

Summary
Chronic, low-grade inflammation, particularly in adipose tissue, is an important modulator of obesity-induced insulin resistance and the toll-like receptor 4 (Tlr4) is a key initiator of inflammatory responses in macrophages. We performed bone marrow transplantation (BMT) of Tlr4lps-del or control C57Bl/10J bone marrow cells into irradiated wild type C57Bl6 recipient mice to generate hematopoietic cell specific Tlr4 deletion mutant (BMT-Tlr4-/-) and control (BMT-wt) mice. When mice were fed a high-fat diet (HFD) for 16 weeks, BMT-wt mice developed obesity, hyperinsulinemia, glucose intolerance and insulin resistance. In contrast, BMT-Tlr4-/- mice became obese, but did not develop fasting hyperinsulinemia, and had improved hepatic and skeletal muscle insulin sensitivity during euglycemic clamp studies compared to HFD BMT-wt mice. The HFD BMT-Tlr4-/- mice showed markedly reduced adipose tissue inflammatory markers and macrophage content compared to HFD BMT-wt mice. In summary, our results indicate that Tlr4 signaling in hematopoietic-derived cells is important for the development of hepatic and adipose tissue insulin resistance in obese mice.
Introduction
Insulin resistance is a major metabolic defect in obesity, and is associated with increased risk of various diseases, such as type 2 diabetes, hypertension and coronary heart disease (Facchini et al., 2001). In recent years, chronic, low-grade inflammation has emerged as an important contributor to the etiology of insulin resistance in obesity, and because the expansion of adipose tissue mass is an obvious corollary of obesity, much research has focused on adipose tissue as a potential site of this inflammation. Indeed, obese adipose tissue is characterized by increased expression of inflammatory genes, such as tumor necrosis factor (TNF)-α, interleukin (IL)-6, Regulated upon Activation Normal T-cell Expressed and Secreted (RANTES), and monocyte chemoattractant protein (MCP)-1, as well as increased infiltration by immune cells, particularly macrophages (Nguyen et al., 2007; Weisberg et al., 2003; Xu et al., 2003). Macrophages are an important modulator of inflammation, through their capacity to secrete a variety of proinflammatory chemokines and cytokines. In fact, adipose tissue macrophages (ATMs) appear to be responsible for much of the increase in inflammation in adipose tissue with obesity (Weisberg et al., 2003; Xu et al., 2003). Consistent with a role for macrophages and inflammation in the pathogenesis of insulin resistance, we have recently shown that deletion of the two primary inflammatory pathways in macrophages, namely the inhibitor of IκB kinase/nuclear factor κB (IKK/NFκB) (Arkan et al., 2005) and c-Jun NH2 terminal kinase 1/activator protein (JNK1/AP1) (Solinas et al., 2007) pathways attenuates obesity-induced insulin resistance. Thus, preventing the propagation of inflammatory signals within macrophages is sufficient to mitigate obesity-induced insulin resistance. Nonetheless, the upstream components or pathways that detect, initiate and activate the proinflammatory IKK/NFκB and JNK/AP1 pathways remain to be fully elucidated.
Potential “sensors” that may link obesity to inflammation are the toll-like family of receptors (Tlr's); the pattern recognition receptors that play critical roles in innate immunity (Aderem and Ulevitch, 2000; Wolowczuk et al., 2008). Relevant to obesity and inflammation, Tlr's, particularly Tlr2 and Tlr4, are highly expressed in macrophages and adipose tissue. Indeed, we (Nguyen et al., 2007) and others (Lee et al., 2001; Lee et al., 2003), have shown that fatty acids (particularly saturated fatty acids) can activate Tlr2/4 resulting in activation of the IKK/NFκB and JNK1 pathways, with enhanced secretion of pro-inflammatory chemokines and cytokines (e.g. TNF-α). In contrast, in vitro fatty acid-induced activation of JNK and IKK, or induction of proinflammatory cytokine expression or secretion, is prevented by siRNA-mediated knockdown of Tlr2/4 in the RAW264.7 macrophage cell line, or in macrophages from Tlr4 knockout mice (Nguyen et al., 2007; Shi et al., 2006).
Since obesity is characterized by elevated fatty acid levels (Horowitz et al., 1999; Jensen et al., 1989), the fact that fatty acids can stimulate a receptor that, activates inflammatory pathways provides a potentially important link between obesity, inflammation and insulin resistance. In support of this, Tlr4 expression is increased in adipose tissue in obesity, and in proinflammatory macrophages (Nguyen et al., 2007; Shi et al., 2006). In addition, obesity due to high-fat diet (HFD) feeding and insulin resistance caused by a lipid-plus-heparin infusion were attenuated in Tlr4 knockout mice, in parallel with decreased inflammation in both liver and adipose tissue (Shi et al., 2006; Tsukumo et al., 2007). However, while these studies clearly implicate Tlr4 in the development of lipid and obesity-induced insulin resistance, the specific tissue(s) in which Tlr4 depletion works to protect mice from insulin resistance remains to be defined.
Considering the clear role of macrophages in the propagation of inflammatory signals in adipose tissue and liver (i.e. through the liver-specific macrophage cell type, the Kupffer cell), we hypothesized that knockout of Tlr4 signaling in hematopoietic-derived cells (which includes macrophages), would reduce obesity-related increases in macrophage infiltration and inflammation and subsequently prevent in vivo insulin resistance. To address this hypothesis, we generated mice with Tlr4 deleted exclusively in hematopoietic cells. Our results reveal that mice deficient in Tlr4 in their hematopoietic compartment are protected from high-fat diet (HFD) and obesity-induced insulin resistance, in parallel with reduced macrophage infiltration in adipose tissue, reduced chemokine and lymphokine secretion, and a marked reduction in inflammation in adipose and liver.
Results
Deletion of Tlr4 exclusively in immune cells does not alter peripheral blood hematopoietic lineage distributions
To generate mice with a complete knockout of Tlr4 in macrophages and other immune cells, irradiated wild type (wt) C57BL6 mice were transplanted with bone marrow from Tlr4 lps-del (Poltorak et al., 1998) or wt C57BL/10J mice. This adoptive transfer approach yielded chimeric mice that were deficient in Tlr4 (BMT-Tlr4-/-) in all hematopoietic derived cells, but which had normal Tlr4 expression in all non-hematopoietic tissues such as skeletal muscle, liver, and adipose tissue. Using this technique, eight weeks following bone marrow transplantation (BMT), >95% of white blood cells from BMT-Tlr4-/- mice lacked Tlr4 (Figure 1A). Mice transplanted with bone marrow from wild type C57BL/10J mice (BMT-wt) displayed normal Tlr4 expression in all hematopoietic derived cells and non-hematopoietic cells/tissues. The loss of Tlr4 did not alter hematopoietic cell lineage distribution, since monocyte, lymphocyte, and neutrophil counts were normal in the BMT-Tlr4-/- and BMT-wt mice (Figure 1B and C), though the proportions of the three cell types differed slightly between normal chow diet (NCD) and high fat diet (HFD) fed mice.
Figure 1. Hematopoietic homeostasis is maintained in mice following bone marrow transplantation of Tlr4 deleted hematopoietic cells.
