

Abstract
Background:
Bapineuzumab, a humanized anti-amyloid-beta (Aβ) monoclonal antibody for the potential treatment of Alzheimer disease (AD), was evaluated in a multiple ascending dose, safety, and efficacy study in mild to moderate AD.
Methods:
The study enrolled 234 patients, randomly assigned to IV bapineuzumab or placebo in 4 dose cohorts (0.15, 0.5, 1.0, or 2.0 mg/kg). Patients received 6 infusions, 13 weeks apart, with final assessments at week 78. The prespecified primary efficacy analysis in the modified intent-to-treat population assumed linear decline and compared treatment differences within dose cohorts on the Alzheimer's Disease Assessment Scale–Cognitive and Disability Assessment for Dementia. Exploratory analyses combined dose cohorts and did not assume a specific pattern of decline.
Results:
No significant differences were found in the primary efficacy analysis. Exploratory analyses showed potential treatment differences (p < 0.05, unadjusted for multiple comparisons) on cognitive and functional endpoints in study “completers” and APOE ε4 noncarriers. Reversible vasogenic edema, detected on brain MRI in 12/124 (9.7%) bapineuzumab-treated patients, was more frequent in higher dose groups and APOE ε4 carriers. Six vasogenic edema patients were asymptomatic; 6 experienced transient symptoms.
Conclusions:
Primary efficacy outcomes in this phase 2 trial were not significant. Potential treatment differences in the exploratory analyses support further investigation of bapineuzumab in phase 3 with special attention to APOE ε4 carrier status.
Classification of evidence:
Due to varying doses and a lack of statistical precision, this Class II ascending dose trial provides insufficient evidence to support or refute a benefit of bapineuzumab.
GLOSSARY
- AD
= Alzheimer disease;
- ADAS-Cog
= Alzheimer's Disease Assessment Scale–Cognitive subscale;
- AE
= adverse event;
- APP
= amyloid precursor protein;
- CDR-SB
= Clinical Dementia Rating–Sum of Boxes;
- CI
= confidence interval;
- DAD
= Disability Assessment for Dementia;
- mITT
= modified intent-to-treat;
- MMSE
= Mini-Mental State Examination;
- NTB
= Neuropsychological Test Battery;
- RM
= repeated measures;
- SMC
= Safety Monitoring Committee;
- VE
= vasogenic edema.
Alzheimer disease (AD) is a progressive dementing disease characterized by cerebral neuronal loss, deposits of extracellular β-amyloid (Aβ) plaques, and intraneuronal neurofibrillary tangles.1 Studies in transgenic mice producing excess Aβ have shown that antibodies directed against the N-terminal of Aβ reduce amyloid deposits in both brain tissue and cerebral vasculature.2,3 These antibodies also block the synaptotoxic effects of Aβ oligomers and improve cognitive performance in amyloid precursor protein (APP) transgenic mice.4,5
Previous Aβ immunotherapy studies in humans utilizing active immunization with the full length Aβ42 peptide suggested clinical benefits.6,7 However, meningoencephalitis developed in 6% of patients,8 likely due to a proinflammatory T-cell response against the Aβ peptide.9
One potential means of eliminating the proinflammatory T-cell response is by passively infusing anti-Aβ antibodies. Bapineuzumab, an antibody targeted against the N-terminus of Aβ, is a passive Aβ immunotherapy being tested for AD. Bapineuzumab is hypothesized to bind to Aβ in the brain and facilitate its removal, yielding beneficial clinical effects. A phase 1 study with bapineuzumab determined a half-life of 24 days (unpublished data), leading to phase 2 dosing every 13 weeks. The current multiple ascending dose study was initially designed and powered to evaluate the safety of bapineuzumab within individual dose cohorts. Although the study design remained unchanged, the protocol was amended to evaluate efficacy as the primary objective based partly on preliminary results from the phase 1 study. It was recognized that the small cohorts in this study would provide sufficient power to detect only very large treatment differences; however, if successful, the urgency of delivering an effective treatment to patients with AD argued for making efficacy the primary outcome.
METHODS
This phase 2, multicenter, randomized, double-blind, placebo-controlled, multiple ascending dose study was conducted at 30 sites in the United States between April 2005 and March 2008.
Patients.
Eligible patients were aged 50 to 85 years inclusive, met criteria for probable AD,10 and had an MRI consistent with AD. Additional inclusion criteria were a Mini-Mental State Examination (MMSE) score of 16–2611 and a Rosen Modified Hachinski Ischemic score ≤4.12 Patients were excluded for clinically significant neurologic disease other than AD; a major psychiatric disorder, history of stroke or seizures, a Hamilton Rating Scale score for Depression >1213; current anticonvulsant, antiparkinsonian, anticoagulant, or narcotic medications; recent immunosuppressive or cancer chemotherapy medications; or cognitive enhancers other than acetylcholinesterase inhibitors or memantine at a stable dose for at least 120 days before screening.
Standard protocol approvals, registrations, and patient consents.
The study (ClinicalTrials.gov number NCT00112073) was approved by each site's local institutional review board, and written informed consent was obtained from each patient (or legally authorized representative).
Study design and treatment.
A total of 234 patients were randomly assigned to receive either IV bapineuzumab or placebo, in an 8:7 ratio, in 1 of 4 sequential dose cohorts (0.15, 0.5, 1.0, or 2.0 mg/kg). Adaptive stratified randomization was used to achieve a balance of baseline acetylcholinesterase inhibitor or memantine use and screening MMSE score (low = 16–21 vs high = 22–26). Patients received study drug as a 1-hour IV infusion every 13 weeks for 6 infusions during the 18-month study. An independent Safety Monitoring Committee (SMC) assessed the safety of treatment throughout the trial. The 0.5 mg/kg dose cohort was first to enroll. The 0.15 mg/kg dose cohort was added by protocol amendment after the 0.5 mg/kg cohort completed enrollment to evaluate dose effects more fully after vasogenic edema (VE) was observed on brain MRI in the phase 1 study. Each dose cohort was enrolled after the SMC had reviewed safety in the preceding cohort. The final assessment was at week 78.
Outcome measures.
