium-based technologies have been developed for analysis of antibodies binding soluble proteins. These include antigen arrays to detect serum auto-antibody responses to soluble antigens immobilized on a microarray [92]. These arrays simultaneously detect femtomolar concentrations of antibodies recognizing up to 230 antigens while utilizing very small volumes of patient samples [93]. Other emerging technologies include multiplexed assays using fluorescent microspheres, technology developed by Luminex and which has been licensed by three companies for lupus characterization [94]. Auto-antibody binding patterns of soluble antigens have primarily been characterized, because these are readily accessible with the current technology. However, auto-antibodies also recognize intact cells and thus membrane proteins. For instance, antigen microarrays have identified unique patterns of antibodies binding lipids in patient samples which predict disease pathology in Alzheimers and multiple sclerosis (MS) [95]. Analysis of auto-antibody binding to membrane protein antigens via a high-throughput nanopore SPR array would further enhance the power of this approach by monitoring not just patterns of binding but also the kinetics, as clinical relevance may correspond to high- or low-affinity auto-antibodies.
Autoantibody-based therapeutics binding membrane proteins
Not only do the binding patterns of auto-antibodies have diagnostic potential, some of these auto-antibodies are mechanistically involved in repair of disease and may ameliorate disease when administered therapeutically. IgM antibodies binding asialo-Gm1 glycolipids have been successfully characterized using SPR and Gm1 containing liposomes [96]. Approved antibody based therapeutics are used to treat cancer, inflammatory diseases, transplantation recipients, infections and cardiovascular disease and have a high rate of approval as drugs compared to small molecule based drugs [97].
By isolating mAbs from humans with monoclonal gammopathy, a condition in which the individual carries the mAb in high concentration for long periods of time, and focusing only on those individuals free of antibody-based disease, candidate mAbs can be isolated that have already been tested for long term, high dose, toxicity in at least one human [98, 99]. Human mAbs from serum were selected based on cell surface binding and then assayed for efficacy in models of disease [100–102]. Mouse and human mAbs have been isolated that promote CNS protection and repair, bind specifically to surface plasma membrane antigens, activate intracellular signals that promote neuron or glial cell survival [103] and cross the blood-brain-barrier to accumulate within injured regions of the CNS [101]. This process results in candidate mAbs with proven in vivo efficacy and a degree of toxicology data, but without an identified antigen or mechanism of action. In order to transition to clinical trials, data regarding the antigen and mechanism of protection would greatly increase the probability of a molecule’s regulatory approval.
Some of the target antigens bound by reparative mouse mAbs are known and all antigens are lipids or carbohydrates [104]. Our data suggests that the reparative IgMs, which have a total of 10 antigen binding sites, do not bind to a single membrane molecule, but to a membrane micro-domain complex composed of multiple antigens. If this native membrane complex is disrupted IgM binding to the target cell is lost. Cell and tissue specificity of the reparative IgMs is maintained only when bound to intact plasma membranes. When candidate antigens are presented in isolated form such as an ELISA, the IgMs often bind non-specifically. Therefore, a new antigen screening technology is required to study these difficult, but critical lipid and carbohydrate molecules of the plasma membrane antigens in their native state to preserve appropriate antibody binding kinetics. It has not been possible to use a commercial BIAcore™ system to model the complex interactions of these natural autoantibodies with cell surface antigens because studying individual proteins, which the BIAcore™ does well, does not allow the study of a multiple antigen complex. The combination of SPR with suspended lipid bilayers spanning nanopores has the potential to identify the individual components of the membrane complex recognized by the IgMs. One example of a therapeutic antibody is an IgM auto-antigen whose binding to white matter in the CNS promotes remyelination in in vitro and in vivo models of MS (see Figure 9). SPR analysis would facilitate the study of reconstituted myelin membranes isolated from glycolipid knock out mice and allow the introduction of candidate antigens back into these membranes for IgM binding studies.
Figure 9.

Application of antigen identification in reconstituted membrane binding screening assays. A, A human IgM that binds to the surface of neurons (green label) promotes neurite extension from rat cerebellar granule cells when presented as a substrate. This IgM was identified by screening for biologic properties; while the antigens recognized are unknown, preliminary evidence suggests the antigens are lipids [105]. B and C, A recombinant human IgM that promotes repair in models of demyelination such as multiple sclerosis binds specifically to the central white matter in an unfixed slice of mouse cerebellum, immunocytochemistry (B), whole tissue visualized by phase contrast (C). This IgM binding specificity is maintained only on live tissue, and is lost using frozen or fixed specimens, suggesting intact cell membrane is critical for IgM binding. D, Proposed model of reparative IgM binding which could be tested by SPR on supported membranes. The pentameric IgM binds to the surface of a target cell; antibody multivalency initiates clustering of plasma membrane molecules and activation of signaling which can lead to cellular responses such as proliferation, differentiation, or increased resistance to apoptosis.
Basic membrane biology
The union of SPR and SLB technologies will illuminate many fundamental issues in biology, from the basic physics of lipid membranes to membrane biogenesis and the molecular details of cellular interactions [106]. For instance, the fine details of HIV fusion with cell membranes [107], bacterial outer membrane protein transport, assembly and insertion into the outer membrane [106], membrane protein diffusion [108, 109] and formation of lipid raft structures [110] are all important questions which are beginning to be quantitatively addressed with SPR monitoring of supported lipid bilayers.