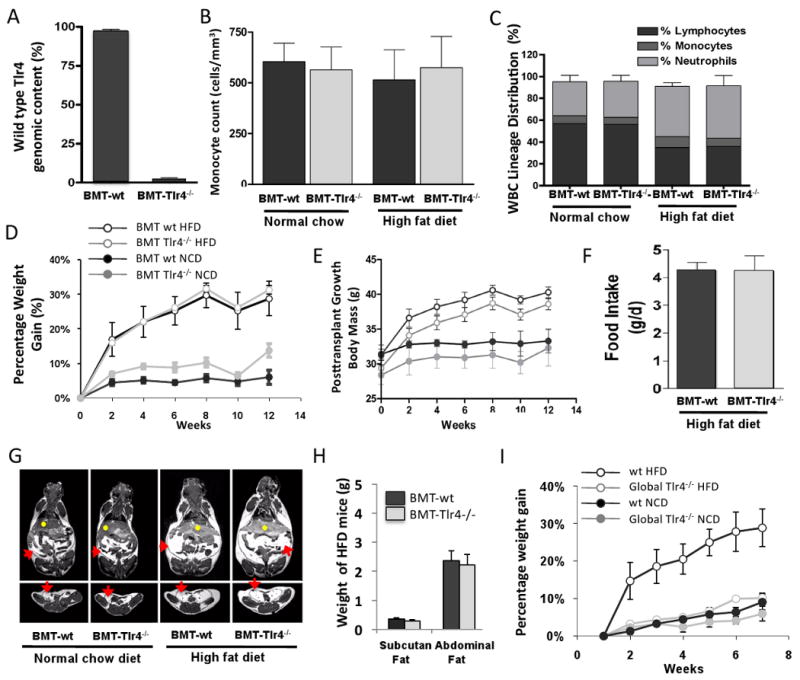
Panel A shows real-time PCR analysis for the wt Tlr4 genomic locus in peripheral blood mononuclear cells from transplanted mice, demonstrating near complete reconstitution of white blood cells in bone marrow transplant recipient mice with Tlr4 deletion mutant hematopoietic cells (BMT-Tlr4-/-) (n=8). Panels B, and C, show hematogram analyses demonstrating normal blood cell lineage distributions including monocytes, lymphocytes and neutrophils, in mice BMT-Tlr4-/-), as compared with control mice transplanted with wild type Tlr4 (BMT-wt) cells. High fat diet fed mice had slightly altered lineage distributions with slightly increased neutrophil counts and decreased lymphocyte counts, but this was similar for both the BMT-wt and BMT-Tlr4-/- mice. Panel D, E, and F show weight gain of mice given high fat diet (HFD) or normal chow diet (NCD) over 12 weeks following BMT, with no statistical difference in weight gain or food intake/day between BMT-Tlr4-/-and BMT-wt mice. Panel G and H shows cross section and coronal section views using 3D magnetic resonance imaging (MRI) of mice and software based analyses of tissue volumes to determine the total weight of abdominal adipose tissues and subcutaneous fat tissues demonstrating similar distribution of adipose tissue in the BMT-wt and BMT-Tlr4-/- mice. Red arrows signify visceral fat, and yellow dots signify hepatic tissue. Panel I shows weight gain results of a control experiment where the global Tlr4 knockout mice and wild type mice were placed on HFD. No weight gain was seen in the global Tlr4 knockout mice on a HFD.
Hematopoietic cell deletion of Tlr4 does not prevent obesity, but ameliorates high-fat-diet/obesity-induced hyperinsulinemia
As expected, body weight gain in mice fed a high fat diet (HFD) significantly outpaced mice fed normal chow diet (NCD). There were no significant body weight differences between BMT-wt or BMT-Tlr4-/- mice and no differences in food intake were detected (Figure 1D, 1E, 1F, and 1G). In vivo volumetric analysis of body composition using magnetic resonance imaging (MRI) revealed a comparable increase in visceral and subcutaneous adipose deposition in HFD versus NCD mice, irrespective of BMT donor type cells (Figure 1G). These results demonstrate that the loss of Tlr4 did not affect the ability of the BMT mice to become obese; moreover, it did not affect the distribution of fat in the obese animal (Figure 1H). The percent weight gain on HFD is nearly identical in the wt versus the Tlr4-/- BMT groups, as is the absolute body weight gain (9.2 versus 9.0 gms, BMT Tlr4-/- versus the BMT wt mice, respectively. Since the BMT-Tlr4-/- group started at a slightly lower weight (29.4 ± 1.1 versus 31.3 ± 1.4 gms) the absolute body weight at the end of the 12 week period is slightly (1.7 gms) less in the BMT Tlr4-/- mice. Thus, the BMT-wt and BMT-Tlr4-/- groups are well matched for body weight, obesity, and all other aspects of adiposity. In contrast, on HFD, body weight gain was markedly reduced in the global Tlr4 knockout mice (i.e. non transplanted mice with knockout of Tlr4 in all tissues) compared to wt controls (Figure 1I). Absolutely body weights were 29.3 ± 0.6 vs 28.2 ± 0.7 at week 0 and 32.1 ± 0.9 vs 36.0 ± 0.8 at week 6, for a wt gain of 2.8 vs 7.8 gms in the Tlr4-/- vs wt, respectively, and these findings are comparable to the results of Tsukomo et al. (15).
The glucose response during a glucose tolerance test (GTT) was increased in HFD fed mice, and there were no statistical differences between the BMT-wt and BMT-Tlr4-/- mice (Figure 2A). On HFD, mice become hyperinsulinemic, but the insulin response during the GTT was significantly lower in HFD BMT-Tlr4-/- versus BMT-wt mice (Figure 2B and C), indicating an overall improvement in insulin action as a result of Tlr4 deletion from hematopoietic cells. Since both groups remain glucose intolerant, there must be a functional β cell defect in both, but we did not perform further islet studies. We next performed an insulin tolerance test (ITT), which shows that while HFD caused insulin resistance in BMT-wt mice, the hypoglycemic response in the HFD BMT-Tlr4-/- group was comparable to NCD fed mice (Fig 2D), directly demonstrating protection from insulin resistance in these mice. Thus, the deleterious effects of HFD/obesity on glucose metabolism and insulin sensitivity were significantly improved in HFD BMT-Tlr4-/- mice, suggesting that Tlr4 in immune cells plays an important role in mediating the effects of obesity on insulin action.
Figure 2. Insulin tolerance tests in BMT-Tlr4-/- mice on HFD show normal insulin sensitivity.
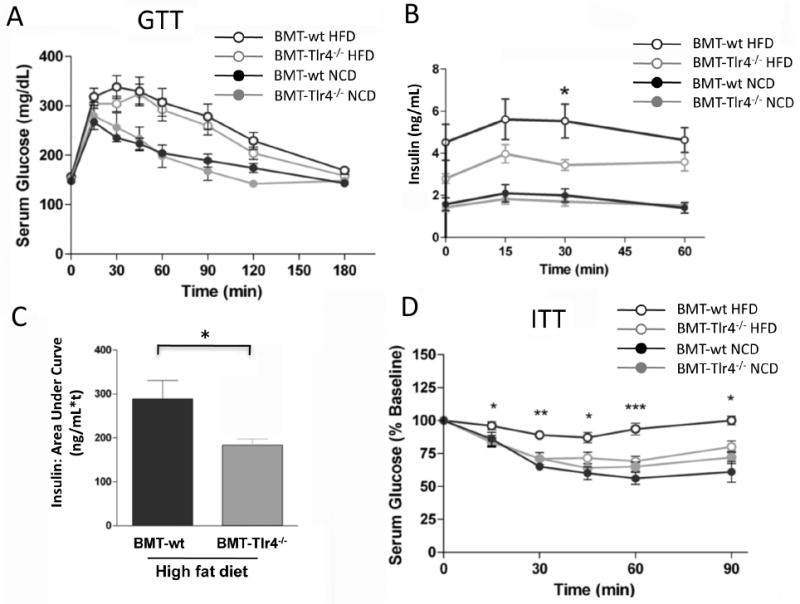
The insulin tolerance test (ITT) performed on bone marrow transplanted mice reveals a significantly increased insulin sensitivity in BMT-Tlr4-/-mice fed a high fat diet (HFD) compared to BMT-wt mice also fed HFD, Panel A. The glucose tolerance test (GTT) reveals less efficient glucose clearing over time in obese mice compared to normal weight control mice for either BMT group, Panel B. * and ** signifies statistical significance between BMT-Tlr4-/- and BMT-wt glucose levels for p<0.05 and p<0.01, respectively, using t-test. Panels C and D reveal the partial correction of insulin levels in obese BMT-Tlr4-/- mice compared to BMT-wt mice. Area-under-curve analysis of insulin data from Panel C shows a statistical difference between BMT-Tlr4-/- and BMT-wt mice both fed HFD.