The Alzheimer's Disease Assessment Scale–Cognitive subscale (ADAS-Cog)14,15 and Disability Assessment for Dementia (DAD)16 scales were co-primary outcomes. The ADAS-Cog/12-item (score range 0–80) and DAD scales (score range 0%–100%) were administered before the first treatment, at each treatment visit, and at week 78. Except for the prespecified analyses, ADAS-Cog results are reported for the standard 11-item scale (without delayed word list recall; range 0–70) for comparability to other studies (ADAS-Cog/12-item scale results are presented in table e-1 on the Neurology® Web site at www.neurology.org).
The Neuropsychological Test Battery (NTB)17 and MMSE (range 0–30) were evaluated at the same intervals as the primary measures, and the Clinical Dementia Rating-Sum of Boxes (CDR-SB; range 0–18)18 was administered every 6 months. In patients consenting to lumbar puncture, CSF was obtained before treatment and at week 52. CSF biomarkers were measured by sandwich ELISAs for total tau,19 phospho-tau (P-tau181),20 and Aβ4221 (with the 4G8 antibody replacing 3D6 to make it specific for AβX-42). Volumetric and safety MRI scans were performed before treatment, at week 6, and then at 13-week intervals through week 71. Exploratory MRI outcomes included change in whole brain and ventricular volumes from baseline to week 71 as measured by the boundary shift integral method.22
Statistical analysis.
Prespecified.
The primary efficacy analysis compared bapineuzumab to placebo within the 0.5, 1.0, and 2.0 mg/kg cohorts based on change from baseline through week 78 using a repeated measures (RM) linear mixed effects model in the mITT population. Model terms were included for baseline MMSE score stratum, the baseline value of the efficacy variable, treatment, time as a continuous variable, and the interaction between treatment and time. The mean response was assumed to progress linearly with time. The 0.15 mg/kg cohort was analyzed with the same model; however, analyses in this group were exploratory and not adjusted for multiple comparisons. Analyses were performed with SAS version 9.1.
Exploratory analyses.
The prespecified model assumed a linear rate of change throughout the study. The treatment difference at week 78 was however of primary interest and therefore, exploratory analyses that did not assume a specific pattern of decline over time were conducted. Nonlinear decline was also apparent in some outcomes, cohorts, and treatment groups. The exploratory analyses included time (study visit) as a categorical rather than continuous variable in the RM model. This model estimates covariate-adjusted means for each group at each time point (with week 78 being of primary interest), taking into account all observed data at all time points from all subjects including those with missing data. In addition to baseline score, MMSE stratum, and treatment, since VE occurred with greater frequency in APOE ε4 carriers than noncarriers, the revised model also included terms for APOE ε4 carrier status and for the APOE ε4 carrier status by time, treatment, and treatment-by-time interaction. Given the small sample size of each cohort and lack of a clear efficacy dose response, the 4 cohorts were combined. Positive treatment differences for efficacy variables indicate less decline in the bapineuzumab group. Treatment differences with p values between 0.05 and 0.10 were considered trends. Reported p values were not adjusted for multiple comparisons.
Populations.
The modified intent-to-treat (mITT) population included patients who received at least one dose of study drug and had one or more postbaseline, co-primary efficacy evaluations. The “completer” population was defined as patients who completed all 6 infusions and a week 78 efficacy assessment.
Sample size.
This study was not designed or powered as an efficacy study. The sample size was calculated to ensure ≥80% probability of detecting an adverse event (AE) that occurred with a rate of at least 5% within a single bapineuzumab-treated dose cohort.
RESULTS
Subject disposition.
Patient disposition is summarized by treatment groups combined across the 4 dose cohorts (figure 1). Of 317 screened patients, 234 were randomized (124 bapineuzumab vs 110 placebo). All randomized patients received at least one dose of bapineuzumab or placebo and were included in the safety population. Among those randomized, 122 bapineuzumab and 107 placebo patients were included in the mITT population. The percentage of patients who had a week 78 assessment was similar (74% bapineuzumab vs 79% placebo; figure 1). Eighty patients (65%) in the bapineuzumab group and 78 (71%) in the placebo group were completers. Fewer carriers in the bapineuzumab group were completers than noncarriers (42/72 [58%]) vs 36/47 [77%]). Two completers in the bapineuzumab group did not have APOE genotyping.

Figure 1 Patient disposition
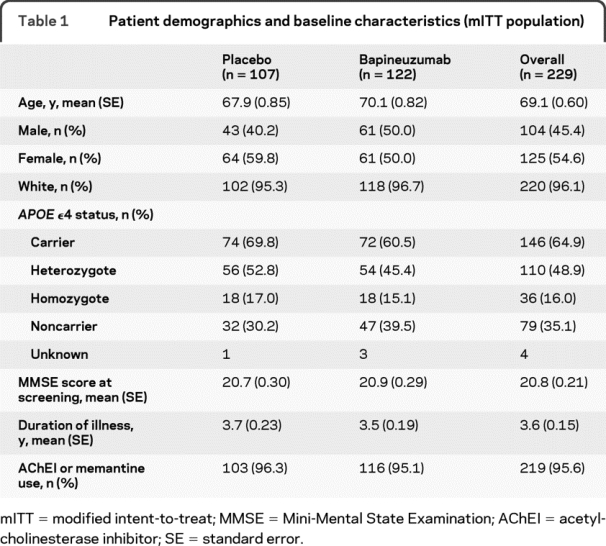
Demographics and baseline characteristics.
Demographic characteristics, summarized by treatment group in table 1, showed no significant differences.
Table 1 Patient demographics and baseline characteristics (mITT population)
Efficacy.
Prespecified analyses for the 0.5, 1.0, and 2.0 mg/kg dose cohorts.
In the prespecified analyses, neither of the primary outcome measures showed significant differences for any dose cohort and no clear dose trend was apparent (table 2). The modeled treatment differences (δ) for the 0.5, 1.0, and 2.0 mg/kg dose groups were 1.2 (95% confidence interval [CI] −4.3, 6.7), 0.0 (95% CI −3.6, 3.6), and 0.5 (95% CI −3.9, 5.0) on the ADAS-Cog and −4.6 (95% CI −14.9, 5.7), −0.8 (95% CI −9.1, 7.4), and 5.7 (95% CI −2.8, 14.1) on the DAD.
