

Abstract
The retrotrapezoid nucleus (RTN) contains chemically defined neurons (ccRTN neurons) that provide a pH-regulated excitatory drive to the central respiratory pattern generator. Here we test whether ccRTN neurons respond to stimulation of the perifornical hypothalamus (PeF), a region that regulates breathing during sleep, stress and exercise. PeF stimulation with gabazine increased blood pressure, phrenic nerve discharge (PND) and the firing rate of ccRTN neurons in isoflurane-anaesthetized rats. Gabazine produced an approximately parallel upward shift of the steady-state relationship between ccRTN neuron firing rate and end-tidal CO2, and a similar shift of the relationship between PND and end-tidal CO2. The central respiratory modulation of ccRTN neurons persisted after gabazine without a change in pattern. Morphine administration typically abolished PND and reduced the discharge rate of most ccRTN neurons (by 25% on average). After morphine administration, PeF stimulation activated the ccRTN neurons normally but PND activation and the central respiratory modulation of the ccRTN neurons were severely attenuated. In the same rat preparation, most (58%) ccRTN neurons expressed c-Fos after exposure to hypercapnic hyperoxia (6–7% end-tidal CO2; 3.5 h; no hypothalamic stimulation) and 62% expressed c-Fos under hypocapnia (∼3% end-tidal CO2) after PeF stimulation. Under baseline conditions (∼3% end-tidal CO2, hyperoxia, no PeF stimulation) few (11%) ccRTN neurons expressed c-Fos. In summary, most ccRTN neurons are excited by posterior hypothalamic stimulation while retaining their normal response to CNS acidification. ccRTN neurons probably contribute both to the chemical drive of breathing and to the feed-forward control of breathing associated with emotions and or locomotion.
Introduction
Any attempt to explain the homeostatic regulation of breathing must account for two fundamental facts that were already well known at the beginning of the 20th century: (1) arterial  remains constant in the face of large changes in the metabolic production of this gas and (2) hypercapnia produces a powerful stimulation of breathing (Haldane & Priestley, 1905). Moreover, if one disregards the special case of strenuous exercise, the very stability of arterial
remains constant in the face of large changes in the metabolic production of this gas and (2) hypercapnia produces a powerful stimulation of breathing (Haldane & Priestley, 1905). Moreover, if one disregards the special case of strenuous exercise, the very stability of arterial  indicates that the breathing adjustments that accompany various behaviours, notably locomotion, are driven to a minimal degree by changes in blood or brain acidity. Instead, these breathing adjustments and their associated cardiovascular changes are primarily triggered by feed-forward inputs from higher brain centres to the brainstem respiratory pattern generator (CPG) with some contribution from reflexes of muscle, joint or vestibular origin when the behaviour involves locomotion (Eldridge et al. 1985; Eldridge, 1994; Saper, 2002; Bell, 2006; Waldrop et al. 2006).
indicates that the breathing adjustments that accompany various behaviours, notably locomotion, are driven to a minimal degree by changes in blood or brain acidity. Instead, these breathing adjustments and their associated cardiovascular changes are primarily triggered by feed-forward inputs from higher brain centres to the brainstem respiratory pattern generator (CPG) with some contribution from reflexes of muscle, joint or vestibular origin when the behaviour involves locomotion (Eldridge et al. 1985; Eldridge, 1994; Saper, 2002; Bell, 2006; Waldrop et al. 2006).
The rat retrotrapezoid nucleus (RTN) contains about 2000 neurons that have a well-defined phenotype characterized by the presence of vesicular glutamate transporter 2 (VGLUT2) mRNA, a paired-like homeobox 2b (Phox2b)-immunoreactive (ir) nucleus and the absence of both tyrosine hydroxylase (TH) and choline acetyltransferase (ChAT) (Stornetta et al. 2006). We have called these neurons the chemically coded RTN neurons (ccRTN neurons) to distinguish them from other types of neurons in this area (Lazarenko et al. 2009). ccRTN neurons are activated by hypercapnia in anaesthetized rats, are uniformly activated by acidification in slices and the selective activation of ccRTN neurons in vivo stimulates breathing (Guyenet, 2008; Abbott et al. 2009; Lazarenko et al. 2009). Their acute inhibition or chronic destruction eliminates or markedly attenuates breathing and the ability of CO2 to elicit breathing under anaesthesia (Takakura et al. 2006; Takakura et al. 2008). Mutations that affect the normal embryonic development of the ccRTN neurons, albeit with varying degrees of selectivity, produce a lethal impairment of breathing and the loss of CO2 sensitivity at birth (Dubreuil et al. 2008; Pagliardini et al. 2008). Collectively, this evidence suggests that the ccRTN neurons provide a substantial fraction of the excitatory drive to the CPG at rest and make an important contribution to the homeostatic regulation of breathing by CO2.
The present experiments are designed to test the possibility that the activity of the ccRTN neurons is regulated not only by chemical drives (CNS acidification, carotid bodies) but also by inputs from higher brain centres. The second objective is to determine whether and how these descending inputs interact with the ability of the ccRTN neurons to encode changes in brain pH. For this test case we chose to stimulate the perifornical region of the hypothalamus based on prior evidence that activating this region increases blood pressure, respiration and locomotion (Waldrop et al. 1988; Iwamoto et al. 1996; Dimicco et al. 2002; McDowall et al. 2007; Tanaka & McAllen, 2008; Kuwaki, 2008; Zhang et al. 2009). Using single-unit recording and c-Fos expression as indicators of the activity of the ccRTN neurons we show here that, under anaesthesia, these cells are activated equally powerfully by hypothalamic stimulation and by CNS acidification and that there is no apparent interaction between the two stimuli. These results suggest that the ccRTN neurons are a site of integration between the chemical drive to breathe and the feed-forward command of breathing. The results also suggest that the ccRTN neurons retain their ability to respond to changes in brain pH regardless of their level of activity.
Methods
All experiments were done in the Department of Pharmacology at the University of Virginia, Charlottesville, Virginia. A total of 35 Sprague–Dawley rats weighing between 250 and 350 g were used. Procedures were in accordance with National Institutes of Health Animal Care and Use Guidelines and were approved by the Animal Care and Use Committee of the University of Virginia. These procedures are also in compliance with the policies and regulations of The Journal of Physiology as elaborated by Drummond (2009).
Neurophysiological experiments in vivo
General anaesthesia was induced with 5% isoflurane in pure oxygen. Rectal temperature was maintained close to 37.5°C with a servo-controlled heating pad. Surgery was done under artificial ventilation with 3% isoflurane in pure oxygen (∼1 ml (100 g)−1, 70–80 cycles min−1). End-tidal CO2 was monitored with a capnometer (Columbus Instruments, Columbus, OH, USA). This capnometer is a low-volume high-flow device that registers normal atmospheric CO2 during inspiration when no CO2 is added to the breathing mixture (Fig. 1). The end-expiratory CO2 reported in this paper is the peak CO2 value registered by the capnometer at the end of expiration averaged over 10 breaths. Our previous measurements performed under anaesthesia with 1% halothane anaesthesia in oxygen revealed a linear relationship between end-expiratory CO2 measured with this instrument and arterial  measured simultaneously by blood gas analysis (Guyenet et al. 2005). We also noted that, at low levels of CO2, the microcapnometer readings somewhat underestimated arterial partial pressure of CO2 (
measured simultaneously by blood gas analysis (Guyenet et al. 2005). We also noted that, at low levels of CO2, the microcapnometer readings somewhat underestimated arterial partial pressure of CO2 ( ) measured by blood gas analysis (
) measured by blood gas analysis (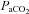 of 23 mmHg at ‘end-tidal CO2’ of 2%). This error became gradually smaller percentage-wise as CO2 increased, with
of 23 mmHg at ‘end-tidal CO2’ of 2%). This error became gradually smaller percentage-wise as CO2 increased, with  registering 75 mmHg at a capnometer reading of 10% end-tidal CO2. Similar comparative measurements were not performed in the present study and we make the assumption that the relationship between end-expiratory CO2 values measured by our capnometer and actual
registering 75 mmHg at a capnometer reading of 10% end-tidal CO2. Similar comparative measurements were not performed in the present study and we make the assumption that the relationship between end-expiratory CO2 values measured by our capnometer and actual 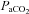 are the same under isoflurane as under halothane.
are the same under isoflurane as under halothane.
Figure 1. Hypothalamic stimulation activates PND and RTN neurons: representative experiment.

A, experimental approach: the phrenic nerve discharge (PND) and a single RTN unit were simultaneously recorded. The caudal hypothalamus was stimulated by injecting the selective GABAA receptor antagonist gabazine (5 mm, 30 nl; 7, facial nucleus). B, centre of gabazine injection site in experiment shown in C (f, fornix; ic, internal capsule; mt, mamillary tract). Ca, representative experiment. From top to bottom: per cent CO2 measured in the expired air where the top of the trace represents the end-tidal CO2, arterial pressure, rectified and integrated PND and the discharge rate of a single RTN neuron. Gabazine was injected where indicated by the filled arrowhead. Cb and Cc, excerpts taken at the times indicated by the open arrowheads in Ca showing original traces for PND and the RTN unit recorded at the same end-tidal CO2 level before and after gabazine. D, steady-state relationship between RTN discharge rate and end-tidal CO2 (etCO2) before (filled circles) and after gabazine (open circles). E, steady-state relationship between PND (product of amplitude and frequency, in arbitrary units) and end-tidal CO2 before (filled circles) and after gabazine injection (open circles).
The rats were subjected to the following previously described surgical procedures (Mulkey et al. 2004; Guyenet et al. 2005): bilateral vagotomy in the neck, femoral artery cannulation for arterial pressure (AP) measurements, femoral vein cannulation for fluid and drug administration, phrenic nerve dissection via a dorsolateral approach, a burr hole in the occipital plate to insert a recording electrode into the left medulla oblongata and another burr hole in the parietal bone to insert a drug-delivery pipette into the left hypothalamus. Finally, a bipolar stimulation electrode was inserted next to the mandibular branch of the facial nerve to map the facial motor nucleus with antidromic field potentials as a guide to locate the immediately adjacent retrotrapezoid nucleus.