Hematopoietic cell specific deletion of Tlr4 improves insulin sensitivity in liver and adipose tissue
To further quantify whole-body insulin sensitivity and to better delineate the tissue-specific site(s) responsible for the improved glucose homeostasis in BMT-Tlr4-/- mice, hyperinsulinemic-euglycemic clamp studies were performed. With this procedure, we measure the rate of glucose infusion (GINF) required to keep a constant level of blood glucose during a simultaneous infusion of insulin, and the higher the GINF, the greater the overall insulin sensitivity. The clamp results showed that the GINF required to maintain euglycemia (∼125 mg/dL) was not significantly different between NCD fed BMT-wt and BMT-Tlr4-/-mice. As expected, BMT-wt mice fed HFD had markedly decreased GINF values, confirming insulin resistance. In contrast, GINF values were ∼70% higher in the HFD BMT-Tlr4-/- mice compared to BMT-wt (Figure 3A) demonstrating partial protection from HFD-induced insulin resistance. During the clamp studies, the steady-state insulin and glucose concentrations were the same between groups.
Figure 3. The hyperinsulinemic euglycemic clamp test shows obese BMT-Tlr4-/- mice have normal insulin sensitivity of hepatocytes and adipocytes.
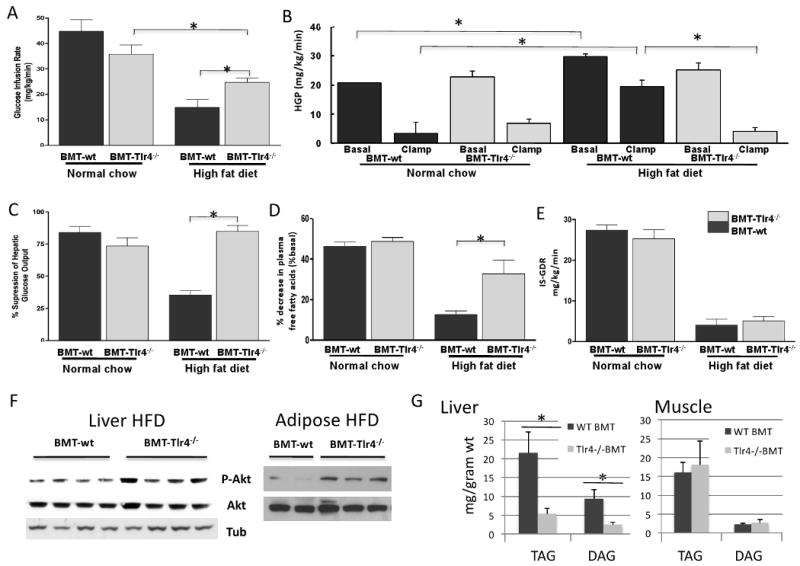
Panel A shows glucose infusion rates during clamp test of mice with the BMT-Tlr4-/- mice showing partial correction of insulin function as measured by partially restored glucose infusion rates (n=8). Panel B shows the hepatic glucose production (HGP) both at basal levels and during the clamp test. On HFD, HGP in the BMT-Tlr4-/- mice was not different from HGP on chow diet, whereas, HFD led to an increase in HFP in the BMT-wt group. The percent suppression of HGP is shown in Panel C. Panel D demonstrates the percentage suppression of free fatty acid secretion from adipose tissue, with obese BMT-Tlr4-/- mice showing normalized adipocyte insulin sensitivity. Panel E shows the insulin stimulated glucose disposal rate (IS-GDR) on NCD or HFD for the two genotypes. * signifies statistical significance between mice groups p<0.05, using t-test, n>8. Panel F shows Western blot analyses of adipose and hepatic tissue for makers of insulin signaling. Analysis demonstrates the up regulated Phosphorylated Akt in the adipose and liver of Tlr4-/- BMT mice compared to the wt BMT, all mice fed the high fat diet, suggesting increased insulin sensitivity. Data shows representative wells with Tubulin as a loading control. Panel G and H show lipid analyses of Liver and muscle cell lysates from BMT mice. The triacylglycerol (TAG) and diacylglycerol (DAG) levels were determined via lipid extraction by organic solvents and separation via thin layer chromoatography. Consistent with the in vivo BMT data, where increase insulin sensitivity was seen in the Tlr4-/- mice, liver lysates from these mice showed reduced TAG and DAG. At least 8 mice were analyzed per group and the measurements were averaged from triplicate wells. Statistical analyses were performed on the samples using student's T-test; blue bar with asterix indicates p value less than 0.1.
To determine the contribution of hepatic glucose production, a simultaneous infusion of tracer labeled glucose was infused to provide a measure of the rate of glucose disposal (Rd). Subtracting the GINF from the total Rd yields the endogenous glucose production rate (mainly from liver). As compared to NCD mice, HFD BMT-wt mice, displayed increased basal hepatic glucose production (HGP) with a markedly impaired ability of insulin to suppress HGP during the clamp (Figure 3B and C), demonstrating hepatic insulin resistance. In contrast, in HFD BMT-Tlr4-/- mice, basal HGP and insulin suppression of HGP was completely normalized to values seen in NCD mice (Figure 3B and C). Adipose tissue insulin sensitivity was assessed by measuring the percent decrease in plasma free fatty-acid concentration during the clamp study, and was also normalized in the HFD BMT-Tlr4-/- mice (Figure 3D). Consistent with these changes in in vivo insulin sensitivity, we also observed increased Akt phosphorylation in insulin stimulated liver and adipose tissue from the BMT Tlr4-/- mice compared to BMT wt controls (Fig 3F). Notably, the HFD induced impairment in insulin-stimulated glucose disposal rate (IS-GDR) in BMT-wt, which primarily reflects skeletal muscle insulin sensitivity, was not prevented in BMT-Tlr4-/- mice (Figure 3E). Taken together, these data demonstrate that Tlr4 deficiency in hematopoietic cells prevents HFD-induced glucose intolerance and insulin resistance, primarily via effects in the liver and adipose tissue.
The lipid content of liver and muscle lysates was also analyzed (Figure 3G). We observed reduced TAG and DAG concentrations in the livers of Tlr4-/- BMT mice with no change in muscle. These results are consistent with our findings of increased insulin sensitivity in liver but not muscle in the BMT-Tlr4-/- mice. We did not perform measures of lipogenesis or fat oxidation, and, although ketone body levels were the same between the two genotypes (Supplemental Fig. S4), this is a relatively non-specific assessment of fat oxidation. Therefore, we cannot be sure whether the decrease in hepatic TAGs and DAGs is due to increased fat oxidation or decreased lipogenesis.
Inflammatory cytokine signaling is mediated by macrophages in obese HFD BMT-Tlr4-/- mice
Given that macrophages are the cell type with the highest surface expression of Tlr4, and, as such, are potent sensors and effectors for Tlr4 mediated inflammatory responses, we assessed various inflammatory markers in liver, adipose tissue and skeletal muscle. Consistent with the MRI data in Figure 1G, HFD led to an increase in liver weights (Figure 4A). Since Kuppfer cells express CD11b, we demonstrated that the liver macrophage population (Kupffer cells) was derived from the transplanted bone marrow, by enriching for CD11b+ cells using magnetic bead absorption according to the method of Seki et al. (Seki et al., 2007). Although peripheral blood mononuclear cells can also express CD11b, the number of these cells is very low in these liver tissues as determined by the q-PCR data showing normal Tlr4 content in cells lacking CD11b. More importantly, CD11b positive cells were almost completely devoid of Tlr4 (Figure 4B), indicating near complete replacement of the recipient Kupffer cells with bone marrow donor-derived cells. This is in full agreement with the high level reconstitution of donor derived hematopoietic cells seen in the peripheral blood, Figure 1A.