Table 2 ADAS-Cog/12 and DAD: Change from baseline at week 78 in the mITT population (RM linear model, prespecified analysis by cohort)
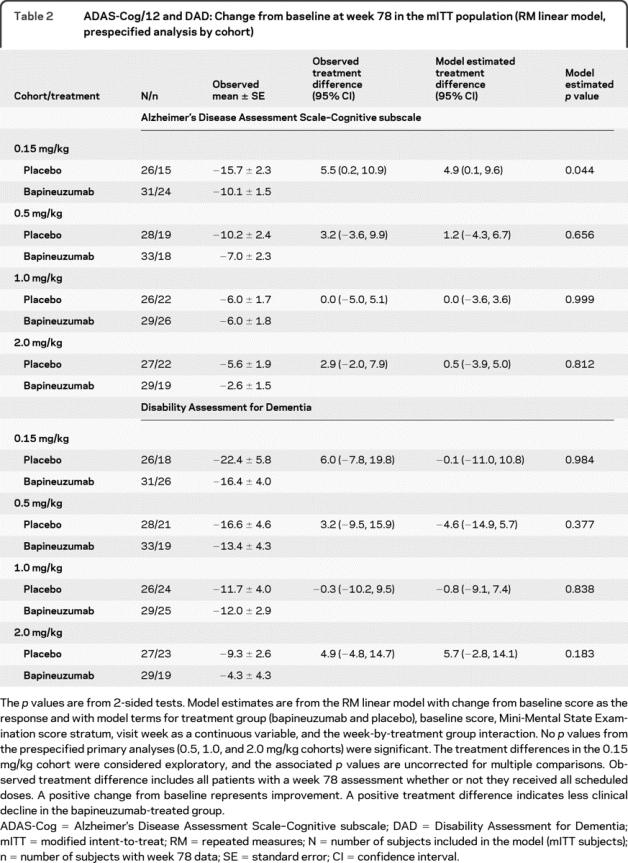
Observed treatment differences at week 78.
The observed treatment differences at week 78 were positive in the 0.15, 0.5, and 2.0 mg/kg cohorts (table 2) for the ADAS-Cog and DAD. The apparent differences between the modeled and observed data were partly explained by nonlinear decline evident in some cohorts and treatment groups. Given the possibility of treatment effects across the range of tested doses and limited power to detect treatment differences within individual cohorts, exploratory analyses combined all dose groups without assuming linear decline over time.
Exploratory analyses by overall treatment group.
Treatment differences (δ) in the mITT population showed trends on the ADAS-Cog (δ = 2.3 [95% CI −0.3, 4.9], p = 0.078; figure 2) and NTB (δ = 0.13 [95% CI −0.01, 0.28], p = 0.068) but not other outcomes (e.g., DAD, figure 2). In the completer population, treatment differences were observed (figure 2) on the ADAS-Cog (δ = 4.3 [95% CI 1.5, 7.2], p = 0.003), NTB (δ = 0.16 [95% CI 0.00, 0.32], p = 0.045), and DAD (δ = 6.1 [95% CI 0.3, 12.0], p = 0.041) but not on the CDR-SB (δ = 0.7 [95% CI −0.3, 1.6], p = 0.159) and the MMSE showed only a trend (δ = 1.5 [95% CI −0.2, 3.3], p = 0.087).

Figure 2 Estimated mean change from baseline over time on Alzheimer's Disease Assessment Scale–Cognitive subscale (ADAS-Cog) and Disability Assessment for Dementia (DAD) for the 4 combined dose cohorts in the modified intent-to-treat (mITT) and completer populations
Error bars represent one standard error. A positive change from baseline represents improvement. The p values are not adjusted for multiple comparisons. (A) ADAS-Cog, mITT; (B) ADAS-Cog, completers; (C) DAD, mITT; (D) DAD, completers.
Exploratory analyses by APOE ε4 carrier status.
For the 79 APOE ε4 noncarriers (47 bapineuzumab vs 32 placebo), treatment differences were observed on the ADAS-Cog (δ = 5.0 [95% CI 0.6, 9.3], p = 0.026), NTB (δ = 0.35 [95% CI 0.10, 0.59], p = 0.006), MMSE (δ = 2.7 [95% CI 0.1, 5.4], p = 0.043), and CDR-SB (δ = 1.5 [95% CI 0.1, 3.0], p = 0.040) but not on the DAD (δ = 6.9 [95% CI −2.2, 16.0], p = 0.137). Completer analyses (36 bapineuzumab vs 21 placebo) showed somewhat greater treatment differences on all measures.
For the 146 APOE ε4 carriers (72 bapineuzumab vs 74 placebo), no treatment differences were observed on any endpoint including the ADAS-Cog (δ = 0.9 [95% CI −2.3, 4.1], p = 0.588) and DAD (δ = −1.2 [95% CI −7.8, 5.4], p = 0.726). Although somewhat larger differences were present in completer analyses in carriers (42 bapineuzumab vs 57 placebo) [ADAS-Cog (δ = 2.6 [95% CI −0.9, 6.1], p = 0.141) and DAD (δ = 5.0 [95% CI −2.3, 12.3], p = 0.182)], a trend was not observed.
CSF biomarkers.
The change in CSF biomarkers from baseline to week 52 was evaluated in a small substudy (20 bapineuzumab and 15 placebo). No differences were observed between bapineuzumab- and placebo-treated patients for AβX-42 or total-tau. Phospho-tau181 trended toward greater reduction (δ = −9.1 pg/mL [95% CI −18.5, 0.3], p = 0.056) in the bapineuzumab group.
MRI volumetric analyses.
Exploratory MRI analyses in the mITT population showed no treatment differences in brain or ventricular volume change. APOE ε4 noncarriers showed 10.7 mL less brain volume loss in the bapineuzumab group compared with placebo (95% CI 3.4, 18.0; p = 0.004). No difference in ventricular volume was noted. APOE ε4 carriers showed no treatment difference in brain volume; however, greater ventricular enlargement was observed in the bapineuzumab group compared with placebo (2.6 mL; 95% CI 0.2, 5.0; p = 0.037).
Safety.
