

Abstract
Ever since the significance of pathological 43-kDa transactivating responsive sequence DNA-binding protein (TDP-43) for human disease has been recognized in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration with ubiquitin positive inclusions (FTLD-U), a number of publications have emerged reporting on this pathology in a variety of neurodegenerative diseases. Given the heterogeneous and, in part, conflicting nature of the recent findings, we here review pathological TDP-43 and its relationship to human disease with a special focus on ALS and FTLD-U. To this end, we propose a classification scheme in which pathological TDP-43 is the major disease defining pathology in one group, or is present in addition to other neurodegenerative hallmark pathologies in a second category. We conclude that the TDP-43 proteinopathies represent a novel class of neurodegenerative disorders akin to α-synucleinopathies and tauopathies, with the concept of ALS and FTLD-U to be widened to a broad clinico-pathological multisystem disease, i.e., TDP-43 proteinopathy.
Keywords: Amyotrophic lateral sclerosis (ALS), Frontotemporal dementia, TDP-43 proteinopathy
Introduction
In 2006, the notion that pathological TDP-43 is involved in human disease was raised when it was identified as the major disease protein in frontotemporal lobar degeneration with ubiquitinated inclusions (FTLD-U), FTLD-U with motor neuron disease (FTLD-MND), and amyotrophic lateral sclerosis (ALS), suggesting a common pathogenesis linked to TDP-43 abnormalities in these disorders [3, 75]. Recently, detailed clinico-pathological studies on the whole spectrum of TDP-43 related neurodegeneration have become available, and have contributed to establishing the significance of pathological TDP-43 for ALS, FTLD-U and other neurodegenerative diseases [32].
TDP-43 (TAR DNA Binding Protein 43) is a 414 amino acid nuclear protein encoded by the TARDBP gene on chromosome 1 (for reviews, see [11, 15, 64, 94, 105]); its functions are not yet completely understood. TARDBP was first identified as a gene encoding a 43 kD protein that binds to the transactive response (TAR) DNA sequence of human immunodeficiency virus type 1. Subsequently, TDP-43 was also shown to be involved in the splicing of the cystic fibrosis transmembrane conductance regulator gene, the apolipoprotein A-II gene and possibly others [1, 6, 15–17, 67, 79, 95, 102–105]. TDP-43 is a highly conserved protein ubiquitously expressed in many tissues including the central nervous system (CNS) where it is present in neuronal and glial nuclei and to a lesser extent in the cytoplasm. It contains two RNA-recognition motifs and a glycine-rich carboxy terminal region that may be required for exon skipping and splicing inhibitory activity. This is consistent with the finding that the carboxy terminal domain binds to several proteins of the heterogeneous nuclear ribonucleoprotein family involved in the biogenesis of mRNA. Other studies also identified TDP-43 as a multi-functional RNA binding protein involved in: (1) exon-skipping of cystic fibrosis transmembrane conductance regulator and apolipoprotein A-II genes [15–17, 67]; (2) exon-inclusion of the survival of motor neuron gene [12]; (3) stabilization of low molecular weight neurofilament protein mRNA through a direct interaction with its 3′UTR [95]; (4) modulation of cyclin-dependent kinase 6 expression [6, 7] and microRNA biogenesis [37]. TDP-43 has also been shown to bind to the proximal promoter of the mouse SP-10 gene (acrosomal vesicle protein 1), which is involved in spermatogenesis, thereby implicating TDP-43 in the regulation of its expression [1]. Finally, other reported functions of TDP-43 include: (1) acting as scaffold for nuclear bodies (i.e. Gemini of coiled bodies) through interaction with survival of motor neuron protein [103]; (2) cell cycle regulation and apoptosis [6, 7] and (3) mRNA transport and regulation of local translation at synapses [102]. Thus, the physiological functions of TDP-43 are diverse but incompletely characterized, and they likely involve the regulation of multiple biological processes through TDP-43 binding to DNA, RNA, and/or proteins.
Since pathological TDP-43 also occurs in disorders other than ALS and FTLD-U, it is useful to separate these disorders, in which TDP-43 aggregations are the main feature, from other neurodegenerative diseases with secondary TDP-43 pathologies, yielding the following two groups: First, the major TDP-43 proteinopathies or the “TDP-43 multisystem diseases”, which include FTLD-U, FTLD-MND, ALS with (ALS-D) or without dementia, and others; and second, the neurodegenerative disorders with TDP-43 inclusions being present to a lesser degree or more localized, representing “additional” or “secondary” pathology to other protein aggregations such as tau, α-synuclein, or others. In addition to this dichotomy, we include a third group encompassing all those relevant neurodegenerative diseases characterized by minor or no TDP-43 pathology. Table 1 summarizes the current status of pathological TDP-43 related diseases, which shall be reviewed in this article. We focus especially on the spectrum of the major TDP-43 proteinopathies with ALS and FTLD-U being the conceptual starting points, as the clinical significance of the secondary TDP-43 pathology in other neurodegenerative diseases is yet to be fully established.