T cell receptor-pMHC interactions
Antibodies are currently one of the most rapidly growing classes of therapeutic molecules [111, 112], able to treat solid and circulating tumors and limit inflammation associated with autoimmune reactions by virtue of specific, high affinity ligand recognition [113]. In contrast, the exclusively membrane-bound cellular immune system, which plays a central role in defense against cancer and viral infections and the pathology of auto-immune diseases such as diabetes, is much less well understood [114–116]. T cell discrimination between self versus non-self occurs based on the tri-partite binding kinetics between a T cell receptor (TCR) and peptide antigens presented by major histocompatibility complex (MHC) proteins on a cell. Proteins produced intracellularly or ingested from the external milieu are proteolyzed into short peptides approximately nine amino acids residues long and complexed with MHC proteins. After trafficking of the peptide-MHC (pMHC) complex to the cell membrane, the composite surface is surveyed by αβ T cell receptors (for review, [117]). The outcome of a productive TCR-pMHC binding interaction depends on the subclass of T cell involved, but can include induction or repression of immune responses or lysis of the pMHC-bearing cell, with direct relevance to vaccine design and anti-cancer therapeutics.
Key issues in TCR-pMHC recognition include: (1) identification of immunodominant peptides bound by both the MHC and TCR; (2) identification of TCRs binding known pMHC complexes; and (3) characterization of the TCR-pMHC binding kinetics resulting in T cell activation or de-activation. Currently there is a paucity of methods for characterizing TCR molecules and peptides associated with disease; in general these approaches study the interaction indirectly using whole cells or quantitatively using artificial soluble pMHC variants. Recombinant systems for production of soluble variants of TCRs and pMHCs have been developed [17, 18, 118, 119], but still require significant effort, including identification of solubilizing mutations for each unique receptor studied. The resulting soluble pMHC tetramers have been immobilized on arrays and used to capture T cells in order to identify activating peptides and characterize T cell responses to a peptide vaccine against melanoma [23–25, 120, 121]. Supported lipid bilayer technology was first developed to study the TCR-pMHC binding interaction under more physiologic conditions [27]. Peptide-MHC molecules were purified from APCs, immobilized on a solid support and T cells allowed to bind, which was monitored by fluorescence. This technology has been extended in conjunction with modern photolithographic techniques and multi-parameter fluorescent protein labeling to visualize the coordinated movement of TCR-pMHC and co-stimulatory molecules during formation of the “immune synapse,” a pre-requisite for T cell activation [122–125]. The ability to monitor TCR-pMHC interactions both quantitatively and in the context of a native lipid membrane would represent a major advance.
Conclusions
For two driving industrial biological needs, ligand screening and biomarker discovery with membrane proteins, as well as fundamental research in membrane biology, currently available quantitative screening technologies such as BIAcore™ have limitations. The systems have been retrofit to accommodate the unique needs of membrane proteins, but still suffer from, for instance, the poorly defined form of the lipid bilayer coupled to L1 BIAcore chips™. The problem is compounded for transmembrane proteins such as GPCR because proteins in direct contact with a solid substrate (in particular the gold substrate in BIAcore™) often lose their functionality or denature. The nanopore-based dynamic sensing architecture in development by several groups has the unique potential to overcome this challenge, since each nanopore sits on a glass substrate and forms a tiny well in which to confine supported lipid membranes, while the surrounding gold film provides SPR effects to dynamically measure the binding kinetics of molecules onto the membrane. Furthermore, this nanopore geometry offers the intriguing possibility of suspending lipid bilayers over metallic nanopore arrays to mimic the structure of natural biological membranes as proposed herein. This new platform is beginning to be used and we anticipate a number of breakthroughs in biological research.
Supplementary Material
Acknowledgements
J.A.M. was supported by the NIH (AI #066239), the Packard Foundation (#2005-098). S.-H.O., A.E.W. and M.R. acknowledge support by the Minnesota Partnership Award for Biotechnology and Medical Genomics. M.R. was supported by grants from the NIH (NS RO1 32129, NS RO1 24180), the National Multiple Sclerosis Society (R63172, CA 1011A8) and the Applebaum and Hilton Foundations.