Most patients reported AEs over the course of 18 months (94% bapineuzumab vs 90% placebo). The rate of AEs was higher for bapineuzumab (7.5 vs 5.7 events per patient), but over 90% were mild to moderate in severity. AEs reported in >5% of bapineuzumab patients and at a rate twofold higher than placebo included VE (9.7% vs 0%), back pain (12.1% vs 5.5%), anxiety (11.3% vs 3.6%), paranoia (6.5% vs 0.9%), vomiting (9.7% vs 3.6%), hypertension (8.1% vs 3.6%), weight loss (6.5% vs 1.8%), skin laceration (5.6% vs 2.7%), gait disturbance (5.6% vs 1.8%), and muscle spasm (5.6% vs 0.9%). Except for VE, AEs were not dose-related. Other potentially important AEs reported in <5% of bapineuzumab-treated patients but more frequently than with placebo included deep vein thrombosis (3.2% vs 0%), syncope (4.8% vs 1.8%), seizures (3.2% vs 0.9%), pulmonary embolism (0.8% vs 0%), and cataracts (4.0% vs 0.9%). With respect to the AEs listed above, only vomiting and gait disturbance occurred in temporal association with VE. Serious AEs were reported in 37 (30%) bapineuzumab-treated patients and 22 (20%) placebo patients. VE in the high-dose bapineuzumab group accounted for much of this difference. Three deaths occurred in bapineuzumab-treated subjects (one case each of AD progression, obstructive renal failure, and pneumonia after a thoracic aortic dissection). A fourth death (progression of AD) was reported in a bapineuzumab-treated subject after the 78-week treatment period. None of the deaths were considered treatment or VE-related by the respective investigators or the SMC. The deaths were not associated with APOE ε4 or dose. No deaths were reported in the placebo group.
Vasogenic edema.
VE was detected on MRI in 12/124 patients (9.7%) treated with bapineuzumab and in 0/110 (0%) with placebo. Ten of these cases were detected on MRI scans specified by the protocol, and 11/12 occurred after the initial or second dose of study drug. VE (figure 3) appeared on the fluid-attenuated inversion recovery MRI sequence with high signal intensity in the white matter, leptomeninges, or sulci, and was frequently associated with gyral swelling and cortical T2 hyperintensity in the adjacent cortex. Six cases reported no clinical symptoms. In symptomatic patients, the most common AEs reported within 1 month of VE detection on MRI were headache, confusion, vomiting, and gait disturbance. One patient required treatment with dexamethasone. After cessation of dosing, these symptoms generally resolved over several weeks, while the MRI findings resolved over several months.

Figure 3 Serial MRI scans (fluid-attenuated inversion recovery) in a patient with vasogenic edema (VE)
This 69-year-old woman is an APOE ε4 homozygote who was treated with bapineuzumab 1.0 mg/kg IV. She remained asymptomatic despite the appearance of multiple areas of VE evident on the MRI. The VE was apparent on MRI by 7 weeks after her first infusion and resolved by 19 weeks. The patient was redosed at 0.5 mg/kg of bapineuzumab IV and followed for over 2 years without recurrence of VE.
VE increased with increasing bapineuzumab dose; VE rates were 3.2% for 0.15 mg/kg, 0% for 0.5 mg/kg, 10.0% for 1.0 mg/kg, and 26.7% for 2.0 mg/kg. Eleven of the 12 VE cases occurred at doses ≥1.0 mg/kg. Ten of the 12 VE cases occurred in APOE ε4 carriers with a higher rate observed in APOE ε4 carriers (13.5%; 10/74) than noncarriers (4.3%; 2/47). VE increased with APOE ε4 gene dose, with VE rates of 4.3% (2/47) in patients with 0 copies of the allele, 7.1% (4/56) with 1 copy, and 33.3% (6/18) with 2 copies. Redosing was instituted in 6/12 patients starting at 0.15 mg/kg and titrating up to 50% of the originally assigned dose. No recurrence of VE has been observed to date in these patients.
DISCUSSION
This clinical trial explored the long-term safety and efficacy of bapineuzumab for the treatment of AD. The relatively low discontinuation rate during the 18-month study demonstrates that IV administration of serial doses of bapineuzumab is feasible and generally well-tolerated. The prespecified within-dose cohort analyses did not demonstrate significant treatment differences. Exploratory analyses with all dose cohorts combined showed favorable trends on the ADAS-Cog and NTB in the mITT population, while completer analyses showed differences on these endpoints and the DAD.
We also found possible differences by APOE ε4 carrier status. In noncarriers, potential treatment differences favoring bapineuzumab were observed on some clinical measures. No treatment differences were demonstrated in APOE ε4 carriers. A number of reasons might account for these findings: first, greater efficacy was found in subjects who completed the study and a greater proportion of noncarriers were completers; second, more advanced Aβ pathology in APOE ε4 carriers23 may have affected the clinical response; finally, the differences observed in these exploratory analyses could be due to chance.
The development of VE primarily at higher doses and in APOE ε4 carriers suggests that carriers be evaluated at a lower dose range in future studies. The etiology of VE is unknown but may be related to vascular amyloid burden. Amyloid deposition in cerebral blood vessels is more extensive in APOE ε4 carriers than noncarriers.23 VE can occur spontaneously with cerebral amyloid angiopathy24,25 and with agents that alter vascular permeability, e.g., some cases of posterior reversible encephalopathy syndrome.26,27 VE may result from transient increases in vascular permeability associated with Aβ removal from cerebral blood vessels, or other mechanisms related to amyloid clearance.28 VE resolved on MRI after discontinuation of bapineuzumab and was generally manageable with careful monitoring and dose adjustment. Other than VE, no major safety concerns were found. The deaths in the bapineuzumab group were not considered treatment related and were within the expected range for an 18-month AD study of this size.29–31
The reduction in brain volume loss observed in the APOE ε4 noncarriers, relative to the placebo group, paralleled the clinical differences observed and may indicate slowing of brain atrophy. In carriers, no treatment-related difference in brain volume was observed. The cause and meaning of the increase in ventricular volume is uncertain. The trend toward reduced CSF phospho-tau181, a potential indicator of neurofibrillary pathology,32 may suggest downstream effects of bapineuzumab on tau pathology similar to those seen with anti-Aβ immunotherapy in other studies,7,33,34 but requires replication.
This trial had several limitations. The sequential recruitment of small dose cohorts to evaluate safety, and the variable rate of decline in the treated and placebo groups within cohorts, restricted the statistical power to demonstrate efficacy and assess dose response. The prespecified linear model, motivated by the supposition that divergent slopes might argue for disease modification, lacked consistency with some of the observed data. VE and its relationship to APOE ε4 status and dose were not anticipated and limited treatment exposure in some patients. The exploratory analyses attempted to circumvent some of these limitations by 1) combining dose cohorts to increase sample size; 2) relaxing the assumption of linear disease progression; 3) assessing potential treatment differences by APOE ε4 status; and 4) evaluating results separately in completers. The exploratory efficacy analyses were not prespecified or controlled for multiple comparisons. These results must therefore be interpreted cautiously and require replication in more definitive trials.