Figure 4. Insulin sensitivity in hepatocytes of obese mice correlates with reduced inflammatory signaling of Tlr4-/- hepatic macrophages.
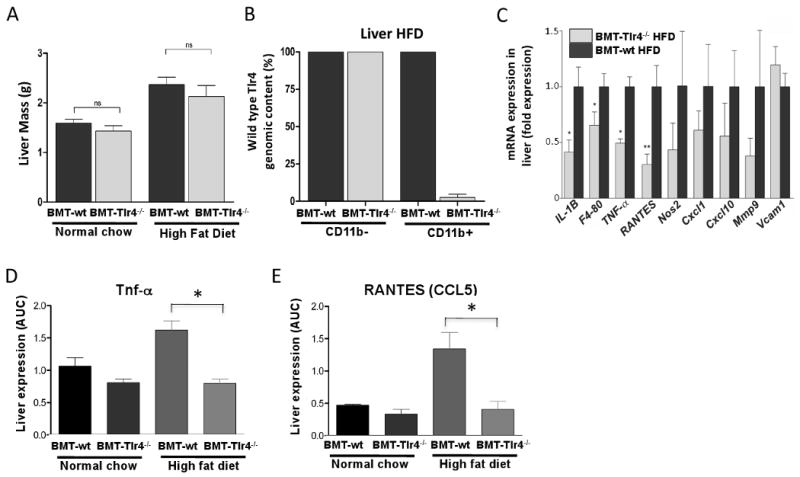
CD11b liver macrophages (predominantly Kupffer cells) play an important role in regulating inflammation in the liver. Panels A and B show the increase in liver weight of HFD fed mice compared to NCD mice, and the near complete reconstitution of CD11b positive cells of the Tlr4-/- genotype in the liver of obese mice following bone marrow transplantation. Homogenized liver tissue from bone marrow transplanted mice was enriched for the macrophage specific marker CD11b and real time PCR performed to assay for wild type Tlr4 genomic DNA content. Panel C shows the fold reductions of macrophage specific and non-specific pro-inflammatory cytokines, chemoattractants, and signaling molecules of BMT-Tlr4-/- mice compared to BMT-wt mice fed a HFD. Panels D and E confirm levels of TNF-α and the chemoattractant RANTES via ELISA.
In liver tissue, mRNA expression analyses of macrophage-specific immune activators, such as IL-1β and F4/80 were significantly reduced by 60% and 30%, respectively, in HFD BMT-Tlr4-/- versus HFD BMT-wt mice (Figure 4C). Similarly, levels of other macrophage expressed inflammatory genes, such as TNF-α, and RANTES (Ccl5) were also significantly reduced (50% and 70%, respectively). Moreover, immune regulators such as Nos2, Cxcl1, Cxcl10 and Mmp9 tended towards reduced expression. As a control, the endothelial cell specific marker VCAM1 showed no statistically significant change in level of expression between BMT-wt and BMT-Tlr4-/- mice. Complementing the changes in gene expression, the protein content in liver tissue of TNF-α, and RANTES was also significantly reduced (Figure 4D and E) in BMT-Tlr4-/- mice. Western blot analysis of c-Jun N-terminal kinase 1 and 2 (JNK1/2) revealed that JNK signaling was also reduced in the livers and adipose tissues of HFD fed BMT-Tlr4-/- versus BMT-wt mice (Figure S1). Since TNFα and IL-6 stimulate JNK activity, which can interfere with IRS and insulin signaling, and tissue levels of these cytokines are reduced in the BMT-Tlr4-/- mice, these results are fully consistent with the enhanced insulin sensitivity in the BMT-Tlr4-/- groups. This connection between IL-6, JNK1, and insulin sensitivity is consistent with the work of Sabio et al (Sabio et al., 2008).
In adipose tissue, the protein amounts of TNF-α, IL-6 and IL-12p70 were also significantly reduced (Figure 5A and Supplemental Figure S3). Because macrophages are an important source of TNF-α and IL-6 in adipose tissue (Weisberg et al., 2003; Xu et al., 2003), we measured macrophage infiltration in adipose tissue from HFD BMT-wt and BMT-Tlr4-/- mice. Analysis of adipose tissue from HFD BMT-wt mice demonstrated increased staining with the macrophage specific antibody, MAC2, as compared to NCD mice (Figure 5D, compare to panels B and C). Interestingly, this was markedly reduced in BMT-Tlr4-/- mice (Figure 5E), as measured by the number of crown-like structures (macrophages) present in the extracellular space between adipocytes (Figure 5F). These findings are consistent with the aforementioned reductions in gene expression and protein content of proinflammatory signaling molecules in both liver and adipose tissue.
Figure 5. Reduced inflammatory cell recruitment to adipose tissue in BMT-Tlr4-/- mice on HFD.
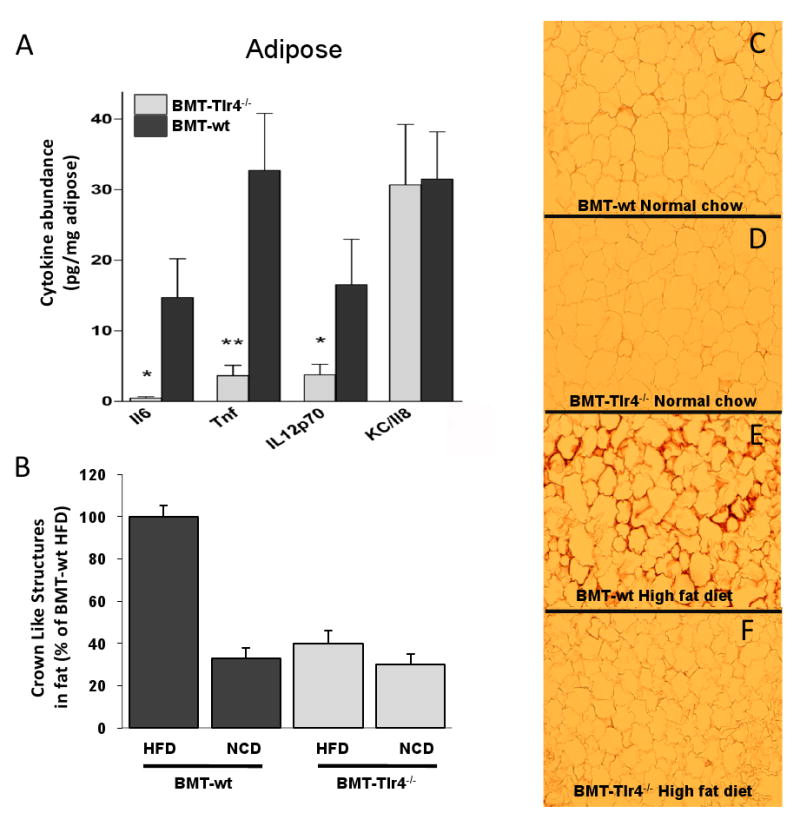
Panel A shows decreased expression of common proinflammatory cytokines in adipose tissue of BMT-Tlr4-/- mice compared to BMT-wt mice fed a HFD. Panels B-E show histological analysis of adipose tissue stained with MAC2 antibody for macrophage detection. Significantly increased macrophage infiltration between adipocytes is detectable and visually quantitated as crown-like-structures with significant reductions in BMT-Tlr4-/- mice compared to BMT-wt mice fed HFD (Panel F). * and ** signifies statistical significance between BMT-Tlr4-/- and BMT-wt levels for p<0.05 and p<0.01, respectively, using student's t-test.