Abbreviations used
- GPCR
G protein coupled receptor
- ELISA
enzyme linked immuno-sorbent assay
- EOT
extraordinary optical transmission
- LSPR
localized surface plasmon resonance
- MS
multiple sclerosis
- SLB
supported lipid bilayer
- SPR
surface plasmon resonance
- SUV
small unilamellar vesicle
References
- 1.Wu H, Pfarr DS, Johnson S, Brewah YA, et al. Development of motavizumab, an ultra-potent antibody for the prevention of respiratory syncytial virus infection in the upper and lower respiratory tract. J Mol Biol. 2007;368:652–665. doi: 10.1016/j.jmb.2007.02.024. [DOI] [PubMed] [Google Scholar]
- 2.Cunningham BC, Wells JA. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science. 1989;244:1081–1085. doi: 10.1126/science.2471267. [DOI] [PubMed] [Google Scholar]
- 3.Rickert M, Boulanger MJ, Goriatcheva N, Garcia KC. Compensatory energetic mechanisms mediating the assembly of signaling complexes between interleukin-2 and its alpha, beta, and gamma(c) receptors. J Mol Biol. 2004;339:1115–1128. doi: 10.1016/j.jmb.2004.04.038. [DOI] [PubMed] [Google Scholar]
- 4.Cooper MA. Advances in membrane receptor screening and analysis. Journal of Molecular Recognition. 2004;17:286–315. doi: 10.1002/jmr.675. [DOI] [PubMed] [Google Scholar]
- 5.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 7.Day PW, Rasmussen SG, Parnot C, Fung JJ, et al. A monoclonal antibody for G protein-coupled receptor crystallography. Nat Methods. 2007;4:927–929. doi: 10.1038/nmeth1112. [DOI] [PubMed] [Google Scholar]
- 8.Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 9.Burg TP, Godin M, Knudsen SM, Shen W, et al. Weighing of biomolecules, single cells and single nanoparticles in fluid. Nature. 2007;446:1066–1069. doi: 10.1038/nature05741. [DOI] [PubMed] [Google Scholar]
- 10.Cui Y, Wei QQ, Park HK, Lieber CM. Nanowire nanosensors for highly sensitive and selective detection of biological and chemical species. Science. 2001;293:1289–1292. doi: 10.1126/science.1062711. [DOI] [PubMed] [Google Scholar]
- 11.Armani AM, Kulkarni RP, Fraser SE, Flagan RC, et al. Label-free, single-molecule detection with optical microcavities. Science. 2007;317:783–787. doi: 10.1126/science.1145002. [DOI] [PubMed] [Google Scholar]
- 12.Edidin M. Lipids on the frontier: a century of cell-membrane bilayers. Nat Rev Mol Cell Biol. 2003;4:414–418. doi: 10.1038/nrm1102. [DOI] [PubMed] [Google Scholar]
- 13.Grisshammer R, White JF, Trinh LB, Shiloach J. Large-scale expression and purification of a G-protein-coupled receptor for structure determination -- an overview. J Struct Funct Genomics. 2005;6:159–163. doi: 10.1007/s10969-005-1917-6. [DOI] [PubMed] [Google Scholar]
- 14.Palczewski K, Kumasaka T, Hori T, Behnke CA, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 15.Link AJ, Skretas G, Strauch EM, Chari NS, et al. Efficient production of membrane-integrated and detergent-soluble G protein-coupled receptors in Escherichia coli. Protein Sci. 2008;17:1857–1863. doi: 10.1110/ps.035980.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O'Malley MA, Lazarova T, Britton ZT, Robinson AS. High-level expression in Saccharomyces cerevisiae enables isolation and spectroscopic characterization of functional human adenosine A2a receptor. J Struct Biol. 2007;159:166–178. doi: 10.1016/j.jsb.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scott CA, Garcia KC, Stura EA, Peterson PA, et al. Engineering protein for X-ray crystallography: the murine Major Histocompatibility Complex class II molecule I-Ad. Protein Sci. 1998;7:413–418. doi: 10.1002/pro.5560070222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maynard J, Adams EJ, Krogsgaard MK, Petersson K, et al. High level bacterial secretion of single-chain T cell receptors. J Immunol Methods. 2005;306:51–67. doi: 10.1016/j.jim.2005.07.022. [DOI] [PubMed] [Google Scholar]
- 19.Rich RL, Miles AR, Gale BK, Myszka DG. Detergent screening of a G-protein-coupled receptor using serial and array biosensor technologies. Anal Biochem. 2009;386:98–104. doi: 10.1016/j.ab.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Slovic AM, Stayrook SE, North B, Degrado WF. X-ray structure of a water-soluble analog of the membrane protein phospholamban: sequence determinants defining the topology of tetrameric and pentameric coiled coils. J Mol Biol. 2005;348:777–787. doi: 10.1016/j.jmb.2005.02.040. [DOI] [PubMed] [Google Scholar]
- 21.Dexlin L, Ingvarsson J, Frendeus B, Borrebaeck CA, et al. Design of recombinant antibody microarrays for cell surface membrane proteomics. J Proteome Res. 2008;7:319–327. doi: 10.1021/pr070257x. [DOI] [PubMed] [Google Scholar]
- 22.Ghatnekar-Nilsson S, Dexlin L, Wingren C, Montelius L, et al. Design of atto-vial based recombinant antibody arrays combined with a planar wave-guide detection system. Proteomics. 2007;7:540–547. doi: 10.1002/pmic.200600485. [DOI] [PubMed] [Google Scholar]