During surgery the adequacy of the anaesthesia was gauged by the fact that nociceptive stimuli applied to the tail and hind legs produced no movement or change in blood pressure. During the recording period, isoflurane was reduced to 1.7–1.8% in pure oxygen. After a 15 min period of equilibration during which we verified that the previous anaesthesia criteria were still in effect, the rats were given the muscle relaxant pancuronium at the initial dose of 1 mg kg−1 intravenously. The level of anaesthesia was thereafter gauged by the stability of the arterial pressure, the absence of phrenic nerve activation during the application of a firm tail or toe pinch and a blood pressure response of less than 10 mmHg to these stimuli. The experiments were terminated with an overdose of isoflurane (5%) immediately followed by intracardiac perfusion with aldehyde fixatives.
Hypothalamic stimulation was done by injecting the GABAA receptor antagonist gabazine (5 mm in sterile saline containing 1% fluorescent microbeads (Lumafluor Inc., Durham, NC, USA) for identification of injection sites) through a glass pipette with a 25 μm external diameter. The electrode was inserted into the hypothalamus at a 28 deg angle pointing towards the back to reach the perifornical hypothalamic region (8.6 mm ventral, 0.6 mm lateral and 3.1 mm caudal to bregma). Typically 30 to 60 nl of gabazine solution were injected using multiple 5–9 ms pressure pulses (60 pounds per square inch).
The phrenic nerve discharge (PND, right side) was recorded with a bipolar wire electrode (200–3000 Hz), rectified and integrated as described previously (Mulkey et al. 2004; Guyenet et al. 2005). Unit recordings were made with glass pipettes filled with 2 m NaCl (7–12 MΩ). All analog data (end-tidal CO2, PND, unit activity) were acquired via a micro-1401 digitizer from Cambridge Electronic Design (Cambridge, UK) and were processed off-line using version 5 of the Spike 2 software (Cambridge Electronic Design) as described previously (Mulkey et al. 2004; Takakura et al. 2006; Moreira et al. 2007). Processing included action potential discrimination, measurement of neuronal discharge rate and PND ‘integration’ consisting of full-wave rectification and smoothing (time constant 0.015 or 0.03 s). The central respiratory modulation of the RTN neurons was determined by means of phrenic nerve-triggered histograms as described previously (Guyenet et al. 2005).
Prior to the recording session, the ventilation parameters (rate and tidal volume) were increased to lower end-tidal CO2 sufficiently to silence the phrenic nerve. Ventilation parameters were usually kept constant thereafter and various amounts of CO2 were bled into the breathing mixture to increase end-expiratory CO2 in four to five steps of approximately 1% each (∼7 mmHg). Each CO2 level was maintained for 5 min which was sufficient to reach steady state (RTN neurons and PND). Measurements (RTN firing rate, PND rate and amplitude) were made during the last 30 s of each step.
C-Fos expression experiments
These experiments were done in 14 vagotomized rats. These rats were anaesthetized and subjected to the same surgical procedures as the rats described above except that no burr hole was drilled in the occipital plate and no stimulation electrode was inserted next to the facial nerve. End-tidal CO2 was maintained at 2.5–3% throughout surgery in all rats. Isoflurane was then reduced from 3% (in oxygen) to 1.7–1.9% (still in oxygen) and after stabilization and verification of the adequacy of the anaesthesia, the muscle relaxant was administered (see previous section for details). In all rats ventilation was very briefly reduced to ascertain that PND was being properly monitored and then ventilation was readjusted to set end-tidal CO2 below the phrenic discharge threshold (2.5–3% CO2). End-tidal CO2 was left at this low level in the control rats (N= 5) which also received a 60 nl injection of saline into the left hypothalamus and a second one 90 min later (for details of placement and volumes see previous section). These rats were maintained for a total of 3.5 h under the same condition of ventilation before terminating the experiments with an overdose of isoflurane (5%) followed by intracardiac perfusion with aldehyde fixatives (see histology below). In the second group of rats (high CO2 group; N= 5), extra CO2 was bled into the oxygen–isoflurane breathing mixture to raise end-tidal CO2 to 6–7% corresponding to the approximate saturation of the chemoreflex in this preparation (Abbott et al. 2009). These animals were ventilated with this mixture for 3.5 h before terminating the experiment as described for the control group. The last group (gabazine group; N= 4) was maintained for the whole experiment at 2.5–3% end-tidal CO2. These rats received two injections of gabazine (5 mm; 30–60 nl) into the hypothalamus, the first at the beginning of the 3.5 h period and the second approximately 90 min later. The experiment was terminated as described for the other two groups.
Histological procedures
All histology was performed using brain tissue from rats deeply anaesthetized with 5% isoflurane and perfused transcardially with 100 ml of buffered saline followed by 500 ml of freshly prepared 4% paraformaldehyde in 100 mm phosphate buffer, pH 7.4. Brains were removed and stored in the perfusion fixative at 4°C before being cut into 30-μm-thick coronal slices. Tissue was kept in cryoprotectant solution (20% glycerol plus 30% ethylene glycol in 50 mm phosphate buffer, pH 7.4) at −20°C until processed. We identified ccRTN neurons as Phox2b-ir neurons that are devoid of tyrosine hydroxylase (TH) immunoreactivity and located within a defined region of the ventrolateral medulla (Takakura et al. 2008). C-Fos immunoreactivity was detected with a goat anti-c-Fos polyclonal IgG (1: 500, Santa Cruz Biotechnology, Santa Cruz, CA, USA) followed by Alexa Fluor 488 donkey anti-goat IgG (1: 200; Invitrogen, Carlsbad, CA, USA). TH was detected with a mouse anti-TH monoclonal antibody (1: 1000; Chemicon, Temecula, CA, USA) followed by Alexa Fluor 488 donkey anti-mouse IgG (1: 200; Invitrogen). Phox2b was detected using a rabbit anti-Phox2B (1: 800, gift from Dr J.-F. Brunet, Ecole Normale Supérieure, Paris, France) followed by Cy3 AffiniPure donkey anti-rabbit IgG (1: 200; Jackson ImmunoResearch, West Grove, PA, USA). The tissue was mounted in 0.1 m phosphate buffer, dehydrated and coverslips were affixed using DPX (Sigma-Aldrich, USA). Note that the same reporter, Alexa Fluor 488, was used to detect TH and c-Fos. In this manner, the ccRTN neurons could be most easily identified as cells with a Phox2b-ir nucleus (Alexa 488-positive, green) but without Alexa 488 in their cytoplasm (TH-negative). The c-Fos-expressing ccRTN neurons could thus be identified as TH-negative neurons with nuclei that contained both Alexa 488 and Cy3.
A Zeiss Axioskop 2 microscope (Oberkochen, Germany) was used for all observations. Immunoreactive nuclei or neurons were mapped using a motor-driven microscope stage controlled by the Neurolucida software (Stornetta & Guyenet, 1999). The Neurolucida files were exported to the NeuroExplorer software (MicroBrightField, Williston, VT, USA) to count the various types of neuronal profiles within defined areas. In all experiments, a one-in-six series of 30 μm brain sections (i.e. every 180 μm) were arranged on slides in sequential order. Alignment of coronal sections between brains was done relative to a reference section (bregma −11.6 mm, according to the atlas of Paxinos and Watson (1998)) selected as the most caudal section containing an identifiable cluster of facial motor neurons. For each animal, 12 sections were counted starting with the section 360 μm caudal to the reference section (2 sections behind the reference section).
The gabazine injection sites were identified using adjacent 30 μm sections counterstained with thionin and rapidly dehydrated through a graded series of alcohols. The Texas Red-coated microbeads were identified by their fluorescence. The centre of each injection was plotted using the Neurolucida software and located in relation to a series of nearby landmarks revealed by the thionin counterstaining.
Statistics
Statistical analysis was done with Sigma Stat version 3.1 (Jandel Corporation, Point Richmond, CA, USA). Data are reported as means ± standard error of the mean (s.e.m.). The paired t test, one- or two-way parametric ANOVA were used as appropriate. The Student–Newman–Keuls method was used for group comparisons following one-way ANOVA (c-Fos experiments). The Holm–Sidak method was used for all pair-wise multiple comparisons procedures following two-way ANOVA (unit and PND data). Significance was set at P < 0.05.
Results
Hypothalamic stimulation activates PND and RTN neurons
We recorded phrenic nerve discharge (PND) and the activity of a single RTN unit simultaneously and determined how these two variables were affected by stimulating the caudal hypothalamus with an injection of the selective GABAA receptor antagonist gabazine (Fig. 1A). This experiment required continuous recording of the unit for at least an hour and was accomplished with 10 neurons in seven rats (gabazine was injected a second time in three rats following complete recovery from the first trial). The drug was targeted to the perifornical region of the hypothalamus, close to the dorsomedial nucleus (Fig. 1B) based on prior evidence that stimulating this region increases blood pressure, respiration and locomotion (Hilton & Redfern, 1986; Waldrop et al. 1988; Iwamoto et al. 1996; Dimicco et al. 2002; McDowall et al. 2007; Tanaka & McAllen, 2008; Kuwaki, 2008; Zhang et al. 2009). Figure 1C–E describes a representative experiment. Typical of its class, this RTN neuron was located below the caudal end of the facial motor nucleus (Fig. 1A), was silenced by hypocapnia (Fig. 1Ca and D), had a robust response to increasing levels of CO2, (Fig. 1Ca and D) and a modest degree of respiratory modulation (Fig. 1Cb and c). This particular unit was least active during the early part of the phrenic discharge (early-inspiratory dipper). CO2 was stepped up to the level that caused the maximum PND in this preparation to determine the relationship between end-tidal CO2 and PND and that between end-tidal CO2 and the RTN unit (Fig. 1Ca, D and E). These curves were generated prior to administration of gabazine into the hypothalamus and during the 30–40 min period following drug administration. Hypothalamic stimulation increased blood pressure, PND (amplitude and frequency) and the discharge rate of the RTN unit (Fig. 1Ca–c). These effects occurred synchronously. The relationship between end-tidal CO2 and the activity of the RTN neuron was shifted upward in approximately parallel fashion (Fig. 1D). A similar upward shift was observed in the relationship between PND (product of amplitude and frequency) and end-tidal CO2 (Fig. 1E). After gabazine, neither the unit nor the PND could be silenced by hyperventilating the rat down to 2% end-tidal CO2. However, both the cell and the PND remained responsive to changes in arterial  .