This limited phase 2 trial did not demonstrate efficacy on its primary outcomes, but exploratory analyses found potential treatment differences in completers and APOE ε4 noncarriers. These preliminary findings support continued evaluation of bapineuzumab for AD in phase 3 with consideration to possible treatment differences by APOE ε4 carrier status.
ACKNOWLEDGMENT
The authors thank the following individuals for their assistance: Erika Jones and Robert Schilling (study management); Kristen Morris (pharmacovigilance); Rezi Zawadski and Jenny Wei (statistical analyses); John Baer and Kay Jing (clinical study summary); and Shona Clegg (MRI volumetric analyses and figure 3).
DISCLOSURE
This study was sponsored by Elan Pharmaceuticals (JANSSEN Alzheimer Immunotherapy acquired the Alzheimer immunotherapy program from Elan in September 2009) and Wyeth Research (which was acquired by Pfizer in October 2009). Dr. Salloway serves on the scientific advisory boards of Elan Pharmaceuticals, Sanofi-Aventis, Pfizer Inc, and Bristol-Myers Squibb. He served on the scientific advisory for Eisai Inc.; serves as Associate Editor for Journal of Neuropsychiatry and Clinical Neurosciences; receives publishing royalties for The Frontal Lobes and Neuropsychiatric Illness (American Psychiatric Press Inc., 2001), The Neuropsychiatry of Limbic and Subcortical Disorders (American Psychiatric Press Inc., 1997), and Vascular Dementia (Humana Press, 2004); receives honoraria from Eisai Inc., Pfizer Inc, Novartis, Forest Laboratories Inc., Elan Pharmaceuticals, and Athena Diagnostics, Inc.; holds corporate appointments with Merck Serono and Medivation, Inc.; receives research support from Elan Pharmaceuticals, Wyeth, Bristol-Myers Squibb, and Eisai Inc.; received research support from Myriad Genetics, Inc., GlaxoSmithKline, Neurochem-Alzhemed, Cephalon, Inc., Forest Laboratories Inc., and Voyager; receives research support from the Alzheimer's Disease Neuroimaging Initiative, Dominantly Inherited Alzheimer's Network [NIA 1U01AG032438-01]; received research support from Aging Brain: DTI, Subcortical Ischemia and Behavior [NIA 1 R03 AG023916-01A1]; and receives research support from The Norman and Rosalie Fain Family Foundation. Dr. Sperling serves on the scientific advisory boards of Link Pharmaceuticals, SAB; served as an editor for Alzheimer's Disease and Associated Disorders; received honoraria for Grand Rounds from Pfizer Inc, Forest Pharmaceuticals; serves as a consultant to Elan Pharmaceuticals, Wyeth, Link, and Merck Serono, and Eisai Pharmaceuticals; Dr. Sperling's husband served as consultant to GE Healthcare; Dr. Sperling receives research support from Elan Pharmaceuticals, Wyeth, Neurochem, Pfizer, Novartis, Bristol-Myers-Squibb; receives research support from the NIA [R01AG027435 (PI), P50-AG005134 Massachusetts Alzheimer's Disease Research Center (Project Leader)]; receives research support from the Alzheimer's Disease Neuroimaging Initiative, and the Dominantly Inherited Alzheimer's Network; receives research support from Fidelity Foundation and the Alzheimer's Association. Dr. Gilman serves on the scientific advisory board of GlaxoSmithKline, China Research, Adamas Pharmaceuticals, and Longitude Capital Funding; received funding for travel from Ann Arbor to Shanghai, China funded by GlaxoSmithKline; serves as Editor-in-Chief of Experimental Neurology, Neurobiology of Disease, MedLink Neurology, and Contemporary Neurology Series; receives royalties from Contemporary Neurology Series (Oxford University Press, 1982-present), MedLink Neurology (MedLink Corporation, 1992-present), Experimental Neurology (Elsevier, 2003-present), Neurobiology of Disease (Elsevier, 2005-present); receives research support from NIH-NIA [P50 AG08671,PI], Michigan Alzheimer's Disease Research Center [AG08671 (Program director, PI, and Co-Investigator)], NIH [U01-AG16976 (PI)] and NIHNINDS [P01 NS15655 (PI)]. Dr. Fox served on the scientific advisory boards of Alzheimer's Research Forum, GE Healthcare, Elan Pharmaceuticals, Wyeth, and Eisai Inc.; serves on the editorial boards of Alzheimer's Disease and Associated Disorders, Neurodegenerative Diseases, and Biomed Central: Alzheimer's Research and Therapy; holds a patent for QA Box that may accrue revenue; received honoraria for his research group from GE Healthcare for speaking; received honoraria for reviewing from Lancet Neurology; received honoraria for his research group for consulting from Eli Lilly, Abbott Laboratories, Eisai, Elan Pharmaceuticals, Wyeth, GE Healthcare, Sanofi-Aventis, and Lundbeck; received research support from GlaxoSmithKline, Elan Pharmaceuticals, Wyeth, Lundbeck, Sanofi-Aventis, IXICO, and Pfizer Inc. for contracted image analysis research; receives research support from MRC [G0801306 (PI), G0601846 (PI)] NIH [U01 AG024904 (Co-Investigator (subcontract))], Alzheimer Research Trust [ART/RF/2007/1 (PI)]; and received research support from EPSRC [GR/S48844/01 (PI)]. Dr. Blennow served on the scientific advisory boards of AdLyfe Corporation, Bayer Schering Pharma AG, Bristol-Myers Squibb, Merz Pharmaceuticals GmbH; received honoraria from Pfizer Inc AB for a lecture; holds corporate appointment with Wyeth Research, AstraZeneca, Bristol-Myers Squibb and Eli Lilly; received research support from Pfizer Inc Sweden (2007) and Innogenetics (ongoing); receives research support from The Research Council, Sweden [14002 (2007–2009) (Main Investigator)]and LUA/ALF project/Västra Götalandsregionen [ALFGBG-11019, (Main Investigator)], The Swedish Brain Power project and The Swedish Council for Working Life and Social Research, The Swedish Alzheimer Foundation, Stiftelsen för Gamla Tjänarinnor, and The King Gustaf V and Queen Victoria Foundation. Dr. Raskind serves on the scientific advisory boards of Hoechst-Roussel, Schering-Plough, Elan Pharmaceuticals; serves on the speakers' bureau of Bristol-Myers Squibb, Parke Davis (Janssen) and Forest Laboratories, Inc.; serves as member of editorial boards for the Journal of Geriatric Psychiatry and Neurology; receives research support from Elan Pharmaceuticals (Site Investigator); Eli Lilly (Site Investigator); receives research support from ADRC [P50 AG005136 (PI); COMRP PRO54292 (PI); and PTSD WB1XWH-08-2-(PI)]; served as an expert witness for Johnson & Johnson. Dr. Sabbagh serves as the Chair of the scientific advisory board of Amerisciences; serves as an editor for Clinical Neurology News and Journal of Alzheimer's Disease; receives royalties from The Alzheimer's Answer: Reduce Your Risk and Keep Your Brain Healthy (Wiley 2008); receives honoraria from advisory boards from Lilly, Wyeth, GSK, and Elan Pharmaceuticals; serves as a consultant to Elan Pharmaceuticals; served in the past on the speakers' bureaus of Eisai Inc., Pfizer Inc, Novartis, and Forest Laboratories Inc. but is no longer active; receives research support from Elan Pharmaceuticals, Wyeth, Novartis, Medivation, Avid, Abbott, Baxter, GlaxoSmithKline, Bristol-Myers Squibb, Eli Lilly; receives research support from NIAADCC P30 [NIA P3-AG 019610]; received research support from the Alzheimer's disease cooperative study, ADNI; received royalty payments from Amerisciences; pays royalties on the commercial product Cognivite. Dr. Honig served on the scientific advisory board for Forest Laboratories Inc.; serves as an Associate Editor for Archives of Neurology/AMA Journals; served as a consultant for Bayer Healthcare and Dainippon Pharmaceuticals, and serves as a consultant for Elan Pharmaceuticals; serves on the speakers' bureau for Eisai Inc., Pfizer Inc, Johnson & Johnson, Ortho McNeil Pharmaceuticals, and Novartis Pharmaceuticals; served on the speakers' bureau for Forest Laboratories Inc.; serves as a consultant for the City College of NY; receives research support from Avid Pharmaceuticals, Bayer Healthcare, Bristol-Myers Squibb, Elan Pharmaceuticals, Eli Lilly, Forest Laboratories Inc., GlaxoSmithKline, Myriad Genetics, Inc., Novartis, Wyeth; receives research funding from NIH/NIA [R43AG033474 (Subcontract PI), P50AG08702 (Core Leader), R01AG14763 (Co-Investigator), R01AG007370 (Co-Investigator) R01AG026114 (Co-Investigator), R01AG017761, Co-Investigator)], NIH/NHLBI [R01HL091099 (Co-Investigator)]; received research funding from NIH/NIA [P01AG07232 (Senior Investigator)], NIH/NINDS [R01NS042859, (Co-Investigator)]; receives research support from Alzheimer's Association, (PI) and Henry Panasci Foundation (Investigator). Dr. Doody serves on the medical and scientific advisory boards of Medivation and Sonexa; attended ad hoc advisory board meetings for Zapaq, Comentis, Athenagen, Astellas, Neurochem, Myriad Genetics, Inc and Elan Corporation/Wyeth; served on steering committees for Sanofi-Synthelabo and Ono; served on a data safety monitoring board for Debiopharm, serves on a data safety monitoring board for GlaxoSmithKline; serves on the Pfizer Inc Scholars selection committee; has received travel funding to attend some of the above listed advisory board meetings and some consulting meetings with companies listed; serves on the editorial boards of Alzheimer's Disease and Associated Disorders, Dementia and Geriatric Cognitive Disorders, and BioMed Central: Alzheimer's Research and Therapy; receives royalties from publishing from Alzheimer's Dementia (Carma Publishing, 2008); received honoraria for lectures from Eisai Inc., Lundbeck, Pfizer Inc, and Forest Laboratories Inc.; served as a consultant for Abbott, Amgen, AstraZeneca, Bristol-Meyers Squibb, Eisai Inc., Epix, Forest Laboratories Inc., Fujisawa, GlaxoSmithKline, Janssen, Lilly, Merck Serono, Merz, Novartis, Ocera, Pfizer Inc, Saegis, Sanofi-Synthelabo, Takeda, Teva, and Voyager; serves as a consultant for Astellas, Dainippon, Noven, Schering-Plough, Suven, Varinel, and Transition; received research support from Eisai Inc., Eisai Inc./Teva Forest Laboratories Inc., GlaxoSmithKline, Myriad Genetics, Inc., Sanofi-Synthelabo; receives research support from Elan Corporation/Wyeth, and Pfizer Inc; received research support from NIDDK IR21DK062098 (Co-Investigator); NIH/NCRR [P20 RR020626 (Co-Investigator); receives research support from NIH [UO1AGO24904(Site PI); AF10483(Site PI)]; received the Zenith Award, Alzheimer's Association (PI); holds stock options in Medivation and Sonexa; served as a consultant to a legal firm representing Eisai Inc. in a patent challenge case and a legal firm representing Forest Laboratories Inc. in a patent challenge case. Dr. van Dyck's wife (Amy Arnsten, PhD) holds or has applied for the following patents: [Use of guanfacine in the treatment of behavioral disorders, Use of lofexidine in the treatment of behavioral disorders, Chelerythrine, analogs thereof and their use in the treatment of bipolar disorder and other cognitive disorders] application no. 