- 23.Kwong GA, Radu CG, Hwang K, Shu CJ, et al. Modular nucleic acid assembled p/MHC microarrays for multiplexed sorting of antigen-specific T cells. J Am Chem Soc. 2009;131:9695–9703. doi: 10.1021/ja9006707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen DS, Soen Y, Stuge TB, Lee PP, et al. Marked differences in human melanoma antigen-specific T cell responsiveness after vaccination using a functional microarray. PLoS Med. 2005;2:e265. doi: 10.1371/journal.pmed.0020265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stone JD, Demkowicz WE, Jr, Stern LJ. HLA-restricted epitope identification and detection of functional T cell responses by using MHC-peptide and costimulatory microarrays. Proc Natl Acad Sci U S A. 2005;102:3744–3749. doi: 10.1073/pnas.0407019102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karlsson R. Real-time competitive kinetic analysis of interactions between low-molecular-weight ligands in solution and surface-immobilized receptors. Anal Biochem. 1994;221:142–151. doi: 10.1006/abio.1994.1390. [DOI] [PubMed] [Google Scholar]
- 27.Brian AA, McConnell HM. Allogeneic stimulation of cytotoxic T cells by supported planar membranes. Proc Natl Acad Sci U S A. 1984;81:6159–6163. doi: 10.1073/pnas.81.19.6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Groves JT, Ulman N, Boxer SG. Micropatterning fluid lipid bilayers on solid supports. Science. 1997;275:651–653. doi: 10.1126/science.275.5300.651. [DOI] [PubMed] [Google Scholar]
- 29.Boxer S. Molecular transport and organization in supported lipid membranes. Current opinion in chemical biology. 2000;4:704–709. doi: 10.1016/s1367-5931(00)00139-3. [DOI] [PubMed] [Google Scholar]
- 30.Sackmann E. Supported membranes: scientific and practical applications. Science. 1996;271:43–48. doi: 10.1126/science.271.5245.43. [DOI] [PubMed] [Google Scholar]
- 31.Salafsky J, Groves JT, Boxer SG. Architecture and function of membrane proteins in planar supported bilayers: a study with photosynthetic reaction centers. Biochemistry. 1996;35:14773–14781. doi: 10.1021/bi961432i. [DOI] [PubMed] [Google Scholar]
- 32.Przybylo M, Sykora J, Humpolickova J, Benda A, et al. Lipid diffusion in giant unilamellar vesicles is more than 2 times faster than in supported phospholipid bilayers under identical conditions. Langmuir. 2006;22:9096–9099. doi: 10.1021/la061934p. [DOI] [PubMed] [Google Scholar]
- 33.Merzlyakov M, Li E, Gitsov I, Hristova K. Surface-supported bilayers with transmembrane proteins: role of the polymer cushion revisited. Langmuir. 2006;22:10145–10151. doi: 10.1021/la061976d. [DOI] [PubMed] [Google Scholar]
- 34.Wagner ML, Tamm LK. Tethered polymer-supported planar lipid bilayers for reconstitution of integral membrane proteins: silane-polyethyleneglycol-lipid as a cushion and covalent linker. Biophys J. 2000;79:1400–1414. doi: 10.1016/S0006-3495(00)76392-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chan YH, Lenz P, Boxer SG. Kinetics of DNA-mediated docking reactions between vesicles tethered to supported lipid bilayers. Proc Natl Acad Sci U S A. 2007;104:18913–18918. doi: 10.1073/pnas.0706114104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kiessling V, Crane JM, Tamm LK. Transbilayer effects of raft-like lipid domains in asymmetric planar bilayers measured by single molecule tracking. Biophys J. 2006;91:3313–3326. doi: 10.1529/biophysj.106.091421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lang HL, Duschl C, Vogel H. A new class of thiolipids for the attachment of lipid bilayers on gold surfaces. Langmuir. 1994;10:197–210. [Google Scholar]
- 38.Reimhult E, Kumar K. Membrane biosensor platforms using nano- and microporous supports. Trends in Biotechnology. 2008;26:82–89. doi: 10.1016/j.tibtech.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 39.Steltenkamp S, Muller MM, Deserno M, Hennesthal C, et al. Mechanical properties of pore-spanning lipid bilayers probed by atomic force microscopy. Biophys J. 2006;91:217–226. doi: 10.1529/biophysj.106.081398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lorenz B, Mey I, Steltenkamp S, Fine T, et al. Elasticity mapping of pore-suspending native cell membranes. Small. 2009;5:832–838. doi: 10.1002/smll.200800930. [DOI] [PubMed] [Google Scholar]
- 41.Danelon C, Perez JB, Santschi C, Brugger J, et al. Cell membranes suspended across nanoaperture arrays. Langmuir. 2006;22:22–25. doi: 10.1021/la052387v. [DOI] [PubMed] [Google Scholar]
- 42.Maglia G, Heron AJ, Hwang WL, Holden MA, et al. Droplet networks with incorporated protein diodes show collective properties. Nat Nanotechnol. 2009;4:437–440. doi: 10.1038/nnano.2009.121. [DOI] [PubMed] [Google Scholar]