.
This experiment was reproduced with a similar outcome in nine other neurons (6 rats). The effects of gabazine on PND and RTN units were highly significant overall (Fig. 2A and B; P < 0.001 by two-way ANOVA). However, there was no statistically significant interaction between the CO2 level and the effect of gabazine (P= 0.96; two-way ANOVA) suggesting that the ability of the cells to respond to increases in CO2 was not changed by hypothalamic stimulation. In fact, hypothalamic stimulation produced an approximately parallel and upward shift of the relationship between RTN activity and end-tidal CO2 (Fig. 2A).
Figure 2. Hypothalamic stimulation activates PND and RTN neurons: group data.

A, relationship between the discharge rate of RTN units (10 cells, 7 rats) and end-tidal CO2 before (filled circles) and after gabazine (open circles). Gabazine produced an approximately parallel upward shift of the relationship between RTN discharge rate and end-tidal CO2. The effect of gabazine was significant (P < 0.001 by ANOVA) but there was no statistically significant interaction between the CO2 level and the effect of gabazine (P= 0.96; two-way ANOVA). B, relationship between the PND (product of amplitude and frequency; 7 rats) and end-tidal CO2 before (filled circles) and after gabazine (open circles). The effect of gabazine was significant (P < 0.001 by ANOVA) but there was no statistically significant interaction between the CO2 level and the effect of gabazine (two-way ANOVA). C, computer-assisted mapping of the centre of the gabazine injection sites. All the sites were collapsed on a single transverse section located at the geographic centre of the injection sites (bregma −3.3 mm after (Paxinos & Watson, 1998)). Abbreviations: Arc, arcuate nucleus; DMC, compact part of the dorsomedial hypothalamic nucleus; f, fornix; ic, internal capsule; mt, mamillothalamic tract; opt, optical tract; PeF, perifornical region; VMH, ventromedial hypothalamic nucleus. The grey circle is meant to represent a sphere that contained the centre of all the injection sites in any direction.
The gabazine injection sites were clustered in the perifornical region at the level of the dorsomedial hypothalamic nucleus (Fig. 2C). In Fig. 2C the dots represent the centre of the injection sites. The effective spread of the drug was not determined. The 1 mm diameter grey circle illustrates the scatter of the injection sites. A similar spread was observed in the rostrocaudal direction.
Morphine depresses the PND but has little effect on the activation of RTN neurons by hypothalamic stimulation
This experiment was performed to test whether RTN activation by hypothalamic stimulation could be a mere consequence rather than a cause of the activation of the CPG. To address this point we took advantage of the known fact that μ-opiate receptor agonists such as morphine have powerful depressant effects on the CPG but have little effect on the activity of RTN neurons (Sato et al. 1992; Manzke et al. 2003; Mulkey et al. 2004; Onimaru et al. 2006).
PND was recorded simultaneously with a single RTN unit as described in the previous section. Before administering morphine, we determined the steady-state relationship between RTN neuron activity, PND (product of frequency and amplitude) and end-tidal CO2 (Fig. 3A–C). Morphine was then slowly injected intravenously at an initial dose of 6.7 mg kg−1 while end-tidal CO2 was maintained constant. After a few minutes of equilibration the steady-state relationship between PND, the RTN unit and end-tidal CO2 was determined a second time. Finally, gabazine was injected into the perifornical region of the hypothalamus and the relationship between the dependent variables and end-tidal CO2 was determined for the third and final time (Fig. 3A).
Figure 3. Activation of RTN neurons by posterior hypothalamic stimulation persists after administration of morphine: representative experiment.

A, traces are in the same order as in Fig. 1. The RTN neuron was identified before morphine administration by its location, robust response to changing levels of inspired CO2 and mild respiratory modulation (excerpt a shows integrated PND and original recording of the unit). Morphine (6.7 mg kg−1i.v. at arrow) eliminated PND but only slightly decreased the activity of the RTN unit (excerpt b) which became much more regular. After a short equilibration period, unit and PND were challenged again with varying levels of CO2. Finally, gabazine was injected into the DMH/PeF region of the hypothalamus and the relationship between the dependent variables and end-tidal CO2 was determined for the third time. Excerpt c illustrates that, after gabazine and morphine, the RTN unit discharged at a much higher rate than in the control situation (excerpt a) whereas PND was much smaller. Excerpts a–c were obtained at the same level of end-tidal CO2. B, steady-state relationship between the discharge rate of the RTN unit and end-tidal CO2 before morphine (control), after morphine sulfate (MS) and after morphine plus gabazine (gabazine). C, steady-state relationship between PND (product of amplitude and frequency) and end-tidal CO2 before morphine (control), after morphine (MS) and after gabazine and morphine (gabazine). Grey areas in A–C represent the end-tidal CO2 range in which the RTN unit was active but no PND was observed after combined administration of morphine and gabazine.
Morphine caused the anticipated complete inhibition of PND (Fig. 3A). Except in two rats, this inhibition could not be overcome by raising end-tidal CO2 up to 8.6%, a level well beyond that which saturated the chemoreflex in the absence of morphine (Fig. 3A and C). The RTN unit was modestly inhibited by morphine (Fig. 3A and excerpts a and b). Morphine caused a downward shift of the relationship between the discharge rate of the unit and end-tidal CO2 (Fig. 3A and B). As a result, the level of end-tidal CO2 necessary for the unit to be active was elevated by morphine. Gabazine injection into the hypothalamus while end-tidal CO2 was maintained constant activated the RTN neuron vigorously without causing a reappearance of the PND. However, unlike before gabazine, a substantial amount of PND could be elicited at higher levels of end-tidal CO2 (Fig. 3A and excerpt c). Nonetheless, as shown in Fig. 3C, gabazine did not restore the chemoreflex to the control (pre-morphine) level and the relationship between PND and end-tidal CO2 was still considerably down-shifted relative to the control pre-morphine condition. In contrast, after gabazine injection in the hypothalamus, the relationship between RTN neuron activity and end-tidal CO2 was up-shifted relative to the control level despite the presence of morphine (Fig. 3A and B). As a result, there was a large window of end-tidal CO2 (from 2.5% or less to 4.5%, represented in grey in Fig. 3B and C) in which the RTN neuron was active and no PND was observed. This is in contrast to the control (pre-morphine) situation during which the CO2 threshold of the RTN neuron and that of the PND were very similar (usually within 0.5%).
These observations were reproducible (N= 9 rats). On average, morphine administration (6.7–10 mg kg−1i.v.) immediately reduced the resting discharge rate of the RTN neurons sampled (from 10.2 ± 1.36 to 7.7 ± 1.04 Hz, N= 9; P < 0.05 by paired t test) although one of the nine neurons tested was activated by the drug. The effect of morphine seemed to be independent of the level of CO2 (Fig. 4A). After morphine administration, gabazine produced effects on RTN neurons that were very similar to those observed in the absence of morphine (compare Figs 2A and 4A). Gabazine increased the discharge of RTN neurons by about 5 Hz at any given level of CO2 regardless of whether morphine had been administered. In contrast, gabazine produced a relatively modest activation of the PND after morphine treatment (Fig. 4B). To be more specific, on average the end-tidal CO2 threshold for PND was always above 4% after combined treatment with morphine and gabazine whereas with gabazine alone the threshold, if any, was at less than 2% end-tidal CO2. Also, the end-tidal CO2 level at which RTN neurons became active was close to the phrenic apnoea threshold in the control situation (Figs 2 and 4) whereas after combined treatment with morphine and gabazine there was around a 2% end-tidal CO2 difference between the two thresholds.
Figure 4. Activation of RTN neurons by DMH/PeF stimulation persists after administration of morphine: group data.

Average effect of i.v. morphine with subsequent gabazine injection on RTN (9 units) and PND (product of amplitude and frequency; 9 rats). Data points from individual cases were grouped into 1–1.5% CO2 bins, averaged and analysed by two-way ANOVA. A, RTN units were mildly inhibited by morphine (NS by ANOVA). Gabazine administered after morphine significantly increased unit activity at all levels of end-tidal CO2 (P < 0.001). B, morphine eliminated PND (two animals recovered some PND at high levels of CO2). Gabazine produced only partial recovery of PND (P < 0.01 vs. morphine alone; P < 0.05 vs. control).
Effect of hypothalamic stimulation of the central respiratory modulation of RTN neurons
We examined the central respiratory modulation of 20 RTN neurons and found it to be approximately the same as under halothane anaesthesia, and more pronounced at high than at low CO2 (Guyenet et al. 2005). Dips in discharge probability occurred during early inspiration in four neurons (early-I dippers; Figs 1Cc, 5Ab,Ca), during expiration in 10 neurons (E dippers Fig. 5Ac, B and Cb) or during both phases in six neurons (double dippers; Fig. 5Aa). The discharge probability of the E dippers was always lowest during the post-inspiratory phase. The discharge rate increased gradually and to a variable degree during the latter part of expiration and, in some cells, increased abruptly at the onset of the PND (e.g. Fig. 5Ac).
Figure 5. Central respiratory modulation of the ccRTN neurons: effect of hypothalamic stimulation in the presence or absence of morphine.

Aa–c, effect of posterior hypothalamic stimulation on the discharge pattern of ccRTN neurons. The peri-event histograms of single neuron activity were triggered on the ascending portion of the rectified and integrated PND. The three cells shown illustrate the various types of central respiratory pattern that were observed (Aa, double dipper; Ab, early inspiratory dipper; Ac, expiratory dipper). The control and the experimental histograms (gabazine) are from the same neuron. Both histograms (control and gabazine) were constructed during periods when the end-tidal CO2 was approximately the same. B, effect of intravenous morphine on the discharge pattern of a ccRTN neuron. The control and the experimental histogram were obtained during recording periods when the end-tidal CO2 was approximately the same (∼7%). Note the loss of the respiratory modulation. Ca and b, effect of hypothalamic stimulation with gabazine following morphine injection. The control histograms were obtained before administration of morphine. The control and the experimental histograms were obtained during periods when the end-tidal CO2 was approximately the same.