10/672,626 (licensed to Marinus Pharmaceuticals and this license has terminated); Dr. van Dyck's wife (Amy Arnsten, PhD) receives royalties for The Neuropharmacology of Stimulant Drugs: Implications for AD/HD (Oxford University Press, New York, NY, 2000); Dr. van Dyck holds a corporate appointment with Bristol-Meyers Squibb and held a corporate appointment with Forest Laboratories Inc.; Dr. van Dyck's wife (Amy Arnsten, PhD) holds a corporate appointment with Shire Pharmaceuticals; Dr. van Dyck receives research support from Medivation, Inc. [Connection (DIM14) HIC 805003830 (PI)]; Abbott Laboratories [HIC 711003285 (PI)], Eli Lilly and Company [HIC 803003585 (PI)], Elan Pharmaceuticals [(AAB-001, ELN115727, HIC 801003387 (PI)], Elan Pharmaceuticals [HIC 801003388 (PI)], Elan Pharmaceuticals [HIC 712003331 (PI)], Wyeth Research [HIC 709003057 (PI)]; received research support from GlaxoSmithKline [HIC 608001709, 711003271 (PI)], Myriad Genetics, Inc. [HIC 608001710 (PI)], Myriad Genetics, Inc. [HIC 0506000112, 608001770 (PI)], Elan Pharmaceuticals [HIC 27280, 610001937 (PI)], Eli Lilly and Company [HIC 0601001050 (PI)], Neurochem, Inc. [HIC 26729 (PI)], Sanofi-Synthelabo [HIC 25926 (PI)], Eisai Inc. [HIC 25925 (PI)], Merck Serono [HIC 25387 (PI)], Forest Laboratories Inc. [01-HIC 23637, 02-HIC 23654, open-label 03-HIC 25388 (PI)], Dr. van Dyck's wife (Amy Arnsten, PhD) receives funding from Shire Pharmaceutical [Mechanism of Action of Medications for ADHD (PI)]; Dr. van Dyck receives research support from NIH/NIA [1-R01-AG030457-01A1 (PI), 5-U01-AG10483 (PI for Yale Site), 1-UO1-AG024904 (PI Yale Site)], NIH/NIMH [1 R01 MH58620-01A2 (PI)]; received research support from NIH/NIA [5-U01-AG10483 (PI for Yale Site)]; Dr. van Dyck's wife Amy Arnsten, PhD received research support from NIA [AG06036-13 (PI)]; NIMH [P50MH068789 (Co-Principal Investigator)]; NINDS [National Research Service Award 5 T32 NS007224-19 (PI)]. National Institute on Alcohol Abuse and Alcoholism [1RL1AA017536-01; in U54RR024350–01 (PI)]; NIA [PO1 AG030004-01 (PI)]; Dr. van Dyck receives research support from the Alzheimer's Association [Investigator-Initiated Research Grant-07-60026 (PI)]; received research support from American Health Assistance Foundation Grant [A2004-216 (PI)], Alzheimer's Association Grant [Investigator-Initiated Research Grant-01-2891 (PI)], National Alliance for Research on Schizophrenia and Affective Disorders (NARSAD) [Independent Investigator Award (PI)]; Dr. van Dyck's wife Amy Arnsten, PhD received research support from Kavli Neuroscience Institute at Yale (PI) and NARSAD Distinguished Investigator Award; Dr. van Dyck's wife Amy Arnsten, PhD received license fee payments from Shire Pharmaceuticals. Dr. Mulnard receives research support from Myriad Genetics, Inc., Elan Corporation Pharmaceuticals [(AAB-001, ELN115727 (PI)], Elan Corporation [NTB, ELN-AIP-901 (PI)], NIH/NIA [5-U01-AG10483 (PI for UC Irvine Site), 1-UO1-AG024904 (PI for UC Irvine Site)]. Dr. Barakos serves as a neuroradiological consultant to Synarc, an imaging contract research organization contracted by Elan Pharmaceuticals; serves as consultant to Elan Pharmaceuticals for non-clinical research activities. Dr. Gregg was employed by Elan Pharmaceuticals at the time of manuscript acceptance and holds stocks and stock options in Elan Pharmaceuticals. Dr. Gregg is now employed by JANSSEN Alzheimer Immunotherapy Research & Development, LLC. Dr. Liu was employed by Elan Pharmaceuticals at the time of manuscript acceptance and holds stock options in Elan Pharmaceuticals. Dr. Liu is now employed by JANSSEN Alzheimer Immunotherapy Research & Development, LLC. Dr. Lieberburg was employed by Elan Pharmaceuticals at the time of manuscript submission and owns stock and stock options in Elan Pharmaceuticals. Dr. Schenk serves as a guest Editor for Experimental Neurology; holds patents associated with bapineuzumab, Elan Pharmaceuticals (receives no revenue); serves as the Chief Scientific Officer for Elan Pharmaceuticals; holds stock and stock options in Elan Pharmaceuticals. Dr. Schenk is an employee of Elan Pharmaceuticals and JANSSEN Alzheimer Immunotherapy Research & Development, LLC. Dr. Black is an employee of Pfizer Inc. Dr. Black was employed by Wyeth at the time of manuscript acceptance and owned stock and stock options in Wyeth. Wyeth was acquired by Pfizer Inc in October 2009. Dr. Grundman is employed by Elan Pharmaceuticals and JANSSEN Alzheimer Immunotherapy Research & Development, LLC, and owns stock and stock options in Elan Pharmaceuticals.
Supplementary Material
Address correspondence and reprint requests to Dr. Stephen Salloway, Butler Hospital, The Warren Alpert Medical School of Brown University, 345 Blackstone Blvd., Providence, RI 02906 SSalloway@Butler.org
Editorial, page 2052
Supplemental data at www.neurology.org
e-Pub ahead of print on November 18, 2009, at www.neurology.org.
*Investigators of the Bapineuzumab Study 201 are listed in appendix e-1 on the Neurology® Web site at www.neurology.org.
Study funding: This study was sponsored by Elan Pharmaceuticals (JANSSEN Alzheimer Immunotherapy acquired the Alzheimer immunotherapy program from Elan in September 2009) and Wyeth Research (which was acquired by Pfizer in October 2009).
Disclosure: Author disclosures are provided at the end of the article.
Received March 20, 2009. Accepted in final form August 3, 2009.