- 43.Liedberg B, Nylander C, Lundstrom I. Surface-Plasmon Resonance for Gas-Detection and Biosensing. Sensors and Actuators. 1983;4:299–304. [Google Scholar]
- 44.Mrksich M, Sigal GB, Whitesides GM. Surface-Plasmon Resonance Permits in-Situ Measurement of Protein Adsorption on Self-Assembled Monolayers of Alkanethiolates on Gold. Langmuir. 1995;11:4383–4385. [Google Scholar]
- 45.Shumaker-Parry JS, Zareie MH, Aebersold R, Campbell CT. Microspotting streptavidin and double-stranded DNA Arrays on gold for high-throughput studies of protein-DNA interactions by surface plasmon resonance microscopy. Analytical Chemistry. 2004;76:918–929. doi: 10.1021/ac034964v. [DOI] [PubMed] [Google Scholar]
- 46.Smith EA, Corn RM. Surface plasmon resonance imaging as a tool to monitor biomolecular interactions in an array based format. Applied Spectroscopy. 2003;57:320A–332A. doi: 10.1366/000370203322554446. [DOI] [PubMed] [Google Scholar]
- 47.Nelson BP, Grimsrud TE, Liles MR, Goodman RM, et al. Surface plasmon resonance imaging measurements of DNA and RNA hybridization adsorption onto DNA microarrays. Analytical Chemistry. 2001;73:1–7. doi: 10.1021/ac0010431. [DOI] [PubMed] [Google Scholar]
- 48.Homola J, Yee SS, Gauglitz G. Surface plasmon resonance sensors: review. Sensors and Actuators B-Chemical. 1999;54:3–15. [Google Scholar]
- 49.Yuan Q, Wang Z, Nian S, Yin Y, et al. Screening of high-affinity scFvs from a ribosome displayed library using BIAcore biosensor. Appl Biochem Biotechnol. 2009;152:224–234. doi: 10.1007/s12010-008-8251-y. [DOI] [PubMed] [Google Scholar]
- 50.Anderluh G, Besenicar M, Kladnik A, Lakey JH, et al. Properties of nonfused liposomes immobilized on an L1 Biacore chip and their permeabilization by a eukaryotic pore-forming toxin. Anal Biochem. 2005;344:43–52. doi: 10.1016/j.ab.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 51.Salamon Z, Wang Y, Soulages J, Brown M, et al. SPR spectroscopy studies of membrane proteins: transducin binding and activation by rhodopsin monitored in thin membrane films. Biophys J. 1996;71:283–294. doi: 10.1016/S0006-3495(96)79224-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bieri C, Ernst OP, Heyse S, Hofmann KP, et al. Micropatterned immobilization of a G protein-coupled receptor and direct detection of G protein activation. Nat Biotechnol. 1999;17:1105–1108. doi: 10.1038/15090. [DOI] [PubMed] [Google Scholar]
- 53.Alves ID, Salamon Z, Varga E, Yamamura HI, et al. Direct observation of G-protein binding to the human delta-opioid receptor using plasmon-waveguide resonance spectroscopy. J Biol Chem. 2003;278:48890–48897. doi: 10.1074/jbc.M306866200. [DOI] [PubMed] [Google Scholar]
- 54.Harding PJ, Hadingham TC, McDonnell JM, Watts A. Direct analysis of a GPCR-agonist interaction by surface plasmon resonance. Eur Biophys J. 2006;35:709–712. doi: 10.1007/s00249-006-0070-x. [DOI] [PubMed] [Google Scholar]
- 55.Karlsson OP, Lofas S. Flow-mediated on-surface reconstitution of G-protein coupled receptors for applications in surface plasmon resonance biosensors. Anal Biochem. 2002;300:132–138. doi: 10.1006/abio.2001.5428. [DOI] [PubMed] [Google Scholar]
- 56.Stenlund P, Babcock GJ, Sodroski J, Myszka DG. Capture and reconstitution of G protein-coupled receptors on a biosensor surface. Anal Biochem. 2003;316:243–250. doi: 10.1016/s0003-2697(03)00046-0. [DOI] [PubMed] [Google Scholar]
- 57.Anker JN, Hall WP, Lyandres O, Shah NC, et al. Biosensing with plasmonic nanosensors. Nature Materials. 2008;7:442–453. doi: 10.1038/nmat2162. [DOI] [PubMed] [Google Scholar]
- 58.Coe JV, Heer JM, Teeters-Kennedy S, Tian H, et al. Extraordinary transmission of metal films with arrays of subwavelength holes. Annual Review of Physical Chemistry. 2008;59:179–202. doi: 10.1146/annurev.physchem.59.032607.093703. [DOI] [PubMed] [Google Scholar]
- 59.Jeanmaire DL, Vanduyne RP. Surface Raman Spectroelectrochemistry .1. Heterocyclic, Aromatic, and Aliphatic-Amines Adsorbed on Anodized Silver Electrode. Journal of Electroanalytical Chemistry. 1977;84:1–20. [Google Scholar]
- 60.Haynes CL, McFarland AD, Van Duyne RP. Surface-enhanced Raman spectroscopy. Analytical Chemistry. 2005;77:338A–346A. [Google Scholar]
- 61.Ebbesen TW, Lezec HJ, Ghaemi HF, Thio T, et al. Extraordinary optical transmission through subwavelength hole arrays. Nature. 1998;391:667–669. [Google Scholar]
- 62.Brolo AG, Gordon R, Leathem B, Kavanagh KL. Surface plasmon sensor based on the enhanced light transmission through arrays of nanoholes in gold films. Langmuir. 2004;20:4813–4815. doi: 10.1021/la0493621. [DOI] [PubMed] [Google Scholar]
- 63.Tetz KA, Pang L, Fainman Y. High-resolution surface plasmon resonance sensor based on linewidth-optimized nanohole array transmittance. Optics Letters. 2006;31:1528–1530. doi: 10.1364/ol.31.001528. [DOI] [PubMed] [Google Scholar]