PND-triggered histograms were obtained for nine RTN neurons both before and after hypothalamic stimulation with gabazine. These histograms were acquired during periods when end-expiratory CO2 was approximately the same to minimize the potential influence of changes in brain acidity on the pattern. The nine cells examined in this fashion included three early-I dippers, three E dippers and three double dippers. Hypothalamic stimulation raised the discharge probability of the neurons by a roughly constant amount throughout the respiratory cycle without changing their central respiratory pattern (Fig. 5Aa–c). The overall shape of the phrenic nerve discharge was not consistently altered by hypothalamic stimulation in the absence of morphine pretreatment (Fig. 5Aa–c).
Morphine administration eliminated or drastically reduced the rate and amplitude of the phrenic nerve discharge (PND). Following morphine administration the RTN neurons fired more regularly (Fig. 3Aa and b). PND-triggered histograms of RTN neuron activity were built in two cases in which a small PND remained at high levels of CO2 following morphine administration. These peri-event histograms were flat (Fig. 5B), confirming that morphine eliminates the central respiratory modulation of the RTN neurons.
In five cases (1 early-I dipper, 3 E dippers, 1 double dipper) we were able to compare the central respiratory modulation of the same RTN neuron in the control state and after combined administration of morphine and hypothalamic stimulation with gabazine. The peri-event histograms were acquired during periods when end-tidal CO2 was high (6–8%) and approximately the same (within 0.5%). In each case, the probability of discharge of the neuron was much more uniform throughout the respiratory cycle (Fig. 5Ca and b). Finally, the PND duration was longer and usually developed a strongly decrementing configuration not seen under control conditions or during hypothalamic stimulation in the absence of morphine (Fig. 5Cb).
Effect of hypercapnia and hypothalamic stimulation on c-Fos expression by a population of chemically coded RTN neurons
The preceding experiments suggested that RTN neurons are being driven to comparably high levels of activity by hypothalamic stimulation delivered under hypocapnia and by hypercapnia in the absence of hypothalamic stimulation. By necessity, this conclusion is based on a small number of observations made on randomly encountered RTN neurons. The next experiments were designed to confirm and extend this interpretation by using the proto-oncogene product c-Fos as a reporter of cell activation. The CO2-sensitive units of the RTN region belong to a cell group with a well-defined phenotype (ccRTN) characterized by the presence of VGLUT2 mRNA, a Phox2b-ir nucleus and the absence of both tyrosine hydroxylase (TH) and choline acetyltransferase (ChAT) (Stornetta et al. 2006). In practice, the ccRTN cells can be identified histologically as Phox2b-positive and TH-negative cells (Phox2b+TH−) within the region of interest because every Phox2b-ir neuron within this region contains VGLUT2 mRNA (Stornetta et al. 2006) and the Phox2b+TH− are spatially segregated from surrounding ChAT-positive neurons, e.g. the facial motor neurons (Takakura et al. 2008). Finally, the above criteria eliminates any confusion between ccRTN neurons and serotonergic neurons because the latter do not express Phox2b (Stornetta et al. 2006). Hypercapnia is already known to produce c-Fos expression in the RTN region of animals (Sato et al. 1992; Teppema et al. 1994) but whether these neurons are the ccRTN neurons has not been tested.
We compared c-Fos expression by the ccRTN neurons in three groups of isoflurane-anaesthetized, paralysed and artificially ventilated rats. The control group (N= 5) was maintained under hypocapnic conditions (the level of end-tidal CO2 where the PND was undetectable, usually 2.5–3% CO2) and received injections of saline into the hypothalamus. The second group (N= 5) was subjected to hypercapnia (6–7% end-tidal CO2) and also received saline. The third rat group (N= 4) was maintained under hypocapnic conditions (2.5–3% CO2) and received gabazine injections into the perifornical region of the hypothalamus. Each rat was assigned a random letter code and the cells were counted by a ‘blinded’ observer.
Figure 6 shows the RTN in a control rat and in a rat that received a gabazine injection in the hypothalamus. In the control rat, the nuclei of most of the Phox2b+TH− neurons (ccRTN neurons; white arrowheads) lacked c-Fos immunoreactivity whereas, in the gabazine-treated rat, a large proportion of these cells had a c-Fos-ir nucleus. The neurons that were positive for both Phox2b and TH (black arrowheads in Fig. 6) are C1 neurons (Stornetta et al. 2006). Under anaesthesia most of these neurons express c-Fos no matter what other conditions prevail and these neurons were ignored. The number of Phox2b+TH− neurons that had a c-Fos-positive nucleus were identified and counted in a one-in-six series of transverse sections (1 section every 180 μm). Counts were made on both sides of the brain and throughout the portion of the ventrolateral medulla depicted in grey in Fig. 7A (the ‘RTN region’). As a control population we also counted the number of c-Fos-ir neurons located in the nearby parapyramidal region, also represented in grey in Fig. 7A. These presumably serotonergic cells were TH-negative and devoid of Phox2b immunoreactivity. Hypercapnia and gabazine caused a large increase in the number of c-Fos-positive ccRTN neurons (Fig. 7B). Summing up the cells identified at all levels of the region of interest represented in Fig. 7B and multiplying this number by 6 produced an uncorrected total number of c-Fos-positive ccRTN neurons counted per rat (287 ± 58 in the control group; 1520 ± 115 in the hypercapnia group and 1606 ± 218 in the gabazine group). After applying a 0.81 Abercrombie correction factor previously determined on identically prepared histological material (Takakura et al. 2008), a more accurate estimate of the actual number of c-Fos-positive ccRTN neurons in the three rat groups was respectively 233 ± 47, 1233 ± 93 and 1302 ± 177. The total population of ccRTN neurons counted in this manner was previously estimated at 2112 ± 71 per rat (Takakura et al. 2008); therefore, on average, roughly 60% of these neurons expressed c-Fos in each experimental rat group vs. only 11% in the control group. The difference between the experimental groups and the control group was highly significant by one-way ANOVA (Fig. 7C). In contrast, the number of c-Fos-ir neurons located in the parapyramidal region was high at rest and was unaffected by either treatment (Fig. 7D). Very few Phox2b-negative neurons expressed c-Fos within the portion of the RTN that resides under the facial motor nucleus (small dots in Fig. 7A). However, at the caudal end of the RTN, many more such neurons were present (Fig. 7A, level 11.96 mm caudal to bregma). This group of c-Fos-ir neurons extended well beyond our region of interest in the caudal direction. These cells were not counted.
Figure 6. Both hypercapnia and hypothalamic stimulation trigger c-Fos expression by the ccRTN neurons.

C-Fos expression in the RTN region under control condition and after hypothalamic stimulation with gabazine. ccRTN neurons were identified by the presence of a Phox2b-ir nucleus and the absence of tyrosine hydroxylase (TH) immunoreactivity (Phox2b+TH−). Phox2b was identified using a red fluorescent reporter (Cy3, left panels). TH (cytoplasmic) and c-Fos (nuclear localization) were identified using the same green fluorescent reporter (Alexa 488, middle panels). In the control case, the nuclei of most of the ccRTN neurons lacked c-Fos immunoreactivity (white arrowheads in top middle panel). In the gabazine-treated rat, the majority of the ccRTN neurons had a c-Fos-ir nucleus (white arrowheads in lower middle panel). The neurons that were positive for both Phox2b and TH are C1 neurons (black arrowheads). These cells were typically Fos-ir in both control and experimental rats presumably because they are highly active under anesthesia, even at rest. The scale bar, 20 μm, applies to all panels. A colour version of this figure is available online.
Figure 7. Quantification of the number of c-Fos-expressing ccRTN neurons in rats subjected to hypercapnia or hypothalamic stimulation.

A, transverse sections of the medulla oblongata illustrating the two regions in which c-Fos-ir neurons were identified and counted. The numbers at the right of the hemisections refer to the location of the sections caudal to the bregma level (in mm) according to the atlas of Paxinos & Watson (1998). The case represented in A is a rat that had been exposed to 7% CO2 for 3.5 h. Cell counts were made bilaterally. The grey area located ventral to and immediately caudal to the facial motor nucleus (7) delineates the ‘RTN region’ within which the ccRTN neurons (Phox2b+TH−) were identified and counted. C-Fos-ir nuclei were also counted within the parapyramidal region (grey area identified as ppyr on the bregma 11.24 mm section). These neurons form a compact superficial cluster of Phox2b−TH− neurons that are presumably serotonergic. The grey area in the section at 11.96 has been reproduced (represented by the dashed lines) at a higher magnification into the dorsal aspect of the section for clarity. B, rostrocaudal distribution of the number of c-Fos-positive ccRTN neurons (Phox2b+TH−) counted per level in three groups of rats (one 30-μm-thick section every 180 μm). The control group (filled circles, N= 5) was maintained under hypocapnic conditions (2.5–3% CO2), the second group (open circles, N= 5) was subjected to hypercapnia (6–7% end-tidal CO2) and the third group (filled triangles, N= 4) was maintained under hypocapnic conditions (2.5–3% CO2) and received gabazine injections into the DMH/PeF region of the hypothalamus. C, total number of c-Fos-positive ccRTN neurons counted per rat in the same 3 groups of rats. D, total number of c-Fos-positive parapyramidal neurons counted per animal.
Discussion
The present study suggests that the ccRTN neurons, which provide a pH-regulated excitatory drive to the respiratory network, also participate in the feed-forward control of breathing that originates from the posterior hypothalamus.