Table 1.
Overview of human diseases according to their pathological 43 kDa nuclear trans-active response DNA-binding protein (TDP-43) status
| Major TDP-43 diseases |
| Amyotrophic lateral sclerosis with/without dementia, frontotemporal degeneration with motor neuron disease, frontotemporal degeneration with ubiquitin positive, tau and α-synuclein negative inclusions |
| Frontotemporal dementia with inclusion body myopathy and Paget disease of bone |
| Perry syndrome |
| Diseases with secondary TDP-43 pathology |
| Parkinsonism-dementia complex/amyotrophic lateral sclerosis on geographic isolates |
| Alzheimer's disease/hippocampal sclerosis |
| Pick's disease |
| Corticobasal degeneration |
| Argyrophilic grain disease |
| Parkinson's diseases, dementia with Lewy bodies, Parkinson's disease dementia |
| Huntington's disease |
| Myopathies (inclusion body myositis, oculopharyngeal muscular dystrophy, distal myopathies with rimmed vacuoles, polymyositis with mitochondrial pathology, polymyositis) |
| Diseases with minor or no TDP-43 pathology |
| Superoxide dismutase-1 linked amyotrophic lateral sclerosis |
| Primary lateral sclerosis |
| TDP-43 negative frontotemporal lobar degeneration with ubiquitin positive inclusions |
| Charged multivesicular body protein 2B linked frontotemporal dementia |
| Progressive supranuclear palsy |
| Multiple system atrophy |
| Basophilic inclusion body disease/motor neuron disease with basophilic inclusions |
| Neuronal intermediate filament inclusion disease |
| Tangle only dementia |
| Hereditary diffuse leucoencephalopathy with spheroids |
| Prion disease |
| Schizophrenia |
| Normal aging |
| Anoxia/Neoplasma |
| Other disorders yet to be studied |
Major TDP-43 diseases
The concept of linking ALS and frontotemporal dementia (FTD) precedes the discovery of TDP-43 based on the shared accumulation of ubiquitin positive cellular inclusions, as well as on the clinical evidence of overlapping symptoms. Initially, in 2006, TDP-43 pathology in FTLD-U, FTLD-MND or ALS was reported only in select CNS areas [3, 75]; however there has been increasing evidence of the deposition of pathological TPD-43 aggregates in multiple brain areas in various studies [13, 23, 25, 33, 66, 77, 78]. Detailed reviews on the significance of clinical overlap and transition forms between ALS, ALS-D, FTLD-MND and FTLD-U have recently been published [26, 58, 93] and consensus criteria on the diagnosis of frontotemporal cognitive and behavioral syndromes in ALS have emerged [94]. Likewise, the spectrum of TDP-43 pathology and the associated recommended neuropathological nomenclature, including the morphological subtypes, have been the subject of detailed reviews [10, 18, 24, 54, 55, 63, 73]. We recently published a study on whole CNS TDP-43 pathology in a large cohort of cases with all the major TDP-43 proteinopathies, both according to their clinical phenotype (pure MND, FTD, or a combination of both), as well as to their morphological subtypes, in order to describe clinico-pathological findings adopting a broad scope on these disorders [32]. Herein, we conclude that there is significant overlap of both clinical and pathological features as denoted schematically in Fig. 1. In fact, ALS, ALS-D/FTLD-MND and FTLD-U may be situated at different points on one continuous and broad clinico-pathological spectrum of multisystem degenerations. We demonstrated that all the clinical groups showed widespread CNS TDP-43 pathology; however, the presence of MND was associated with a higher burden of inclusions in lower motor neurons [32]. Likewise, the presence of cognitive dysfunction was interrelated with the degree of cortical TDP-43 pathology. Upper and lower motor neurons were affected to an overall mild degree in the FTD group relative to the ALS group, and a certain degree of pathology was present in cortical brain areas in the ALS group, pointing toward a subclinical or preclinical involvement in both cases. Other than the defining clinical syndromes in the voluntary motor and cognitive domains, extrapyramidal signs were the most common clinical features, consistent with the robust pathology found in the striatum. Affect disturbances were observed, as well, and appeared to be associated with severe pathology in the amygdala. Further, we illustrated that the clinical syndrome of FTD with and without MND is associated with TDP-43 pathology accompanied by various degrees of neuronal loss and gliosis. Subcortical degeneration, such as pathology in the basal ganglia or amygdala, is usually present to a degree similar to that of cortical pathology, thereby suggesting that FTLD-U is not solely a cortical degeneration as implied by its designation. FTLD-U subtype 1 (characterized by frequent long neuritic profiles predominantly in the superficial cortical layers) appears to represent the most “cortical variant of degeneration” in comparison with subtypes 2 (with neuronal cytoplasmic inclusions in superficial and deep cortical layers) and 3 (with abundance of small neuritic profiles and neuronal cytoplasmic inclusions predominantly in the superficial cortical layers) [85]. In terms of lower motor neuron pathology, the latter two subtypes are closer to the motor neuron disease phenotype when compared with subtype 1. Similarly, it has been shown that cases with predominantly neuronal intracytoplasmic inclusions (subtype 2 [85]) can present with clinical MND in addition to FTD, whereas cases with predominantly dystrophic neurites (subtype 1 [85]) tend to show semantic dementia, and when neuronal cytoplasmic inclusions and dystrophic neurites are coupled with neuronal intranuclear inclusions (subtype 3 [85]), FTD or progressive nonfluent aphasia is common [59]. Further, FTLD-U patients with numerous neuronal cytoplasmic inclusions, as occurring in subtypes 2 or 3, have shorter survival times than those with subtype 1 [32, 38], potentially representing a link to the involvement of the lower motor neurons in decreased survival.