- 64.Stark PRH, Halleck AE, Larson DN. Short order nanohole arrays in metals for highly sensitive probing of local indices of refraction as the basis for a highly multiplexed biosensor technology. Methods. 2005;37:37–47. doi: 10.1016/j.ymeth.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 65.Ji J, O'Connell JG, Carter DJD, Larson DN. High-throughput nanohole array based system to monitor multiple binding events in real time. Analytical Chemistry. 2008;80:2491–2498. doi: 10.1021/ac7023206. [DOI] [PubMed] [Google Scholar]
- 66.Yang JC, Ji J, Hogle JM, Larson DN. Metallic nanohole arrays on fluoropolymer substrates as small label-free real-time bioorobes. Nano Letters. 2008;8:2718–2724. doi: 10.1021/nl801043t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang JC, Ji J, Hogle JM, Larson DN. Multiplexed plasmonic sensing based on small-dimension nanohole arrays and intensity interrogation. Biosensors & Bioelectronics. 2009;24:2334–2338. doi: 10.1016/j.bios.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Henzie J, Lee MH, Odom TW. Multiscale patterning of plasmonic metamaterials. Nature Nanotechnology. 2007;2:549–554. doi: 10.1038/nnano.2007.252. [DOI] [PubMed] [Google Scholar]
- 69.Nagpal P, Lindquist NC, Oh SH, Norris DJ. Ultrasmooth Patterned Metals for Plasmonics and Metamaterials. Science. 2009;325:594–597. doi: 10.1126/science.1174655. [DOI] [PubMed] [Google Scholar]
- 70.Lesuffleur A, Im H, Lindquist NC, Oh SH. Periodic nanohole arrays with shape-enhanced plasmon resonance as real-time biosensors. Applied Physics Letters. 2007;90 [Google Scholar]
- 71.Lesuffleur A, Im H, Lindquist NC, Lim KS, et al. Laser-illuminated nanohole arrays for multiplex plasmonic microarray sensing. Optics Express. 2008;16:219–224. doi: 10.1364/oe.16.000219. [DOI] [PubMed] [Google Scholar]
- 72.Im H, Lesuffleur A, Lindquist NC, Oh SH. Plasmonic Nanoholes in a Multichannel Microarray Format for Parallel Kinetic Assays and Differential Sensing. Analytical Chemistry. 2009;81:2854–2859. doi: 10.1021/ac802276x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lindquist NC, Lesuffleur A, Im H, Oh SH. Sub-micron resolution surface plasmon resonance imaging enabled by nanohole arrays with surrounding Bragg mirrors for enhanced sensitivity and isolation. Lab on a Chip. 2009;9:382–387. doi: 10.1039/b816735d. [DOI] [PubMed] [Google Scholar]
- 74.Ferreira J, Santos MJL, Rahman MM, Brolo AG, et al. Attomolar Protein Detection Using in-Hole Surface Plasmon Resonance. Journal of the American Chemical Society. 2009;131:436-+. doi: 10.1021/ja807704v. [DOI] [PubMed] [Google Scholar]
- 75.Gordon R, Sinton D, Kavanagh KL, Brolo AG. A new generation of sensors based on extraordinary optical transmission. Accounts of Chemical Research. 2008;41:1049–1057. doi: 10.1021/ar800074d. [DOI] [PubMed] [Google Scholar]
- 76.Dahlin A, Zach M, Rindzevicius T, Kall M, et al. Localized surface plasmon resonance sensing of lipid-membrane-mediated biorecognition events. Journal of the American Chemical Society. 2005;127:5043–5048. doi: 10.1021/ja043672o. [DOI] [PubMed] [Google Scholar]
- 77.Hennesthal C, Steinem C. Pore-spanning lipid bilayers visualized by scanning force microscopy. Journal of the American Chemical Society. 2000;122:8085–8086. [Google Scholar]
- 78.Han XJ, Studer A, Sehr H, Geissbuhler I, et al. Nanopore arrays for stable and functional free-standing lipid bilayers. Advanced Materials. 2007;19:4466-+. [Google Scholar]
- 79.Eftekhari F, Escobedo C, Ferreira J, Duan XB, et al. Nanoholes As Nanochannels: Flow-through Plasmonic Sensing. Analytical Chemistry. 2009;81:4308–4311. doi: 10.1021/ac900221y. [DOI] [PubMed] [Google Scholar]
- 80.Lesuffleur A, Lim K, Lindquist NC, Im H, et al. Plasmonic nanohole arrays for label-free kinetic biosensing in a lipid membrane environment. 31st Annual International Conference of the IEEE EMBS, 1481–1484; Minneapolis, MN, USA. 2009. [DOI] [PubMed] [Google Scholar]
- 81.Seifert R, Wenzel-Seifert K, Gether U, Kobilka BK. Functional differences between full and partial agonists: evidence for ligand-specific receptor conformations. J Pharmacol Exp Ther. 2001;297:1218–1226. [PubMed] [Google Scholar]
- 82.Kobilka B, Schertler GF. New G-protein-coupled receptor crystal structures: insights and limitations. Trends Pharmacol Sci. 2008 doi: 10.1016/j.tips.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 83.Thomsen W, Frazer J, Unett D. Functional assays for screening GPCR targets. Curr Opin Biotechnol. 2005;16:655–665. doi: 10.1016/j.copbio.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 84.Waller A, Simons PC, Biggs SM, Edwards BS, et al. Techniques: GPCR assembly, pharmacology and screening by flow cytometry. Trends Pharmacol Sci. 2004;25:663–669. doi: 10.1016/j.tips.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 85.Fang Y, Lahiri J. GPCR microspot assays on solid substrates. Methods Mol Biol. 2009;552:231–238. doi: 10.1007/978-1-60327-317-6_16. [DOI] [PubMed] [Google Scholar]