Knockdown of Tlr4 using lentiviral vector gene transfer in hematopoietic cells causes improved insulin sensitivity in HFD mice
To verify the importance of hematopoietic Tlr4 in mediating obesity-induced insulin resistance in mice, we utilized a novel approach, employing a lentiviral vector to knockdown Tlr4 in autologous hematopoietic stem cells. Bone marrow hematopoietic and progenitor cells from wild-type C57Bl6 donor mice were transduced in vitro with lentiviral vectors expressing a small interfering RNA (siRNA) targeted against endogenous Tlr4 (LV-siTlr4) or a control vector. To ensure high-level transduction efficiency in the bone marrow cells, the marker gene GFP was included in both vectors. Transduced cells were then transplanted into irradiated C57Bl6 recipients and after 8 weeks for bone marrow reconstitution, bone marrow cells from these primary BMT mice were then sorted by flow cytometry for GFP expression. Using this approach, the GFP positive cells represent the bone marrow cell population that was successfully and stably transduced with the LV-siTlr4 or control vector. We then transplanted these GFP-positive bone marrow cells into irradiated C57Bl6, secondary recipient mice. Endogenous levels of Tlr4 in peripheral blood cells was knocked down by ∼80% in LV-siTlr4 mice, as compared to control LV-GFP mice (Figure 6A). As expected, on HFD both transplanted mouse groups gained a comparable amount of body weight (Figure 6B). On normal chow diet, both the LV-siTlr4 mice and the control vector mice displayed insulin sensitivity during ITTs (Figure 6C). When the same mice were placed on HFD, the LV-shTlr4 mice retained normal insulin sensitivity, while the control vector mice became insulin resistant (Figure 6D). These results confirm the role of hematopoietic Tlr4 expression in obesity-induced insulin resistance.
Figure 6. Lentiviral vector mediated knockdown of Tlr4 in a hematopoietic stem cell gene therapy setting maintains insulin sensitivity in HFD mice.
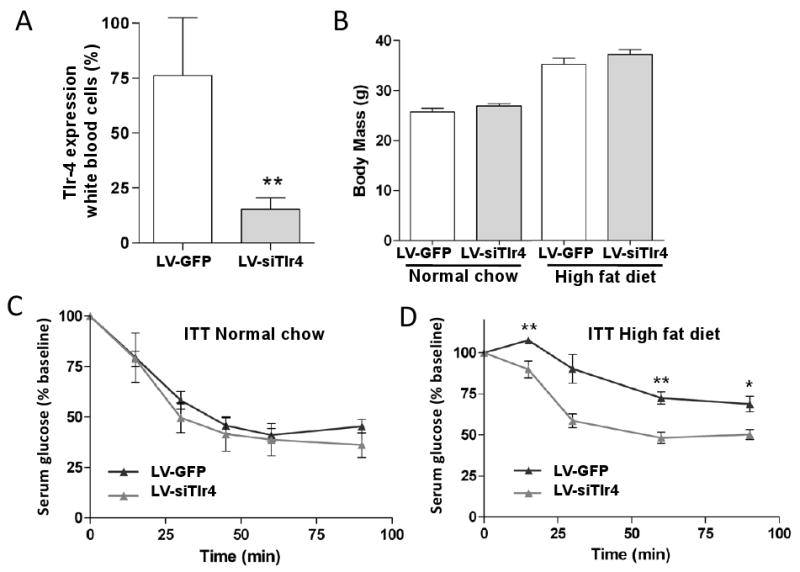
Lentiviral vector driving expression of small interfering RNA (siRNA) targeted against Tlr4 in bone marrow transplanted mice of transduced and sorted hematopoietic stem cells, yields partial but significant knockdown of Tlr4 expression in peripheral blood mononuclear cells of transplanted mice as determined by real time PCR, (n=3), Panel A. Panel B shows average weights of mice fed either a high fat diet or a normal chow diet. Insulin tolerance test (ITT) was performed at 8 weeks posttransplant with LV-siTlr4 and control vector transduced bone marrow mice receiving normal chow (Panel C) or high fat diet (Panel D).
Discussion
Although recent evidence shows that chronic inflammation is a central contributing factor in the development of insulin resistance in obesity, the pathway(s) that transduce the inflammatory signal in obesity are unclear. Here we show that knock out of Tlr4, which is a key receptor involved in activation of the innate immune/inflammatory response, in hematopoietic cells, prevents HFD/obesity-induced hyperinsulinemia, hyperglycemia, and abrogates insulin resistance in liver and adipose tissue. Importantly, the improved insulin action in adipose tissue and liver of these mice occurred in conjunction with reduced macrophage infiltration of adipose tissue, as well as reduced expression of proinflammatory cytokines, such as TNF-α, both in adipose tissue and liver. We further verified the importance of hematopoietic cell Tlr4 in the induction of insulin resistance by using a gene therapy approach to knockdown Tlr4 in autologous hematopoietic stem cells. Considering that Tlr4 is a receptor for fatty acids, our results suggest that Tlr4 acts as an important transducer of the extracellular signal from fatty acids to activation of intracellular inflammatory pathways in hematopoietic cells (most likely macrophages), with subsequent release of proinflammatory cytokines that cause insulin resistance.
Chronic low-grade tissue inflammation has recently garnered considerable attention as a necessary contributor to insulin resistance in obesity. Tlr's play a critical role in activating the innate immune response, and consequently, have been implicated in the induction of insulin resistance in obesity. Tlr4 is an attractive candidate for linking innate immunity to insulin resistance, because it is expressed in most cell types, and Tlr4 is also a receptor for fatty acids, which are increased in obesity (Lee et al., 2001; Lee et al., 2003). Indeed, several recent studies have demonstrated that mice with knockout of Tlr4 (Shi et al., 2006) or a loss-of-function mutation in Tlr4 (Tsukumo et al., 2007) are protected against fatty acid- and obesity-induced insulin resistance. However, since Tlr4 is expressed in many important insulin-responsive cell types (e.g. muscle, adipocytes, hepatocytes), a limitation of these studies is that they do not specifically isolate the contribution of the innate immune system (e.g. macrophage, neutrophils etc.) to changes in insulin sensitivity. To address this and other questions, we used BMT to generate chimeric mice with knockout of Tlr4 specifically in hematopoietic cells. Because innate immune cells are derived from hematopoietic stem cells, our model results in the knockout of Tlr4 in macrophages as well as in other hematopoietic cells. Interestingly, our results demonstrate that BMT-Tlr4-/- mice are protected against HFD/obesity-induced hyperinsulinemia, insulin intolerance, and insulin resistance in adipose tissue and the liver. These results are in line with recent studies in which we found that myeloid-specific knockdown of JNK1 (which is a downstream target of Tlr4 signaling) and IKK improve insulin sensitivity in HFD fed mice.
Two previous studies have examined the issue of insulin resistance in mice in which Tlr4 is either knocked out or disabled (Shi et al., 2006; Tsukumo et al., 2007). In the paper by Shi et al. (Shi et al., 2006), the authors show that the Tlr4 knockout protects animals from the effects of acute lipid infusions to cause insulin resistance, however, the tissues responsible for this systemic effect could not be specified. This finding during acute lipid infusions did not translate that well into the setting of chronic HFD. Thus, they found that the Tlr4 deletion had no effect on body weight or insulin sensitivity in HFD fed male mice, but did lead to increased obesity with insulin sensitivity in females. In contrast, Tsukomo et al. (Tsukumo et al., 2007) studied mice with a loss of function Tlr4-mutation and found that male animals gained less body weight than controls on HFD, and became less insulin resistant. However, in the setting of a lean and insulin sensitive phenotype, it is not clear whether it is the leanness of the mouse, or the knockout of Tlr4, per se, which causes the insulin sensitivity, and the tissue type responsible for the phenotype could not be determined. Since our chimeric mice express Tlr4 deficiency only in bone marrow derived hematopoietic cells, both control and knockout mouse models gained an equal amount of weight on HFD and had equal expansion of both subcutaneous and visceral adipose depots. Thus, differences in adiposity between our two groups (BMT-wt and BMT-Tlr4-/-) is not a confounding factor, making it possible to ascertain the contribution of hematopoietic cell Tlr4 signaling to insulin sensitivity. It is not clear why the Shi et al., and Tsukumo et al. studies are so different from each other, but perhaps it is due to the fact that one group studied Tlr4 null animals (Shi et al., 2006), whereas, the other studied a mouse strain carrying a loss of function mutation in the Tlr4 receptor (Tsukumo et al., 2007). Of course, other strain differences may also be contributing factors. Clearly, the animal model we used is much different, since we employed adoptive transfer to generate chimeric animals all on the same background in which the Tlr4 depletion is only carried in hematopoietic-derived cells with normal Tlr4 in all other tissues.