Fig. 1.
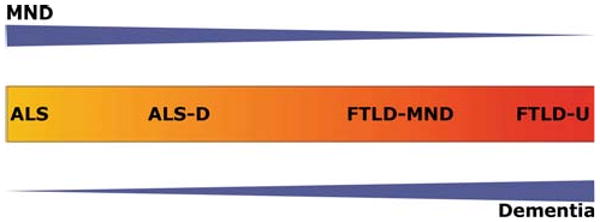
43 kDa nuclear trans-active response DNA-binding protein (TDP-43) multisystem diseases: clinico-pathological spectrum. Schematic illustration of the concept of a clinico-pathological spectrum of the major TPD-43 diseases extending from frontotemporal degeneration with ubiquitin positive, tau and α-synuclein negative inclusions (FLTD-U) at one end to amyotrophic lateral sclerosis (ALS) at the other. Blue arrowhead-like triangles denote clinical syndrome with motor neuron disease decreasing and dementia increasing from left to right. Color change from yellow to red in central box denotes increasing spread and severity of TDP-43 pathology in the brain and spinal cord, as an approximate estimation. Specifically, yellow represents predominant involvement of the spinal cord and red represents predominant involvement of cortical areas. Other brain areas are not as explicitly represented in this color coded diagram. MND motor neuron disease, ALS-D ALS with dementia, FTLD-MND frontotemporal degeneration with MND
From the biochemical perspective, the profile of the TDP-43 proteinopathies has been shown in sporadic and familial FTLD-U and ALS tissue to comprise ubiquitination, variable hyperphosphorylation and N-terminal truncation of the TDP-43 protein. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis of sarkosyl-insoluble extracts isolated from affected cortical regions showed disease-specific bands at ∼45 kD, ∼25 kD, as well as high molecular weight aggregate smears in addition to the normal band at 43 kD [3, 19, 41, 75].
Since pathological TDP-43 is abnormally hyperphosphorylated, we and others have investigated the sites of TDP-43 phosphorylation. Notably, TDP-43 has 41 serine, 15 threonine and 8 tyrosine residues. By predictive in silico analysis some of these are potential phosphorylation sites. Using this approach, Hasegawa and colleagues made phosphorylation-specific antibodies and demonstrated that TDP-43 becomes abnormally phosphorylated at residues 379, 403, 404, 409, and 410 in a small number of cases of FTLD-U and ALS [41, 47]. In our study, we used a similar approach, and we developed and characterized rat monoclonal antibodies (mAbs) 1D3 and 7A9 raised to diphosphopeptide S409/410 of TDP-43 (CSMDSKSpSpGW) [72]. These mAbs were used to study the presence of S409/410 phosphorylation by immunohistochemistry and immunoblots in a large series of FTLD-U cases with/without MND, including familial cases with progranulin (PGRN) or valosin containing protein (VCP) mutations or linkage to chromosome 9p, as well as 18 ALS cases and other neurodegenerative disease cases with or without concomitant TDP-43 pathology. Our data demonstrating that phosphorylation of S409/410 of TDP-43 is a highly consistent feature in TDP-43 inclusions of ALS and FTLD-U confirmed the initial findings of Hasegawa and colleagues [41, 47]. Further, we extended these studies by showing phosphorylated TDP-43 in the inclusions of a more diverse and larger group of sporadic and familial forms of TDP-43 proteinopathies [72]. Physiological nuclear TDP-43 was not detectable with these mAbs by immunohistochemistry, while accumulations of phosphorylated C-terminal fragments were readily seen in Western blots in affected cortical brain regions from ALS and FTLD-U. At least 4 fragments were detected suggesting that they may represent the same C-terminal fragment with different degrees of phosphorylation, different C-terminal fragments with same sites of phosphorylation, or a combination of both. Indeed, these C-terminal fragments were often more abundant than phosphorylated full length TDP-43 in the cortex. However, in the spinal cord the predominate p409/410 species is full length TDP-43 [72], which is consistent with our previous findings that different TDP-43 species may form distinct inclusions in cortical versus spinal cord cells [46].