RTN: definition and cytology
The RTN was originally defined as a sparse collection of neurons located under the facial motor nucleus which innervate the nucleus of the solitary tract and the ventrolateral medulla (Connelly et al. 1989; Smith et al. 1989). This projection pattern and the rough coincidence between the location of these neurons and an area of the ventral medullary surface thought since the 1960s to play a role in chemoreflexes (Loeschcke, 1982) suggested that RTN neurons might be involved in the control of breathing by CO2 (Connelly et al. 1989; Smith et al. 1989). The RTN region had remained poorly defined from a cytological point of view until it was found to contain a distinctive cluster of non-catecholaminergic, non-cholinergic, glutamatergic and Phox2b-expressing neurons (Mulkey et al. 2004; Stornetta et al. 2006; Takakura et al. 2008; Lazarenko et al. 2009). We have called these neurons the chemically coded RTN neurons (ccRTN neurons) (Lazarenko et al. 2009) to distinguish them from other types of neurons that may reside in the same area. The ccRTN neurons are activated by CO2in vivo and uniformly activated by acidification in slices (Lazarenko et al. 2009). Some ccRTN neurons display pre- and post-inspiratory discharges at birth in specific preparations in vitro and are therefore part of a broader collection of respiration-synchronous neurons called the parafacial respiratory group (Onimaru et al. 2008). In the absence of anaesthesia, the RTN region contains late-expiratory neurons whose discharge coincides with the appearance of abdominal nerve activity when the preparation is subjected to CO2 stimulation (Abdala et al. 2009). These expiratory neurons may regulate active expiration (Janczewski & Feldman, 2006; Taccola et al. 2007). They may be a subset of ccRTN neurons that are inactive in anaesthetized preparations or they could be another type of RTN neuron that awaits biochemical characterization.
Relationship between end-expiratory CO2 and RTN discharge rate
The relationship between end-expiratory CO2 and the discharge of ccRTN neurons is curvilinear and saturable (Guyenet et al. 2005). This characteristic has been tentatively attributed to a negative feedback that ccRTN neurons receive from the central pattern generator (Guyenet et al. 2005). The dips in RTN neuron discharge probability that occur during early inspiration, post-inspiration or both in vagotomized preparations have been interpreted as manifestations of this feedback (Guyenet et al. 2005; and present results) because they disappear when the CPG is silenced pharmacologically, for example by morphine administration. These respiratory patterns are cell-specific and vary little with different anaesthetics in rats. Under anaesthesia the PND and, more generally, the activity of the central respiratory pattern generator, are highly dependent on the excitatory drive from ccRTN neurons (Takakura et al. 2008). The negative feedback from the pattern generator to ccRTN neurons should therefore eventually lead to a steady-state discharge of both ccRTN neurons and the phrenic nerve despite increasing levels of brain acidification. This steady state is characteristic of conventional anaesthetised and vagotomised preparations (Eldridge et al. 1984; Takakura et al. 2008), and is also observed in the working heart–brain preparation (e.g. Fig. 6 in Simms et al. 2009).
The CO2 threshold of the RTN neurons presumably depends on the background synaptic drives that these neurons receive in the resting state and on the effect of anaesthetics on their intrinsic properties and their still unidentified pH-sensing mechanism. Anaesthetics and hyperoxia also change cerebrovascular reactivity (Ainslie & Duffin, 2009) which could be another source of variability in the CO2 threshold of RTN neurons and PND between intact animals and various anaesthetized preparations. For unknown reasons, the end-expiratory CO2 threshold for both RTN neuron activity and the PND is notably low under isoflurane anaesthesia (2.5 to 3% by our capnometer measurement which probably corresponds to a  of 25–30 mmHg, see Methods) compared to other anaesthetics including halothane (Guyenet et al. 2005; Takakura et al. 2006).
of 25–30 mmHg, see Methods) compared to other anaesthetics including halothane (Guyenet et al. 2005; Takakura et al. 2006).
ccRTN neurons express c-Fos following hypercapnia
Hypercapnia induces c-Fos expression in the rostral ventrolateral medulla (Sato et al. 1992; Teppema et al. 1994; Okada et al. 2002). We show here that this c-Fos-expressing cell population includes a high percentage of the ccRTN neurons. The Phox2b-ir nuclei that we identified histologically are undoubtedly neurons for the following reasons. These Phox2b-ir nuclei are in precise register with cells that have a characteristic neuronal morphology in two Phox2b-enhanced green fluorescent protein (eGFP) transgenic mice strains (Lazarenko et al. 2009). One of these mice was examined ultrastructurally and eGFP was only detected in neurons (Lazarenko et al. 2009). Secondly, all the CO2-activated neurons that we have been able to record in the RTN in vivo have a Phox2b-positive nucleus and the pH-sensitive neurons recorded in slices in this region contain Phox2b mRNA (Stornetta et al. 2006; Lazarenko et al. 2009). The ccRTN neurons are probably a subset of the cells called type II by Okada et al. (2002), which these authors identified both within and dorsal to the marginal layer. These authors showed that type II cells no longer express c-Fos in an in vitro preparation exposed to CO2 in the presence of TTX and they concluded from this observation that type II cells are not intrinsically chemosensitive. Instead, they suggested that type II cells may be indirectly activated by another type of cell (type I) which also expressed c-Fos under their hypercapnic conditions and retained this property in the presence of tetrodotoxin (TTX). This interpretation may be questioned on two grounds. First, under TTX, calcium entry into neurons would probably be eliminated due to the absence of firing and this would most probably eliminate c-Fos expression regardless of whether type II neurons are directly or indirectly acid-sensitive. Second, the ccRTN neurons retain some acid sensitivity under TTX in the form of an inward current (Mulkey et al. 2004). Under our in vivo conditions, we found no evidence of the tight perivascular clusters of c-Fos-positive small-nucleated cells (‘type I’ cells) described by Okada et al. (2002). These pH-sensitive cells could conceivably be a specialized form of glia; they could also be arterial smooth muscle cells, pericytes or some other CO2-/acid-sensitive vascular component (Wray & Smith, 2004). Our experiments were conducted using a different anaesthetic and we also used hyperoxic conditions in order to eliminate the influence of the carotid bodies. Furthermore, we limited hypercapnia to between 6 and 7% CO2. These experimental differences may explain the absence of perivascular clusters of c-Fos-positive cells in our material.
Fifty-eight per cent of the ccRTN neurons expressed c-Fos after hypercapnia on average. This percentage could be an underestimation of the fraction of the ccRTN neurons that have the potential of expressing c-Fos after hypercapnia. For instance, our immunohistochemical procedure for the detection of c-Fos may lack the sensitivity necessary to detect all the c-Fos-expressing neurons. Also, we examined c-Fos expression at only one time point and we exposed the rats to an end-tidal CO2 level of only 6–7%, conditions that may not have triggered maximal c-Fos expression. Finally, the CO2 level at which the ccRTN neurons start to discharge in vivo is influenced by the synaptic input that they receive, be it serotonergic (Mulkey et al. 2007) or of hypothalamic origin as shown in the present study. Quite possibly, CO2 alone is not a strong enough stimulus to activate the entire population of ccRTN neurons, especially under anaesthesia, and the roughly 40% ccRTN neurons that did not express c-Fos after hypercapnia may be neurons that have a particularly hyperpolarized membrane potential at rest. These hypothetical high-threshold units may be recruited only when breathing needs to be especially intense and could conceivably be the RTN neurons that are suspected to govern the activity of expiratory muscles (Janczewski & Feldman, 2006; Abdala et al. 2009). The alternative explanation is that 40% of the ccRTN neurons lack the ability to respond to CO2 i.e. are acid-insensitive. This interpretation cannot be excluded but it seems less plausible because every ccRTN neuron tested to date responds to acidification in slices (Lazarenko et al. 2009).
The power and limitation of the c-Fos method to analyse neuronal activation patterns in vivo has been discussed many times since its original description (Sagar et al. 1988). Briefly, not all neurons express c-Fos when activated and c-Fos expression denotes an impending increase in protein synthesis which can be elicited by an increase in action potential frequency via a rise in intracellular calcium but may have other triggers. However, the fact that a majority of the ccRTN express c-Fos following hypercapnia adds to a long line of observations suggesting that these neurons are activated by hypercapnia in vivo and by acidification in slices (Guyenet, 2008; Onimaru et al. 2008; Lazarenko et al. 2009).
Within the parapyramidal raphe region, c-Fos-positive neurons were no more numerous in the experimental rats than in the control group. Prior evidence suggests that the serotonergic neurons present in this region are activated by carotid body stimulation but are insensitive to brain acidification in vivo (Erickson & Millhorn, 1994; Mulkey et al. 2004). The present experiment does not provide any additional information regarding their possible pH sensitivity in vivo since these cells also expressed c-Fos under basal hypocapnic conditions.
Finally, according to several prior studies, hypercapnia also induces c-Fos in regions of the ventrolateral medulla that are located caudal to the RTN (Sato et al. 1992; Teppema et al. 1997; Miura et al. 1998; Okada et al. 2002). The nature and function of these other CO2-activated neurons is unknown and the present study was not designed to probe this issue further. The fact that these other neurons also express c-Fos after hypercapnia does not necessarily imply that they are directly sensitive to acidification.
ccRTN neurons and morphine
Sato et al. (1992) first pointed out that RTN neurons must be relatively insensitive to morphine because this drug did not suppress their ability to express c-Fos after hypercapnia. In a prior electrophysiological study performed in halothane-anaesthetized rats, we found that the discharge rate of the ccRTN neurons was not changed appreciably by doses of morphine that profoundly reduced PND (Mulkey et al. 2004). The present and more extensive study performed in isoflurane-anaesthetized rats indicates that most of the ccRTN neurons are in fact inhibited by morphine given intravenously although to a far lesser degree than morphine inhibition of PND. The pre-inspiratory neurons that reside under the facial motor nucleus and are probably a subset of neonatal ccRTN neurons are opiate-insensitive in vitro (Onimaru et al. 2008; Ballanyi et al. 2009). This evidence suggests that the ccRTN neurons do not express μ-opioid receptors at birth but it does not exclude the possibility that such receptors might be present later in life. The inhibition of the ccRTN neurons by morphine in vivo could also result from increased synaptic inhibition or decreased synaptic excitation. Indeed, even under anaesthesia, the resting activity of the ccRTN neurons is controlled to some degree by synaptic drives as suggested by the ability of glutamate or GABA receptor antagonists to change breathing when these drugs are microinjected directly into the RTN (Nattie & Li, 1995; Nattie et al. 2001).