It is of interest, that while we found substantial effects of the hematopoietic Tlr4 knockout to cause systemic insulin sensitivity, these effects were primarily manifested in liver and adipose tissue. With respect to skeletal muscle, we did not observe changes in the insulin stimulated in vivo glucose disposal rate, and since 70-80% of in vivo insulin stimulated glucose disposal is into skeletal muscle, this implies no major changes in skeletal muscle insulin sensitivity. It is well known that on HFD, large increases in macrophage numbers occur in adipose tissue and that, in the liver, Kupffer cell inflammatory activation state is enhanced, and the number of Kupffer cells may also be increased. On the other hand, there are relatively few macrophages that appear in skeletal muscle on HFD, and these cells are mostly present in inter-muscular adipose deposits. Since inflammatory markers were markedly decreased in liver and adipose tissue of the BMT-Tlr4-/- mice, the data indicate that Tlr4 expression in hematopoietic derived cells is an important control point for HFD-induced inflammation in adipose tissue in liver. In skeletal muscle, it is possible that the Tlr4 on the muscle cell itself plays the major role in detecting lipid signals in the setting of HFD-induced skeletal muscle insulin resistance. Indeed, Tsukomo et al. provide evidence for this hypothesis, since they have directly shown that, when studied ex vivo, Tlr4 knockout muscle is protected from fatty acid induced insulin resistance. This is consistent with our own results in which we show that deletion of hematopoietic cell Tlr4 is sufficient to cause a systemic insulin sensitive phenotype, but that this was primarily manifested in liver and adipose tissue and not muscle.
Tlr4 in macrophages is necessary for activation of inflammatory pathways by fatty acids or lipopolysaccharides (LPS). Knockdown of Tlr2 and/or Tlr4 in macrophage cells attenuates fatty acid-induced activation of Jnk1 and abrogates TNF-α secretion into the media. In fact, JNK1 is an obligatory component for the ability of fatty acids and Tlr4 to activate inflammatory pathways, and to increase TNF-α secretion (Nguyen et al., 2007). In support of these findings, we found that the expression of several proinflammatory cytokines, namely, IL-6, TNF-α and IL-12 p70 was markedly reduced in adipose tissue from BMT-Tlr4-/- mice on HFD. It is likely that the reduced expression of TNF-α is directly related to the reduced macrophage infiltration, as macrophages are the primary source of TNF-α in obese adipose tissue (Weisberg et al., 2003; Xu et al., 2003). It is notable that IL-12 p70 was reduced in our mice, since IL-12 is important for the transition of naïve T-cells into Th1 cells (Hsieh et al., 1993). This is relevant to our studies since the main targets of Th1 cells are macrophages, whereby Th1 cells act to induce a macrophage proinflammatory state. Thus, we hypothesize that Tlr4 is an obligate receptor for the transduction of an obesity-derived signal (i.e. increased fatty acids) to macrophages. In turn, activation of inflammation via macrophage Tlr4 potentiates recruitment of additional cells to adipose tissue, with subsequent polarization to a proinflammatory state. This feed-forward process is likely exacerbated by the fact that Tlr4 expression is increased in macrophages in obese adipose tissue (Nguyen et al., 2007). Consistent with this, insulin's effect to suppress circulating FFA levels was much greater in the BMT-Tlr4-/- mice, indicative of improved adipose insulin action.
In the liver, macrophages are present in the form of Kupffer cells. Because Kupffer cells are bone marrow derived, our model allows us to determine the effect of Tlr4 in Kupffer cells on induction of inflammation (and insulin resistance) in the liver of obese animals. Similar to results in adipose tissue, we found that the expression of various proinflammatory markers was markedly reduced in the liver of BMT-Tlr4-/- mice. In fact, the HFD-induced increase in TNF-α and RANTES in liver was completely reversed in BMT-Tlr4-/- mice. In parallel with this decrease in inflammation, the ability of insulin to suppress HGP in BMT-Tlr4-/- mice was normalized to values seen in chow fed mice. These results suggest that activation of inflammatory pathways in immune cells in the liver is necessary for induction of hepatic insulin resistance on HFD.
We extended these findings by demonstrating that transplantation of mice with bone marrow containing lentiviral-driven siRNA knockdown of Tlr4 leads to improved insulin sensitivity on HFD. Interestingly, this occurred despite the fact that we did not achieve as high a level of knockdown as seen in the BMT-Tlr4-/-mice (>95% versus 80% knockdown for BMT-Tlr4-/- and LV-siTlr4, respectively), suggesting that complete knockdown of Tlr4 is not necessary in order for beneficial metabolic effects to occur.
During the review of this manuscript, Coenen et al. (Coenen et al., 2009) published a paper using the BMT Tlr4-/- approach and reported a decrease in ATM content with reduced markers of adipose tissue inflammation on HFD in the KO mice. They also found a decrease in atherosclerosis in the KOs, but no changes in glucose homeostasis. However, this study consisted of only female mice of the LDL KO background, and were non-obese Agouti animals and so were markedly different than those in the current study.
In summary, we have demonstrated that knockout of Tlr4 signaling in macrophages reverses insulin resistance in adipose tissue and liver in HFD fed, obese mice. This protection occurs in parallel with a marked reduction in macrophage infiltration in adipose tissue, and reduced inflammatory markers in adipose and liver. Altogether, these data indicate the importance of innate immunity and hematopoietic derived cells, particularly macrophages and Kupffer cells, in the induction of insulin resistance in obesity. Our data also identify Tlr4 in hematopoietic derived cells as a potential target for the therapeutic treatment of insulin resistance.
Materials and Methods
Bone marrow transplantation
Murine total bone marrow hematopoietic progenitor donor cells were harvested from wild type or Tlr4lps-del C57B10 mice and available through Jackson Laboratories, Bar Harbor, Maine and transplanted via tail vein injection into lethally irradiated C57Bl6J mice (1100 rads; Cobalt-60 source) with a minimum cell dose of 106 mononuclear cells, or 100,000 lineage depleted cells per mouse. Transplanted mice were housed in micro-isolator housing for 6 weeks prior to challenge with high fat diet and subsequent insulin sensitivity analyses. For experiments where lentiviral vectors were used to knockdown Tlr4, bone marrow from wild-type CD45.1 mice (back-crossed to C57Bl6J mice and available through Jackson Laboratories, Bar Harbor, Maine) was lineage depleted for hematopoietic progenitor cell enrichment (as per manufacturers' instruction, Stem Cell Technologies, Vancouver). Transduction of the progenitor and stem cells was performed as described below
Lentiviral vectors
The third generation lentiviral vectors used in these studies have been described, but in brief, contain the self-inactivating deletion, and an internal CMV promoter driving the marker gene GFP (Dull et al., 1998). The small-interfering RNA cassette directed against Tlr4 and driven by human H1 pol III promoter, was place upstream of the CMV promoter, cloning details provided upon request. Lentiviral vector supernatants were prepared as previously described (Miyoshi et al., 1998).