REFERENCES
- 1.Selkoe DJ, Schenk D. Alzheimer's disease: molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol 2003;43:545–584. [DOI] [PubMed] [Google Scholar]
- 2.Schroeter S, Khan K, Barbour R, et al. Immunotherapy reduces vascular amyloid-beta in PDAPP mice. J Neurosci 2008;28:6787–6793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bard F, Cannon C, Barbour R, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 2000;6:916–919. [DOI] [PubMed] [Google Scholar]
- 4.Buttini M, Masliah E, Barbour R, et al. Beta-amyloid immunotherapy prevents synaptic degeneration in a mouse model of Alzheimer's disease. J Neurosci 2005;25:9096–9101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shankar GM, Li S, Mehta TH, et al. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med 2008;14:837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bayer AJ, Bullock R, Jones RW, et al. Evaluation of the safety and immunogenicity of synthetic Abeta42 (AN1792) in patients with AD. Neurology 2005;64:94–101. [DOI] [PubMed] [Google Scholar]
- 7.Gilman S, Koller M, Black RSK, et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 2005;64:1553–1562. [DOI] [PubMed] [Google Scholar]
- 8.Orgogozo JM, Gilman S, Dartigues JF, et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 2003;61:46–54. [DOI] [PubMed] [Google Scholar]
- 9.Pride M, Seubert P, Grundman M, Hagen M, Eldridge J, Black RS. Progress in the active immunotherapeutic approach to Alzheimer's disease: clinical investigations into AN1792-associated meningoencephalitis. Neurodegener Dis 2008;5:194–196. [DOI] [PubMed] [Google Scholar]
- 10.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 11.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 12.Rosen WG, Terry RD, Fuld PA, Katzman R, Peck A. Pathological verification of ischemic score in differentiation of dementias. Ann Neurol 1980;7:486–488. [DOI] [PubMed] [Google Scholar]
- 13.Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry 1960;23:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer's disease. Am J Psychiatry 1984;141:1356–1364. [DOI] [PubMed] [Google Scholar]
- 15.Mohs RC, Knopman D, Petersen RC, et al. Development of cognitive instruments for use in clinical trials of antidementia drugs: additions to the Alzheimer's Disease Assessment Scale that broaden its scope: The Alzheimer's Disease Cooperative Study. Alzheimer Dis Assoc Disord 1997;11 suppl 2:S13–S21. [PubMed] [Google Scholar]
- 16.Gauthier S, Gélinas I, Gauthier L. Functional disability in Alzheimer's disease. Int Psychogeriatr 1997;9 suppl 1:163–165. [DOI] [PubMed] [Google Scholar]
- 17.Harrison J, Minassian SL, Jenkins L, Black RS, Koller M, Grundman M. A neuropsychological test battery for use in Alzheimer disease clinical trials. Arch Neurol 2007;64:1323–1329. [DOI] [PubMed] [Google Scholar]
- 18.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43:2412–2414. [DOI] [PubMed] [Google Scholar]
- 19.Blennow K, Wallin A, Ågren H, Spenger C, Siegfried J, Vanmechelen E. Tau protein in cerebrospinal fluid: a biochemical diagnostic marker for axonal degeneration in Alzheimer's disease? Mol Chem Neuropathol 1995;26:231–245. [DOI] [PubMed] [Google Scholar]
- 20.Vanmechelen E, Vanderstichele H, Davidsson P, et al. Quantification of tau phosphorylated at threonine 181 in human cerebrospinal fluid: a sandwich ELISA with a synthetic phosphopeptide for standardization. Neurosci Lett 2000;285:49–52. [DOI] [PubMed] [Google Scholar]
- 21.Andreasen N, Hesse C, Davidsson P, et al. Cerebrospinal fluid β-amyloid (1-42) in Alzheimer disease: differences between early- and late-onset Alzheimer disease and stability during the course of disease. Arch Neurol 1999;56:673–680. [DOI] [PubMed] [Google Scholar]
- 22.Fox NC, Cousens S, Scahill R, Harvey RJ, Rossor MN. Using serial registered brain magnetic resonance imaging to measure disease progression in Alzheimer disease: power calculations and estimates of sample size to detect treatment effects. Arch Neurol 2000;57:339–344. [DOI] [PubMed] [Google Scholar]
- 23.Chalmers K, Wilcock GK, Love S. APOE ε4 influences the pathological phenotype of Alzheimer's disease by favouring cerebrovascular over parenchymal accumulation of Aβ protein. Neuropathol Appl Neurobiol 2003;29:231–238. [DOI] [PubMed] [Google Scholar]
- 24.Kinnecom C, Lev MH, Wendell L, et al. Course of cerebral amyloid angiopathy-related inflammation. Neurology 2007;68:1411–1416. [DOI] [PubMed] [Google Scholar]
- 25.Oh U, Gupta R, Krakauer JW, Khandji AG, Chin SS, Elkind MS. Reversible leukoencephalopathy associated with cerebral amyloid angiopathy. Neurology 2004;62:494–497. [DOI] [PubMed] [Google Scholar]
- 26.Casey SO, Sampaio RC, Michel E, Truwit CL. Posterior reversible encephalopathy syndrome: utility of fluid-attenuated inversion recovery MR imaging in the detection of cortical and subcortical lesions. AJNR Am J Neuroradiol 2000;21:1199–1206. [PMC free article] [PubMed] [Google Scholar]
- 27.Lamy C, Oppenheim C, Méder JF, Mas JL. Neuroimaging in posterior reversible encephalopathy syndrome. J Neuroimaging 2004;14:89–96. [PubMed] [Google Scholar]
- 28.Boche D, Zotova E, Weller RO, et al. Consequence of Abeta immunization on the vasculature of human Alzheimer's disease brain. Brain 2008;131:3299–3310. [DOI] [PubMed] [Google Scholar]
- 29.Wilcock GK, Black SE, Hendrix SB, Zavitz KH, Swabb EA, Laughlin MA. Efficacy and safety of tarenflurbil in mild to moderate Alzheimer's disease: a randomized phase II trial. Lancet Neurol 208;7:483–493. [DOI] [PubMed]
- 30.Pirttilä T, Wilcock G, Truyen L, Damaraju CV. Long-term efficacy and safety of galantamine in patients with mild-to-moderate Alzheimer's disease: multicenter trial. Eur J Neurol 2004;11:734–741. [DOI] [PubMed] [Google Scholar]
- 31.Neumann PJ, Araki SS, Areculs A, et al. Measuring Alzheimer's disease progression with transition probabilities: estimates from CERAD. Neurol 2001;57:957–964. [DOI] [PubMed] [Google Scholar]
- 32.Blennow K. CSF biomarkers for Alzheimer's disease: use in early diagnosis and evaluation of drug treatment. Expert Rev Mol Diagn 2005;5:661–672. [DOI] [PubMed] [Google Scholar]
- 33.Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 2004;43:321–332. [DOI] [PubMed] [Google Scholar]
- 34.Nicoll JAR, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med 2003;9:448–452. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.