In short, the ccRTN neurons are mildly inhibited by morphine in vivo. The overall effect of morphine on the discharge of RTN neurons is modest (about 25% decrease). However, given that these cells provide an excitatory drive to the respiratory network (Abbott et al. 2009), their inhibition by morphine should contribute to the reduction in respiratory motor activity caused by this drug.
ccRTN neurons contribute to the increase in breathing elicited by hypothalamic stimulation
Chemical stimulation of posterior hypothalamic neurons increases breathing (Hilton & Redfern, 1986; Waldrop et al. 1988; Dimicco et al. 2002; Zhang et al. 2006a; Tanaka & McAllen, 2008). Given that hypothalamic stimulation activates the ccRTN neurons vigorously (this study) and that selective stimulation of the same neurons also activates breathing (Abbott et al. 2009), the activation of the ccRTN neurons must be contributing to the breathing stimulation induced by hypothalamic stimulation. Hypothalamic stimulation in the absence of morphine produced an upward shift of the relationship between RTN neuron activity and CO2. PND-triggered histograms of the activity of single ccRTN neurons (Fig. 5A) revealed that hypothalamic stimulation raised the discharge probability of these neurons uniformly throughout the respiratory cycle. In other words, the activation of the ccRTN neurons produced by hypothalamic stimulation was not respiratory cycle-dependent. This characteristic is also consistent with the notion that hypothalamic stimulation activates the CPG via the ccRTN neurons. The converse interpretation would require that hypothalamic stimulation selectively increases the respiratory phasic component of the discharge of ccRTN neurons.
A CO2 threshold could no longer be determined after hypothalamic stimulation but the RTN neurons appeared to retain a normal sensitivity to changes in CO2 as judged by the approximately parallel shift of the relationship between their discharge rate and end-expiratory CO2 seen both in the presence and the absence of morphine (Figs 2A, 4A). This result suggests that neither hypothalamic stimulation nor morphine interfere detectably with the ability of these neurons to encode changes in brain acidification.
Unit recordings indicated that every ccRTN neuron sampled was vigorously activated by hypothalamic stimulation. However, given the requirement that cells be active to be detected by unit recording, this evidence alone is insufficient to conclude that the entire ccRTN cell group is activated by the type of hypothalamic stimulation that we performed. This interpretation is more plausible given that such stimulation induced c-Fos expression in a majority of these cells (62%).
The present experiments were designed to test the concept of convergence between hypothalamic inputs and chemoreceptor drive at the level of the RTN neurons. The method that we used to stimulate the hypothalamus lacks the selectivity required to identify which particular hypothalamic neurons were responsible for the activation of the RTN. The possible contribution of orexinergic neurons must be considered given that our injections of gabazine were centred in or very near the hypothalamic region that contains the bulk of these cells (Peyron et al. 1998; Kukkonen et al. 2002). Orexin neurons are wake-active, innervate the ventrolateral medulla and facilitate breathing by mechanisms that include RTN activation (Lu et al. 2006; Deng et al. 2007; Adamantidis et al. 2007; Dias et al. 2009). The integrity of the orexin system is required for the full expression of the cardiorespiratory responses to stress and emotions (Zhang et al. 2006b, 2009; Kuwaki et al. 2008). The orexinergic neurons could therefore have contributed to the activation of the ccRTN neurons that we observed. The gabazine injections were also in close proximity to the dorso-medial hypothalamic nucleus (DMH), a structure suspected to cause powerful cardiorespiratory stimulation (McDowall et al. 2007; Tanaka & McAllen, 2008; Dampney et al. 2008) among many other effects (Gooley et al. 2006). Last but not least, stimulation of the posterior hypothalamus induces locomotion (Eldridge et al. 1981; Smith & DeVito, 1984; Smith et al. 1990; Zaretskaia et al. 2008). The seminal observations by Eldridge and collaborators are at the root of the notion of ‘central command’, the feed-forward control of breathing during exercise (Waldrop et al. 2006). The original work on the hypothalamic locomotor region involved electrical stimulation which raised the possibility that the responses could have been at least partially caused by fibres of passage. However, the complete locomotor and cardiorespiratory pattern can be elicited by stimulating the posterior hypothalamus with bicuculline (Waldrop et al. 1988; Zaretskaia et al. 2008). The hypothalamic region targeted by these authors seems close to the region that we explored in rats which raises the possibility that the RTN might be a relay for the central command of respiration during exercise. This hypothesis is congruent with the notion that RTN drives active expiration (Janczewski & Feldman, 2006; Abdala et al. 2009).
The pathways that mediate RTN activation when the posterior hypothalamus is stimulated are still too hypothetical to merit a lengthy discussion. The already mentioned orexin system probably exerts direct effects on RTN neurons (Dias et al. 2009) but these neurons probably influence breathing in many other ways given that they heavily target the serotonergic and catecholaminergic neurons of the brainstem (Bourgin et al. 2000; Brown et al. 2001). The DMH has direct projections to the rostral ventrolateral medulla which are assumed to contribute to the cardiovascular stimulation elicited by activating the posterior hypothalamus (Fontes et al. 2001; Kerman, 2008). The DMH is also polysynaptically connected to skeletal muscle motor neurons and could therefore be responsible for the locomotor aspect of the defense reaction elicited by posterior hypothalamic stimulation (Hilton & Redfern, 1986; Kerman, 2008). The direct projections of the DMH to the ventrolateral medulla could also conceivably be involved in the activation of the ccRTN neurons although in a prior study projections from DMH to RTN were found to be rather light compared to the input from the perifornical region (Rosin et al. 2006).
In conclusion, the ccRTN neurons presumably contribute to the respiratory stimulation associated with the multiple behaviours or physiological responses that are elicited by posterior hypothalamic stimulation (central command, emotions, sleep and thermoregulation).
Role of ccRTN neurons in CO2 homeostasis
The location and neurophysiological properties of the ccRTN neurons suggest that these cells operate both as central chemoreceptors and as a relay for the feed-forward mechanisms that control breathing. From a neuroanatomical point of view, the ccRTN neurons occupy a position in the neuraxis that is similar to that of the C1 neurons which are a well-known relay for the cardiovascular effects elicited by hypothalamic stimulation (Allen, 2002; Stocker et al. 2006; Tanaka & McAllen, 2008; Dampney et al. 2008). The C1 neurons contribute to setting the level of blood pressure via inputs from supramedullary centres including the paraventricular nucleus and the DMH, and are recruited by a variety of somatic reflexes including, presumably, from muscles (Allen, 2002; Dimicco et al. 2002; Potts et al. 2003; Guyenet, 2006; Tanaka & McAllen, 2008). These cells also minimize short-term blood pressure fluctuations because their activity is regulated by the arterial baroreceptors (Guyenet, 2006). The ccRTN neurons may be playing an analogous input–output function with regard to respiratory control. Their central chemoreceptor properties and excitatory inputs from the carotid bodies contribute to the stability of arterial  because these properties operate as respiratory feedback. The hypothalamic inputs to the ccRTN neurons must contribute to setting the intensity of pulmonary ventilation since the direct activation of these cells stimulates breathing (Abbott et al. 2009). As shown in the present study, hypothalamic stimulation and CO2 have roughly additive effects on the discharge of the ccRTN neurons at all levels of CO2. In other words, the ability of the ccRTN neurons to stabilize arterial
because these properties operate as respiratory feedback. The hypothalamic inputs to the ccRTN neurons must contribute to setting the intensity of pulmonary ventilation since the direct activation of these cells stimulates breathing (Abbott et al. 2009). As shown in the present study, hypothalamic stimulation and CO2 have roughly additive effects on the discharge of the ccRTN neurons at all levels of CO2. In other words, the ability of the ccRTN neurons to stabilize arterial  via chemoreceptor feedback persists regardless of their basal level of activity. This characteristic appears well suited a priori to underlie or at least contribute to the feed-forward regulation of breathing envisioned by Eldridge and collaborators (Eldridge et al. 1981; Millhorn et al. 1987; Eldridge, 1994).
via chemoreceptor feedback persists regardless of their basal level of activity. This characteristic appears well suited a priori to underlie or at least contribute to the feed-forward regulation of breathing envisioned by Eldridge and collaborators (Eldridge et al. 1981; Millhorn et al. 1987; Eldridge, 1994).
In summary, the ccRTN neurons are a source of excitatory drive to the central respiratory pattern generator. The level of activity of these neurons is regulated by converging inputs from wake-promoting systems such as the orexin and serotonergic systems (Mulkey et al. 2007; Dias et al. 2009), behaviour-specific inputs from higher centres including the hypothalamus and by a ‘chemical drive’. This chemical drive consists of the presumed direct pH sensitivity of the ccRTN neurons and the polysynaptic excitatory input that these cells receive from the carotid bodies (Takakura et al. 2006; Guyenet, 2008). The chemical drive contributes to the stabilization of arterial  through breathing and seems to operate regardless of the other inputs that these cells receive.
through breathing and seems to operate regardless of the other inputs that these cells receive.
Acknowledgments
This work was supported by the following grants from the National Institutes of Health to P.G.G.: HL74011 and HL28785.
Glossary
Abbreviations
- AP
arterial pressure
- ccRTN
chemically coded neurons of the retrotrapezoid nucleus
- ChAT
choline acetyltransferase
- CPG
central pattern generator
- DMH
dorso-medial hypothalamic nucleus
- eGFP
enhanced green fluorescent protein

arterial partial pressure of CO2
- PeF
perifornical region of the hypothalamus
- Phox2b
paired-like homeobox 2b
- PND
phrenic nerve discharge
- RTN
retrotrapezoid nucleus
- TH
tyrosine hydroxylase
- VGLUT2
vesicular glutamate transporter 2
Author contributions
All authors contributed to (1) conception and design, or analysis and interpretation of data; (2) drafting the article or revising it critically for important intellectual content; (3) final approval of the version to be published.