Transduction
The protocol for efficient transduction of lineage depleted hematopoietic stem cells and progenitors has been previously described (Woods et al., 2006). Briefly, transduction conditions involved 2 days of prestimulation in serum-free expansion medium (SFEM) with 50 ng/ml of each stem cell factor, thrombopoietin, and flt-3 ligand (Stem Cell Technologies, Vancouver, BC), followed by a high multiplicity of infection transduction of blood cells by pelleting up to 500,000 cells and resuspending approximately 30-100 μL of concentrated virus with a titer greater than 10E9 HeLaTU/mL and the volume of virus adjusted to ensure a minimum of 100 infectious units per cell. Incubation was for 1 hour, followed by addition of 150 μL of SFEM medium and cytokines overnight. A second hit was then performed using an additional 30-50 μL of high titer virus directly to the cells in medium and incubated an additional night. All incubations are performed at 37°C with 5% CO2. Expansion during 4-day transduction was approximately 2-3 fold.
Metabolic Studies (ITT and GTT)
Insulin tolerance tests (ITT) were performed pre- and post- diet and following Tlr4 knockdown in all groups of animals. ITT testing allowed us to determine the effectiveness of insulin to reduce fasting glucose levels. Briefly, mice were fasted 6 hours, blood glucose concentrations were assessed before the injection of 0.5 U/kg insulin (intraperitoneal injection) and then 10, 15, 20, 30, 45, 60 and 90 min following injection. At each time point a 5 μ l blood sample was collected via tail nick and glucose assessed with LifeScan OneTouch® glucose monitoring system. One week later, the same group of animals were subjected to a glucose tolerance test (GTT). Here, animals were fasted 6 hours, blood glucose concentrations was assessed before and 10, 15, 20, 30, 45, 60 and 90 min after the injection of 1g/kg 50% dextrose (454 mg/ml).
Hyperinsulinemic-euglycemic clamp
Hyperinsulinemic-euglycemic clamps were conducted to determine insulin stimulated glucose disposal rate (IS-GDR) and the inhibitory effect of insulin on hepatic glucose production (HGP). Briefly, mice were anesthetized with ketamine (80 mg/kg), acepromazine (0.5 mg/kg), and xylazine (16 mg/kg) via IP injection. The jugular vein was cleared of surrounding tissue and two micro-urethane catheters (Type MRE-025) were advanced ∼1 cm into the vessel and secured with 4-0 silk suture. The catheters were tunneled to the mid-scapular region and externalized. The skin was closed with 6-0 suture and the catheters were secured within silastic tubing (0.078″ ID × 0.125″ OD) that had been externalized and secured to the skin with 6-0 silk suture. The mice were allowed to recover for five days before undergoing the clamp protocol. Following a 6 hour fast, blood glucose was assessed via tail nick, body mass was measured, and the mice were placed in a Lucite restrainer (Braintree Scientific, Braintree, MA). Once in the restrainer, ∼75 μl of whole blood was collected for the assessment of plasma insulin and free fatty acids (at t = -60 min). Equilibrating tracer solution (41.6 μCi 3H/ml at 2 μl/min) was infused intravenously for 60 min. At the end of the equilibration period (t = 0 min), 2 × 15 μl of whole blood was collected, and blood was deproteinized for the assessment of tracer specific activity and basal glucose disposal rate. Following the equilibration period, a cocktail containing 8% BSA, insulin and tracer was infused at a constant rate (6.0 mU/kg/min and 41.6 μCi/ml at 2.0 μl/min) along with a variable glucose infusion (50% dextrose, 454 mg/ml). Blood glucose was assessed every 10 min for determination of glucose infusion rate. Glucose infusion rate was adjusted until steady state blood glucose (120 mg/dl, ± 5 mg/dl) was achieved. The clamp was terminated when steady state conditions were maintained for ≥30 min (∼120 min), at which time 2×15 μl of blood was collected for assessment of tracer specific activity and insulin-stimulated glucose disposal rate (t = ∼120 min). At the end of the clamp period the mouse were exsanguinated by cardiac puncture (≥1ml, whole blood collected) and tissues were harvested, mass recorded and preserved as required for future analysis.
Tissue Collection and Analysis
At the end of each clamp study, muscle, liver and fat tissues were harvested for measuring of the mRNA and protein content of insulin signaling molecules, and inflammatory signaling molecules. Real Time RT-PCR was performed using ABI systems 9600 thermal cycler, primers sequences available upon request. MRI analyses including adipose volume determinations were performed using UCSD functional MRI core facility using the 7T system (21 cm bore, Bruker Avance II console). Adipose tissue immunohistochemistry were performed via cryostat sectioning followed by staining with Mac2 antibody (BD Pharmingen) with analysis of crown–like structures as defined previously by Murano et al. (Murano et al., 2008). Kupffer cell enriched fractions were prepared from collagense and pronase digested livers using CD11b magnetic beads (Seki et al., 2007).
The triacylglycerol (TAG) and diacylglycerol (DAG) levels in liver and muscle cell lysates were determined using 50-100 mg of tissue, subjected to lipid extraction by organic solvents as previously described by Bligh EG and Dyer WJ (Can J Biochem Physiol. 37, p911-17, 1959) and Folch J. et al. (JBC 226, p497-509. 1957). The organic solvent layer containing extracted lipids was dried and dissolved in 0.1ml chloroform. TAG and DAG were separated by thin layer chromatography (TLC) and extracted from silica gel by chloroform/methanol. For TAG determination, solvent was evaporated and TAG was resuspended by sonication in TBS containing 0.1% NP-40. The amount of TAG was determined by a commercial kit from WAKO Diagnostic, which measures glycerol level released after hydrolysis of TAG by a lipase. For DAG determination, evaporated lipids were saponified by 1 M KOH for 1h at 55 °C and the mixture was neutralized by 2N HCl. Glycerol level released by hydrolysis of DAG was measured by the same WAKO kit above.
Westerns were performed using 4-12% SDS-Page acrylamide gels Invitrogen CA. The phospho-JNK antibody used targets the threonine 308 amino acid residue: all antibodies were purchased from CellSignalingTechnology, MA.
BioPanel analysis of inflammatory markers in serum from BMT mice for Supplemental Figure S2 (Plasma Insulin, Leptin, Resistin, MCP-1 and Plasminogen activator inhibitor (PAI-1), were measured with a multiplex adipokine assay kit (MADPK-71K, Millipore) according to the instructions of the manufacturer. The multiplex adipokine assay were performed by using a Luminex 200 MAP system (Luminex Technology).
Supplementary Material
Figure S1. Western Blot analysis of Adipose and Hepatic tissue for makers of inflammation. Analysis demonstrates the down regulated Phosphorylated JUNK1/2 in TLR4-/- BMT mice for both adipose and liver tissues in the Tlr4-/- BMT mice compared to wt BMT mice. Data shows representative wells.
Figure S2. BioPanel analysis of inflammatory markers in serum from BMT mice. The Biopanel analysis includes IL-6, Leptin, RANTES, Insulin, tPAI-1, and Resistin of which IL-6 and Leptin showed similar levels in both Tlr4-/- and wt BMT mice. Insulin, tPAI-1 and Resistin all showed trends towards decreases in serum levels in Tlr4-/- BMT mice (blue bar, P<0.1) which is consistent with their increased insulin sensitivity. RANTES showed a trend towards decreased serum levels although not statistically significant. Although IL-6 is not visibly reduced in serum from Tlr4-/- BMT mice, it was seen reduced locally in adipose tissue (Figure 5A) which explains insulin resistance in adipose tissue.
Figure S3. Quantification of Adipocyte cell-size. Although the total weight gain and tissue weights are similar between HFD Tlr4-/- BMT mice and the wt BMT mice, due to differences in inflammation and insulin sensitivity of the adipose from Tlr4-/- BMT mice compared to wt BMT mice we assayed for variances in adipocyte cell-size. For this at least 4-8 microscope fields of view of confluent adipocyte cryostat sections were counted for adipocyte number and the average number compared between the groups. Volumetric analyses were performed following calculation using the average diameter of the cells as determined by total cell number divided by surface area. Error bar denotes standard deviation. Statistical analyses show no difference in cell number or volume between the BMT groups.