- 86.Alves ID, Park CK, Hruby VJ. Plasmon resonance methods in GPCR signaling and other membrane events. Curr Protein Pept Sci. 2005;6:293–312. doi: 10.2174/1389203054546352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Silin VI, Karlik EA, Ridge KD, Vanderah DJ. Development of surface-based assays for transmembrane proteins: selective immobilization of functional CCR5, a G protein-coupled receptor. Anal Biochem. 2006;349:247–253. doi: 10.1016/j.ab.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 88.Sen S, Jaakola VP, Pirila P, Finel M, et al. Functional studies with membrane-bound and detergent-solubilized alpha2-adrenergic receptors expressed in Sf9 cells. Biochim Biophys Acta. 2005;1712:62–70. doi: 10.1016/j.bbamem.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 89.Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349:1526–1533. doi: 10.1056/NEJMoa021933. [DOI] [PubMed] [Google Scholar]
- 90.Darnell RB, DeAngelis LM. Regression of small-cell lung carcinoma in patients with paraneoplastic neuronal antibodies. Lancet. 1993;341:21–22. doi: 10.1016/0140-6736(93)92485-c. [DOI] [PubMed] [Google Scholar]
- 91.Ray S, Britschgi M, Herbert C, Takeda-Uchimura Y, et al. Classification and prediction of clinical Alzheimer's diagnosis based on plasma signaling proteins. Nat Med. 2007;13:1359–1362. doi: 10.1038/nm1653. [DOI] [PubMed] [Google Scholar]
- 92.Robinson WH, Fontoura P, Lee BJ, de Vegvar HE, et al. Protein microarrays guide tolerizing DNA vaccine treatment of autoimmune encephalomyelitis. Nat Biotechnol. 2003;21:1033–1039. doi: 10.1038/nbt859. [DOI] [PubMed] [Google Scholar]
- 93.Chen Z, Tabakman SM, Goodwin AP, Kattah MG, et al. Protein microarrays with carbon nanotubes as multicolor Raman labels. Nat Biotechnol. 2008;26:1285–1292. doi: 10.1038/nbt.1501. [DOI] [PubMed] [Google Scholar]
- 94.Shovman O, Gilburd B, Zandman-Goddard G, Yehiely A, et al. Multiplexed AtheNA multi-lyte immunoassay for ANA screening in autoimmune diseases. Autoimmunity. 2005;38:105–109. doi: 10.1080/08916930400022707. [DOI] [PubMed] [Google Scholar]
- 95.Quintana FJ, Farez MF, Viglietta V, Iglesias AH, et al. Antigen microarrays identify unique serum autoantibody signatures in clinical and pathologic subtypes of multiple sclerosis. Proc Natl Acad Sci U S A. 2008;105:18889–18894. doi: 10.1073/pnas.0806310105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Harrison BA, MacKenzie R, Hirama T, Lee KK, et al. A kinetics approach to the characterization of an IgM specific for the glycolipid asialo-GM1. J Immunol Methods. 1998;212:29–39. doi: 10.1016/s0022-1759(98)00012-x. [DOI] [PubMed] [Google Scholar]
- 97.Carter PJ. Potent antibody therapeutics by design. Nat Rev Immunol. 2006;6:343–357. doi: 10.1038/nri1837. [DOI] [PubMed] [Google Scholar]
- 98.Warrington AE, Rodriguez M. Remyelination-promoting human IgMs: developing a therapeutic reagent for demyelinating disease. Curr Top Microbiol Immunol. 2008;318:213–239. doi: 10.1007/978-3-540-73677-6_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rodriguez M, Warrington AE, Pease LR. Invited Article: Human natural autoantibodies in the treatment of neurologic disease. Neurology. 2009;72:1269–1276. doi: 10.1212/01.wnl.0000345662.05861.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Warrington AE, Asakura K, Bieber AJ, Ciric B, et al. Human monoclonal antibodies reactive to oligodendrocytes promote remyelination in a model of multiple sclerosis. Proc Natl Acad Sci U S A. 2000;97:6820–6825. doi: 10.1073/pnas.97.12.6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pirko I, Ciric B, Gamez J, Bieber AJ, et al. A human antibody that promotes remyelination enters the CNS and decreases lesion load as detected by T2-weighted spinal cord MRI in a virus-induced murine model of MS. Faseb J. 2004;18:1577–1579. doi: 10.1096/fj.04-2026fje. [DOI] [PubMed] [Google Scholar]
- 102.Nguyen LT, Radhakrishnan S, Ciric B, Tamada K, et al. Cross-linking the B7 family molecule B7-DC directly activates immune functions of dendritic cells. J Exp Med. 2002;196:1393–1398. doi: 10.1084/jem.20021466. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 103.Howe CL, Bieber AJ, Warrington AE, Pease LR, et al. Antiapoptotic signaling by a remyelination-promoting human antimyelin antibody. Neurobiol Dis. 2004;15:120–131. doi: 10.1016/j.nbd.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 104.Asakura K, Miller DJ, Pease LR, Rodriguez M. Targeting of IgMkappa antibodies to oligodendrocytes promotes CNS remyelination. J Neurosci. 1998;18:7700–7708. doi: 10.1523/JNEUROSCI.18-19-07700.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Warrington AE, Bieber AJ, Van Keulen V, Ciric B, et al. Neuron-binding human monoclonal antibodies support central nervous system neurite extension. J Neuropathol Exp Neurol. 2004;63:461–473. doi: 10.1093/jnen/63.5.461. [DOI] [PubMed] [Google Scholar]