References
- Abbott SBG, Stornetta RL, Fortuna MG, Depuy SD, West GH, Harris TE, Guyenet PG. Photostimulation of retrotrapezoid nucleus Phox2b-expressing neurons in vivo produces long-lasting activation of breathing in rats. J Neurosci. 2009;29:5806–5819. doi: 10.1523/JNEUROSCI.1106-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdala AP, Rybak IA, Smith JC, Paton JF. Abdominal expiratory activity in the rat brainstem–spinal cord in situ: patterns, origins and implications for respiratory rhythm generation. J Physiol. 2009;587:3539–3559. doi: 10.1113/jphysiol.2008.167502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamantidis AR, Zhang F, Aravanis AM, Deisseroth K, De Lecea L. Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature. 2007;450:420–424. doi: 10.1038/nature06310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ainslie PN, Duffin J. Integration of cerebrovascular CO2 reactivity and chemoreflex control of breathing: mechanisms of regulation, measurement, and interpretation. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1473–R1495. doi: 10.1152/ajpregu.91008.2008. [DOI] [PubMed] [Google Scholar]
- Allen AM. Inhibition of the hypothalamic paraventricular nucleus in spontaneously hypertensive rats dramatically reduces sympathetic vasomotor tone. Hypertension. 2002;39:275–280. doi: 10.1161/hy0202.104272. [DOI] [PubMed] [Google Scholar]
- Ballanyi K, Ruangkittisakul A, Onimaru H. Opioids prolong and anoxia shortens delay between onset of preinspiratory (pFRG) and inspiratory (preBotC) network bursting in newborn rat brainstems. Pflugers Arch. 2009;458:571–587. doi: 10.1007/s00424-009-0645-3. [DOI] [PubMed] [Google Scholar]
- Bell HJ. Respiratory control at exercise onset: an integrated systems perspective. Respir Physiol Neurobiol. 2006;152:1–15. doi: 10.1016/j.resp.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Bourgin P, Huitrón-Reséndiz S, Spier AD, Fabre V, Morte B, Criado JR, Sutcliffe JG, Henriksen SJ, De Lecea L. Hypocretin-1 modulates rapid eye movement sleep through activation of locus coeruleus neurons. J Neurosci. 2000;20:7760–7765. doi: 10.1523/JNEUROSCI.20-20-07760.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RE, Sergeeva O, Eriksson KS, Haas HL. Orexin A excites serotonergic neurons in the dorsal raphe nucleus of the rat. Neuropharmacology. 2001;40:457–459. doi: 10.1016/s0028-3908(00)00178-7. [DOI] [PubMed] [Google Scholar]
- Connelly CA, Ellenberger HH, Feldman JL. Are there serotonergic projections from raphe and retrotrapezoid nuclei to the ventral respiratory group in the rat? Neurosci Lett. 1989;105:34–40. doi: 10.1016/0304-3940(89)90007-4. [DOI] [PubMed] [Google Scholar]
- Dampney RA, Horiuchi J, McDowall LM. Hypothalamic mechanisms coordinating cardiorespiratory function during exercise and defensive behaviour. Auton Neurosci. 2008;142:3–10. doi: 10.1016/j.autneu.2008.07.005. [DOI] [PubMed] [Google Scholar]
- Deng BS, Nakamura A, Zhang W, Yanagisawa M, Fukuda Y, Kuwaki T. Contribution of orexin in hypercapnic chemoreflex: evidence from genetic and pharmacological disruption and supplementation studies in mice. J Appl Physiol. 2007;103:1772–1779. doi: 10.1152/japplphysiol.00075.2007. [DOI] [PubMed] [Google Scholar]
- Dias MB, Li A, Nattie EE. Antagonism of orexin receptor 1 (OX1R) in the retrotrapezoid nucleus (RTN) inhibits the ventilatory response to hypercapnia predominantly in wakefulness. J Physiol. 2009;587:2059–2067. doi: 10.1113/jphysiol.2008.168260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimicco JA, Samuels BC, Zaretskaia MV, Zaretsky DV. The dorsomedial hypothalamus and the response to stress: part renaissance, part revolution. Pharmacol Biochem Behav. 2002;71:469–480. doi: 10.1016/s0091-3057(01)00689-x. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubreuil V, Ramanantsoa N, Trochet D, Vaubourg V, Amiel J, Gallego J, Brunet JF, Goridis C. A human mutation in Phox2b causes lack of CO2 chemosensitivity, fatal central apnoea and specific loss of parafacial neurons. Proc Natl Acad Sci U S A. 2008;105:1067–1072. doi: 10.1073/pnas.0709115105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldridge FL. Central integration of mechanisms in exercise hyperpnea. Med Sci Sports Exerc. 1994;26:319–327. [PubMed] [Google Scholar]
- Eldridge FL, Kiley JP, Millhorn DE. Respiratory effects of carbon dioxide-induced changes of medullary extracellular fluid pH in cats. J Physiol. 1984;355:177–189. doi: 10.1113/jphysiol.1984.sp015413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldridge FL, Millhorn DE, Kiley JP, Waldrop TG. Stimulation by central command of locomotion, respiration and circulation during exercise. Resp Physiol. 1985;59:313–337. doi: 10.1016/0034-5687(85)90136-7. [DOI] [PubMed] [Google Scholar]
- Eldridge FL, Millhorn DE, Waldrop TG. Exercise hyperpnea and locomotion: parallel activation from the hypothalamus. Science. 1981;211:844–846. doi: 10.1126/science.7466362. [DOI] [PubMed] [Google Scholar]
- Erickson JT, Millhorn DE. Hypoxia and electrical stimulation of the carotid sinus nerve induce c-Fos-like immunoreactivity within catecholaminergic and serotinergic neurons of the rat brainstem. J Comp Neurol. 1994;348:161–182. doi: 10.1002/cne.903480202. [DOI] [PubMed] [Google Scholar]
- Fontes MAP, Tagawa T, Polson JW, Cavanagh SJ, Dampney RAL. Descending pathways mediating cardiovascular response from dorsomedial hypothalamic nucleus. Am J Physiol Heart Circ Physiol. 2001;280:H2891–H2901. doi: 10.1152/ajpheart.2001.280.6.H2891. [DOI] [PubMed] [Google Scholar]
- Gooley JJ, Schomer A, Saper CB. The dorsomedial hypothalamic nucleus is critical for the expression of food-entrainable circadian rhythms. Nat Neurosci. 2006;9:398–407. doi: 10.1038/nn1651. [DOI] [PubMed] [Google Scholar]
- Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7:335–346. doi: 10.1038/nrn1902. [DOI] [PubMed] [Google Scholar]
- Guyenet PG. The 2008 Carl Ludwig Lecture: retrotrapezoid nucleus, CO2 homeostasis, and breathing automaticity. J Appl Physiol. 2008;105:404–416. doi: 10.1152/japplphysiol.90452.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG, Mulkey DK, Stornetta RL, Bayliss DA. Regulation of ventral surface chemoreceptors by the central respiratory pattern generator. J Neurosci. 2005;25:8938–8947. doi: 10.1523/JNEUROSCI.2415-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldane JS, Priestley JG. The regulation of the lung-ventilation. J Physiol. 1905;32:225–266. doi: 10.1113/jphysiol.1905.sp001081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton SM, Redfern WS. A search for brain stem cell groups integrating the defence reaction in the rat. J Physiol. 1986;378:213–228. doi: 10.1113/jphysiol.1986.sp016215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto GA, Wappel SM, Fox GM, Buetow KA, Waldrop TG. Identification of diencephalic and brainstem cardiorespiratory areas activated during exercise. Brain Res. 1996;726:109–122. [PubMed] [Google Scholar]
- Janczewski WA, Feldman JL. Distinct rhythm generators for inspiration and expiration in the juvenile rat. J Physiol. 2006;570:407–420. doi: 10.1113/jphysiol.2005.098848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerman IA. Organization of brain somatomotor-sympathetic circuits. Exp Brain Res. 2008;187:1–16. doi: 10.1007/s00221-008-1337-5. [DOI] [PubMed] [Google Scholar]
- Kukkonen JP, Holmqvist T, Ammoun S, Åkerman KEO. Functions of the orexinergic/hypocretinergic system. Am J Physiol Cell Physiol. 2002;283:C1567–C1591. doi: 10.1152/ajpcell.00055.2002. [DOI] [PubMed] [Google Scholar]
- Kuwaki T. Orexinergic modulation of breathing across vigilance states. Respir Physiol Neurobiol. 2008;164:204–212. doi: 10.1016/j.resp.2008.03.011. [DOI] [PubMed] [Google Scholar]
- Kuwaki T, Zhang W, Nakamura A, Deng BS. Emotional and state-dependent modification of cardiorespiratory function: role of orexinergic neurons. Auton Neurosci. 2008;142:11–16. doi: 10.1016/j.autneu.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Lazarenko RM, Milner TA, Depuy SD, Stornetta RL, West GH, Kievits JA, Bayliss DA, Guyenet PG. Acid sensitivity and ultrastructure of the retrotrapezoid nucleus in Phox2b-eGFP transgenic mice. J Comp Neurol. 2009;517:69–86. doi: 10.1002/cne.22136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeschcke HH. Central chemosensitivity and the reaction theory. J Physiol. 1982;332:1–24. doi: 10.1113/jphysiol.1982.sp014397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Sherman D, Devor M, Saper CB. A putative flip-flop switch for control of REM sleep. Nature. 2006;441:589–594. doi: 10.1038/nature04767. [DOI] [PubMed] [Google Scholar]
- McDowall LM, Horiuchi J, Dampney RA. Effects of disinhibition of neurons in the dorsomedial hypothalamus on central respiratory drive. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1728–R1735. doi: 10.1152/ajpregu.00503.2007. [DOI] [PubMed] [Google Scholar]
- Manzke T, Guenther U, Ponimaskin EG, Haller M, Dutschmann M, Schwarzacher S, Richter DW. 5-HT4(a) receptors avert opioid-induced breathing depression without loss of analgesia. Science. 2003;301:226–229. doi: 10.1126/science.1084674. [DOI] [PubMed] [Google Scholar]