Figure S4. Ketone body analysis. Ketone body analysis has been performed using a single-step method whereby both D-3-hydroxybutyrate and acetoacetate are ultimately converted to acetone, which is then measured by gas chromatography (Siegel et al., 1977). The analysis reveals no significant difference in serum levels of ketones in the wt BMT and Tlr4-/-BMT mice suggesting that increased fat oxidation is not responsible for Tlr4 knockout mice having reduced liver TAG and DAG levels, p>0.35 n=8.
Acknowledgments
We thank Anh-Khoi Nguyen and Arezou Amidi for their help with animal maintenance and assistance with ITTs and GTTs. We would also like to thank Gabriela Estepa for assistance with molecular biology techniques. Plasma adipokines were assayed by the University of California at Los Angeles DERC inflammation core (DK-063491-06).
This work was supported by grants from the National Institution of Health grant #'s: DK033651, DK074868, T32 DK 007494, and UC Discovery BioStar grant #bio06-10567. Plasma adipokines were assayed by the University of California at Los Angeles DERC Inflammation Core (DK-063491-06). This research was also supported by the Eunice Kennedy Shriver NICHD/NIH through cooperative agreement of U54 HD 012303-25 as part of the specialized Cooperative Centers Program in Reproduction and Infertility Research. IMV is an American Cancer Society Professor of Molecular Biology, and supported in part by grants from the NIH, Leducq Foundation, Lustgarten Foundation, Ellison Medical Foundation, and the H.N. and Frances C. Berger Foundation. The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, Wynshaw-Boris A, Poli G, Olefsky J, Karin M. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- Coenen KR, Gruen ML, Lee-Young RS, Puglisi MJ, Wasserman DH, Hasty AH. Impact of macrophage toll-like receptor 4 deficiency on macrophage infiltration into adipose tissue and the artery wall in mice. Diabetologia. 2009;52:318–328. doi: 10.1007/s00125-008-1221-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facchini FS, Hua N, Abbasi F, Reaven GM. Insulin resistance as a predictor of age-related diseases. J Clin Endocrinol Metab. 2001;86:3574–3578. doi: 10.1210/jcem.86.8.7763. [DOI] [PubMed] [Google Scholar]
- Horowitz JF, Braudy RJ, Martin WH, 3rd, Klein S. Endurance exercise training does not alter lipolytic or adipose tissue blood flow sensitivity to epinephrine. Am J Physiol. 1999;277:E325–331. doi: 10.1152/ajpendo.1999.277.2.E325. [DOI] [PubMed] [Google Scholar]
- Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O'Garra A, Murphy KM. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science. 1993;260:547–549. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- Jensen MD, Haymond MW, Rizza RA, Cryer PE, Miles JM. Influence of body fat distribution on free fatty acid metabolism in obesity. The Journal of clinical investigation. 1989;83:1168–1173. doi: 10.1172/JCI113997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem. 2001;276:16683–16689. doi: 10.1074/jbc.M011695200. [DOI] [PubMed] [Google Scholar]
- Lee JY, Ye J, Gao Z, Youn HS, Lee WH, Zhao L, Sizemore N, Hwang DH. Reciprocal modulation of Toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. J Biol Chem. 2003;278:37041–37051. doi: 10.1074/jbc.M305213200. [DOI] [PubMed] [Google Scholar]
- Miyoshi H, Blomer U, Takahashi M, Gage FH, Verma IM. Development of a self-inactivating lentivirus vector. J Virol. 1998;72:8150–8157. doi: 10.1128/jvi.72.10.8150-8157.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murano I, Barbatelli G, Parisani V, Latini C, Muzzonigro G, Castellucci M, Cinti S. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J Lipid Res. 2008;49:1562–1568. doi: 10.1194/jlr.M800019-JLR200. [DOI] [PubMed] [Google Scholar]
- Nguyen MT, Favelyukis S, Nguyen AK, Reichart D, Scott PA, Jenn A, Liu-Bryan R, Glass CK, Neels JG, Olefsky JM. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem. 2007;282:35279–35292. doi: 10.1074/jbc.M706762200. [DOI] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Sabio G, Das M, Mora A, Zhang Z, Jun JY, Ko HJ, Barrett T, Kim JK, Davis RJ. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science. 2008;322:1539–1543. doi: 10.1126/science.1160794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13:1324–1332. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. The Journal of clinical investigation. 2006;116:3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel L, Robin NI, McDonald LJ. New approach to determination of total ketone bodies in serum. Clin Chem. 1977;23:46–49. [PubMed] [Google Scholar]
- Solinas G, Vilcu C, Neels JG, Bandyopadhyay GK, Luo JL, Naugler W, Grivennikov S, Wynshaw-Boris A, Scadeng M, Olefsky JM, Karin M. JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity. Cell Metab. 2007;6:386–397. doi: 10.1016/j.cmet.2007.09.011. [DOI] [PubMed] [Google Scholar]
- Tsukumo DM, Carvalho-Filho MA, Carvalheira JB, Prada PO, Hirabara SM, Schenka AA, Araujo EP, Vassallo J, Curi R, Velloso LA, Saad MJ. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes. 2007;56:1986–1998. doi: 10.2337/db06-1595. [DOI] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. The Journal of clinical investigation. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolowczuk I, Verwaerde C, Viltart O, Delanoye A, Delacre M, Pot B, Grangette C. Feeding our immune system: impact on metabolism. Clin Dev Immunol. 2008;2008:639803. doi: 10.1155/2008/639803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods NB, Bottero V, Schmidt M, von Kalle C, Verma IM. Gene therapy: therapeutic gene causing lymphoma. Nature. 2006;440:1123. doi: 10.1038/4401123a. [DOI] [PubMed] [Google Scholar]
- Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. The Journal of clinical investigation. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Western Blot analysis of Adipose and Hepatic tissue for makers of inflammation. Analysis demonstrates the down regulated Phosphorylated JUNK1/2 in TLR4-/- BMT mice for both adipose and liver tissues in the Tlr4-/- BMT mice compared to wt BMT mice. Data shows representative wells.
Figure S2. BioPanel analysis of inflammatory markers in serum from BMT mice. The Biopanel analysis includes IL-6, Leptin, RANTES, Insulin, tPAI-1, and Resistin of which IL-6 and Leptin showed similar levels in both Tlr4-/- and wt BMT mice. Insulin, tPAI-1 and Resistin all showed trends towards decreases in serum levels in Tlr4-/- BMT mice (blue bar, P<0.1) which is consistent with their increased insulin sensitivity. RANTES showed a trend towards decreased serum levels although not statistically significant. Although IL-6 is not visibly reduced in serum from Tlr4-/- BMT mice, it was seen reduced locally in adipose tissue (Figure 5A) which explains insulin resistance in adipose tissue.
Figure S3. Quantification of Adipocyte cell-size. Although the total weight gain and tissue weights are similar between HFD Tlr4-/- BMT mice and the wt BMT mice, due to differences in inflammation and insulin sensitivity of the adipose from Tlr4-/- BMT mice compared to wt BMT mice we assayed for variances in adipocyte cell-size. For this at least 4-8 microscope fields of view of confluent adipocyte cryostat sections were counted for adipocyte number and the average number compared between the groups. Volumetric analyses were performed following calculation using the average diameter of the cells as determined by total cell number divided by surface area. Error bar denotes standard deviation. Statistical analyses show no difference in cell number or volume between the BMT groups.
Figure S4. Ketone body analysis. Ketone body analysis has been performed using a single-step method whereby both D-3-hydroxybutyrate and acetoacetate are ultimately converted to acetone, which is then measured by gas chromatography (Siegel et al., 1977). The analysis reveals no significant difference in serum levels of ketones in the wt BMT and Tlr4-/-BMT mice suggesting that increased fat oxidation is not responsible for Tlr4 knockout mice having reduced liver TAG and DAG levels, p>0.35 n=8.