- 106.de Keyzer J, van der Does C, Kloosterman TG, Driessen AJ. Direct demonstration of ATP-dependent release of SecA from a translocating preprotein by surface plasmon resonance. J Biol Chem. 2003;278:29581–29586. doi: 10.1074/jbc.M303490200. [DOI] [PubMed] [Google Scholar]
- 107.Rich RL, Myszka DG. Spying on HIV with SPR. Trends Microbiol. 2003;11:124–133. doi: 10.1016/s0966-842x(03)00025-8. [DOI] [PubMed] [Google Scholar]
- 108.Segura JM, Guillaume P, Mark S, Dojcinovic D, et al. Increased mobility of major histocompatibility complex I-peptide complexes decreases the sensitivity of antigen recognition. J Biol Chem. 2008;283:24254–24263. doi: 10.1074/jbc.M803549200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Perez JB, Segura JM, Abankwa D, Piguet J, et al. Monitoring the diffusion of single heterotrimeric G proteins in supported cell-membrane sheets reveals their partitioning into microdomains. J Mol Biol. 2006;363:918–930. doi: 10.1016/j.jmb.2006.08.036. [DOI] [PubMed] [Google Scholar]
- 110.Wan C, Kiessling V, Tamm LK. Coupling of cholesterol-rich lipid phases in asymmetric bilayers. Biochemistry. 2008;47:2190–2198. doi: 10.1021/bi7021552. [DOI] [PubMed] [Google Scholar]
- 111.Reichert JM, Rosensweig CJ, Faden LB, Dewitz MC. Monoclonal antibody successes in the clinic. Nat Biotechnol. 2005;23:1073–1078. doi: 10.1038/nbt0905-1073. [DOI] [PubMed] [Google Scholar]
- 112.Pai JC, Sutherland JN, Maynard JA. Progress towards recombinant anti-infective antibodies. Recent Pat Antiinfect Drug Discov. 2009;4:1–17. doi: 10.2174/157489109787236319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Maynard JA, Maassen CB, Leppla SH, Brasky K, et al. Protection against anthrax toxin by recombinant antibody fragments correlates with antigen affinity. Nat Biotechnol. 2002;20:597–601. doi: 10.1038/nbt0602-597. [DOI] [PubMed] [Google Scholar]
- 114.Maynard J, Petersson K, Wilson DH, Adams EJ, et al. Structure of an autoimmune T cell receptor complexed with class II peptide-MHC: insights into MHC bias and antigen specificity. Immunity. 2005;22:81–92. doi: 10.1016/j.immuni.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 115.Feng D, Bond CJ, Ely LK, Maynard J, et al. Structural evidence for a germline-encoded T cell receptor-major histocompatibility complex interaction 'codon'. Nat Immunol. 2007;8:975–983. doi: 10.1038/ni1502. [DOI] [PubMed] [Google Scholar]
- 116.Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol. 2006;24:419–466. doi: 10.1146/annurev.immunol.23.021704.115658. [DOI] [PubMed] [Google Scholar]
- 117.Krogsgaard M, Davis MM. How T cells 'see' antigen. Nat Immunol. 2005;6:239–245. doi: 10.1038/ni1173. [DOI] [PubMed] [Google Scholar]
- 118.Boulter JM, Glick M, Todorov PT, Baston E, et al. Stable, soluble T-cell receptor molecules for crystallization and therapeutics. Protein Eng. 2003;16:707–711. doi: 10.1093/protein/gzg087. [DOI] [PubMed] [Google Scholar]
- 119.Esteban O, Zhao H. Directed evolution of soluble single-chain human class II MHC molecules. J Mol Biol. 2004;340:81–95. doi: 10.1016/j.jmb.2004.04.054. [DOI] [PubMed] [Google Scholar]
- 120.Soen Y, Chen DS, Kraft DL, Davis MM, et al. Detection and characterization of cellular immune responses using peptide-MHC microarrays. PLoS Biol. 2003;1:E65. doi: 10.1371/journal.pbio.0000065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Deviren G, Gupta K, Paulaitis ME, Schneck JP. Detection of antigen-specific T cells on p/MHC microarrays. J Mol Recognit. 2006 doi: 10.1002/jmr.805. [DOI] [PubMed] [Google Scholar]
- 122.Grakoui A, Bromley SK, Sumen C, Davis MM, et al. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285:221–227. [PubMed] [Google Scholar]
- 123.Yamazaki V, Sirenko O, Schafer RJ, Nguyen L, et al. Cell membrane array fabrication and assay technology. BMC Biotechnol. 2005;5:18. doi: 10.1186/1472-6750-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hartman NC, Nye JA, Groves JT. Cluster size regulates protein sorting in the immunological synapse. Proc Natl Acad Sci U S A. 2009;106:12729–12734. doi: 10.1073/pnas.0902621106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mossman KD, Campi G, Groves JT, Dustin ML. Altered TCR signaling from geometrically repatterned immunological synapses. Science. 2005;310:1191–1193. doi: 10.1126/science.1119238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.