- Millhorn DE, Eldridge FL, Waldrop TG, Kiley JP. Diencephalic regulation of respiration and arterial pressure during actual and fictive locomotion in cat. Circ Res. 1987;61:I53–I59. [PubMed] [Google Scholar]
- Miura M, Okada J, Kanazawa M. Topology and immunohistochemistry of proton-sensitive neurons in the ventral medullary surface of rats. Brain Res. 1998;780:34–45. doi: 10.1016/s0006-8993(97)01112-8. [DOI] [PubMed] [Google Scholar]
- Moreira TS, Takakura AC, Colombari E, West GH, Guyenet PG. Inhibitory input from slowly adapting lung stretch receptors to retrotrapezoid nucleus chemoreceptors. J Physiol. 2007;580:285–300. doi: 10.1113/jphysiol.2006.125336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey DK, Rosin DL, West G, Takakura AC, Moreira TS, Bayliss DA, Guyenet PG. Serotonergic neurons activate chemosensitive retrotrapezoid nucleus neurons by a pH-independent mechanism. J Neurosci. 2007;27:14128–14138. doi: 10.1523/JNEUROSCI.4167-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey DK, Stornetta RL, Weston MC, Simmons JR, Parker A, Bayliss DA, Guyenet PG. Respiratory control by ventral surface chemoreceptor neurons in rats. Nat Neurosci. 2004;7:1360–1369. doi: 10.1038/nn1357. [DOI] [PubMed] [Google Scholar]
- Nattie E, Shi J, Li A. Bicuculline dialysis in the retrotrapezoid nucleus (RTN) region stimulates breathing in the awake rat. Resp Physiol. 2001;124:179–193. doi: 10.1016/s0034-5687(00)00212-7. [DOI] [PubMed] [Google Scholar]
- Nattie EE, Li A. Rat retrotrapezoid nucleus iono- and metabotropic glutamate receptors and the control of breathing. J Appl Physiol. 1995;78:153–163. doi: 10.1152/jappl.1995.78.1.153. [DOI] [PubMed] [Google Scholar]
- Okada Y, Chen Z, Jiang W, Kuwana S, Eldridge FL. Anatomical arrangement of hypercapnia-activated cells in the superficial ventral medulla of rats. J Appl Physiol. 2002;93:427–439. doi: 10.1152/japplphysiol.00620.2000. [DOI] [PubMed] [Google Scholar]
- Onimaru H, Ikeda K, Kawakami K. CO2-sensitive preinspiratory neurons of the parafacial respiratory group express Phox2b in the neonatal rat. J Neurosci. 2008;28:12845–12850. doi: 10.1523/JNEUROSCI.3625-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onimaru H, Kumagawa Y, Homma I. Respiration-related rhythmic activity in the rostral medulla of newborn rats. J Neurophysiol. 2006;96:55–61. doi: 10.1152/jn.01175.2005. [DOI] [PubMed] [Google Scholar]
- Pagliardini S, Ren J, Gray PA, Vandunk C, Gross M, Goulding M, Greer JJ. Central respiratory rhythmogenesis is abnormal in lbx1- deficient mice. J Neurosci. 2008;28:11030–11041. doi: 10.1523/JNEUROSCI.1648-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. San Diego: Academic Press; 1998. [Google Scholar]
- Peyron C, Tighe DK, Van Den Pol AN, De Lecea L, Heller HC, Sutcliffe JG, Kilduff TS. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts JT, Paton JF, Mitchell JH, Garry MG, Kline G, Anguelov PT, Lee SM. Contraction-sensitive skeletal muscle afferents inhibit arterial baroreceptor signalling in the nucleus of the solitary tract: role of intrinsic GABA interneurons. Neurosci. 2003;119:201–214. doi: 10.1016/s0306-4522(02)00953-3. [DOI] [PubMed] [Google Scholar]
- Rosin DL, Chang DA, Guyenet PG. Afferent and efferent connections of the rat retrotrapezoid nucleus. J Comp Neurol. 2006;499:64–89. doi: 10.1002/cne.21105. [DOI] [PubMed] [Google Scholar]
- Sagar SM, Sharp FR, Curran T. Expression of c-fos protein in brain: Metabolic mapping at the cellular level. Science. 1988;240:1328–1330. doi: 10.1126/science.3131879. [DOI] [PubMed] [Google Scholar]
- Saper CB. The central autonomic nervous system: conscious visceral perception and autonomic pattern generation. Annu Rev Neurosci. 2002;25:433–469. doi: 10.1146/annurev.neuro.25.032502.111311. [DOI] [PubMed] [Google Scholar]
- Sato M, Severinghaus JW, Basbaum AI. Medullary CO2 chemoreceptor neuron identification by c-fos immunocytochemistry. J Appl Physiol. 1992;73:96–100. doi: 10.1152/jappl.1992.73.1.96. [DOI] [PubMed] [Google Scholar]
- Simms AE, Paton JF, Pickering AE, Allen AM. Amplified respiratory–sympathetic coupling in the spontaneously hypertensive rat: does it contribute to hypertension? J Physiol. 2009;587:597–610. doi: 10.1113/jphysiol.2008.165902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JC, Morrison DE, Ellenberger HH, Otto MR, Feldman JL. Brainstem projections to the major respiratory neuron populations in the medulla of the cat. J Comp Neurol. 1989;281:69–96. doi: 10.1002/cne.902810107. [DOI] [PubMed] [Google Scholar]
- Smith OA, DeVito JL. Central neural integration for the control of autonomic responses associated with emotion. Annu Rev Neurosci. 1984;7:43–66. doi: 10.1146/annurev.ne.07.030184.000355. [DOI] [PubMed] [Google Scholar]
- Smith OA, DeVito JL, Astley CA. Neurons controlling cardiovascular responses to emotion are located in lateral hypothalamus-perifornical region. Am J Physiol Regul Integr Comp Physiol. 1990;259:R943–R954. doi: 10.1152/ajpregu.1990.259.5.R943. [DOI] [PubMed] [Google Scholar]
- Stocker SD, Simmons JR, Stornetta RL, Toney GM, Guyenet PG. Water deprivation activates a glutamatergic projection from the hypothalamic paraventricular nucleus to the rostral ventrolateral medulla. J Comp Neurol. 2006;494:673–685. doi: 10.1002/cne.20835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stornetta RL, Guyenet PG. Distribution of glutamic acid decarboxylase mRNA-containing neurons in rat medulla projecting to thoracic spinal cord in relation to monoaminergic brainstem neurons. J Comp Neurol. 1999;407:367–380. [PubMed] [Google Scholar]
- Stornetta RL, Moreira TS, Takakura AC, Kang BJ, Chang DA, West GH, Brunet JF, Mulkey DK, Bayliss DA, Guyenet PG. Expression of Phox2b by brainstem neurons involved in chemosensory integration in the adult rat. J Neurosci. 2006;26:10305–10314. doi: 10.1523/JNEUROSCI.2917-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taccola G, Secchia L, Ballanyi K. Anoxic persistence of lumbar respiratory bursts and block of lumbar locomotion in newborn rat brainstem spinal cords. J Physiol. 2007;585:507–524. doi: 10.1113/jphysiol.2007.143594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takakura AC, Moreira TS, Colombari E, West GH, Stornetta RL, Guyenet PG. Peripheral chemoreceptor inputs to retrotrapezoid nucleus (RTN) CO2-sensitive neurons in rats. J Physiol. 2006;572:503–523. doi: 10.1113/jphysiol.2005.103788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takakura AC, Moreira TS, Stornetta RL, West GH, Gwilt JM, Guyenet PG. Selective lesion of retrotrapezoid Phox2b-expressing neurons raises the apnoeic threshold in rats. J Physiol. 2008;586:2975–2991. doi: 10.1113/jphysiol.2008.153163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, McAllen RM. Functional topography of the dorsomedial hypothalamus. Am J Physiol Regul Integr Comp Physiol. 2008;294:R477–R486. doi: 10.1152/ajpregu.00633.2007. [DOI] [PubMed] [Google Scholar]
- Teppema LJ, Berkenbosch A, Veening JG, Olievier CN. Hypercapnia induces c-fos expression in neurons of retrotrapezoid nucleus in cats. Brain Res. 1994;635:353–356. doi: 10.1016/0006-8993(94)91462-1. [DOI] [PubMed] [Google Scholar]
- Teppema LJ, Veening JG, Kranenburg A, Dahan A, Berkenbosch A, Olievier C. Expression of c-fos in the rat brainstem after exposure to hypoxia and to normoxic and hyperoxic hypercapnia. J Comp Neurol. 1997;388:169–190. doi: 10.1002/(sici)1096-9861(19971117)388:2<169::aid-cne1>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Waldrop TG, Bauer RM, Iwamoto GA. Microinjection of GABA antagonists into the posterior hypothalamus elicits locomotor activity and a cardiorespiratory activation. Brain Res. 1988;444:84–94. doi: 10.1016/0006-8993(88)90916-x. [DOI] [PubMed] [Google Scholar]
- Waldrop TG, Iwamoto GA, Haouzi P. Point:Counterpoint: Supraspinal locomotor centres do/do not contribute significantly to the hyperpnea of dynamic exercise. J Appl Physiol. 2006;100:1077–1083. doi: 10.1152/japplphysiol.01528.2005. [DOI] [PubMed] [Google Scholar]
- Wray S, Smith RD. Mechanisms of action of pH-induced effects on vascular smooth muscle. Mol Cell Biochem. 2004;263:163–172. doi: 10.1023/B:MCBI.0000041858.78005.d2. [DOI] [PubMed] [Google Scholar]
- Zaretskaia MV, Zaretsky DV, Sarkar S, Shekhar A, Dimicco JA. Induction of Fos-immunoreactivity in the rat brain following disinhibition of the dorsomedial hypothalamus. Brain Res. 2008;1200:39–50. doi: 10.1016/j.brainres.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Sakurai T, Fukuda Y, Kuwaki T. Orexin neuron-mediated skeletal muscle vasodilation and shift of baroreflex during defense response in mice. Am J Physiol Regul Integr Comp Physiol. 2006a;290:R1654–R1663. doi: 10.1152/ajpregu.00704.2005. [DOI] [PubMed] [Google Scholar]
- Zhang W, Shimoyama M, Fukuda Y, Kuwaki T. Multiple components of the defense response depend on orexin: evidence from orexin knockout mice and orexin neuron-ablated mice. Auton Neurosci. 2006b;126–127:139–145. doi: 10.1016/j.autneu.2006.02.021. [DOI] [PubMed] [Google Scholar]
- Zhang W, Zhang N, Sakurai T, Kuwaki T. Orexin neurons in the hypothalamus mediate cardiorespiratory responses induced by disinhibition of the amygdala and bed nucleus of the stria terminalis. Brain Res. 2009;1262:25–37. doi: 10.1016/j.brainres.2009.01.022. [DOI] [PubMed] [Google Scholar]