Ultrastructurally, several studies focusing on the TDP-43 inclusions showed granular and filamentous material (with an average width of ∼15 nm) that labeled for TDP-43 to a variable degree [41, 57, 69, 76, 77, 99].
Recent findings of mutations in the TARDP gene in cases of familial autosomal dominant and rare sporadic ALS patients further corroborate the significance of pathological TPD-43 as being mechanistically implicated in the disease process [9, 22, 35, 36, 39, 50, 52, 84, 91, 101, 108]. However, no mutations in the TARDP gene have yet been reported in either familial or sporadic FTLD-U [9, 35, 83, 87]. Significantly, many of the TARDBP variants display autosomal dominant inheritance in familial ALS patients, suggesting that they may be pathogenic mutations. To date, >25 TARDBP genetic variants have been identified, and the majority of these (including the G290A and G298S mutations we identified) are in the glycine-rich domain of TDP-43, suggesting that they may disrupt the normal exon skipping and splicing functions of TDP-43. Moreover, a number of variants involved the substitution of serine and threonine residues suggesting the possibility of aberrant phosphorylation. PGRN gene mutations cause TDP-43 pathology which is restricted to subtype 3 [8, 18, 19, 21, 90]. Despite the lack of apparent clinical or neuropathological differences between cases with and without PGRN gene abnormalities, a distinct phenotype appears to be present at the molecular level [20]. Given that the proportion of patients with a family history of dementia and/or MND (or other associated clinical features) is higher than that of patients with PGRN or TARDBP mutations, other genetic abnormalities, such as mutations in chromosome 9p or other genetic loci, might play a role as well [32]. Accordingly, TDP-43 pathology in the CNS and muscle have been shown to be associated with mutations in the VCP gene, which presents with FTD, Paget's disease of bone and inclusion body myopathy [18, 19, 74, 106]. Similarly, a recent report on Perry syndrome (characterized by early-onset parkinsonism, depression, severe weight loss and hypoventilation) with TDP-43 pathology in a pallidonigral distribution (with sparing of the cortex, hippocampus and motor neurons) links this rare TDP-43 proteinopathy with pathogenic mutations in the dynactin (DCTN1) gene [27, 107].
Recent studies reporting on elevated TDP-43 plasma levels in clinically diagnosed FTD (and AD) and increased cerebrospinal fluid TDP-43 levels in FTLD-U and ALS patients as compared with controls are intriguing, as they might offer a diagnostic ante-mortem tool and a biomarker for interventional clinical trials, but they need to be verified in larger cohorts and, ideally, confirmed by post-mortem follow up studies [28, 51, 92].
Diseases with secondary TDP-43 pathology
The recognition of TDP-43 proteinopathies as a new class of neurodegenerative disorder has raised the question as to the existence of this pathology in other disorders, since it is known that neurodegenerative diseases frequently exhibit overlapping morphological features (for review see [5]). Indeed, there now are numerous reports on the presence of these alterations as an additional pathology in entities determined by aggregation of other altered proteins. Notably, the degree of the TDP-43 pathology in these disorders is usually lower and its localization usually tends to be somewhat more restricted, as opposed to the widespread distribution found in the major TDP-43 proteinopathies.
Thus, it was shown that TDP-43 pathology is present predominantly in the medial temporal lobe limbic structures in a number of cases of Alzheimer's disease and hippocampal sclerosis [2–4, 19, 44, 45, 100]. The data on the TDP-43 immunoreactivity of Pick's bodies in Pick's disease are conflicting, but TDP-43 pathology is uncommon in this disorder and not yet verified by western blot analysis [3, 29, 43, 44, 57, 100]. For other tauopathies, namely corticobasal degeneration and progressive supranuclear palsy, TDP-43 pathology has been reported to be present in the former [3, 100], but not in the latter [100]. Argyrophilic grain disease also shows concomitant pathological TDP-43 with the distribution of these TDP-43 positive structures being largely overlapping with the tau-positive argyrophilic grains [30]. We and others recently published on pathological TDP-43 in parkinsonism-dementia complex and amyotrophic lateral sclerosis of ethnic Chamorros of Guam [34, 40]. We demonstrated that in contrast to tau pathology, pathological TPD-43 was virtually absent in ethnic Chamorro controls indicating that pathological TPD-43 distinguishes disease from control better than tau-pathology in this cohort. Biochemical analyses showed the presence of FTLD-U-like insoluble TDP-43 in Guam-parkinsonism dementia complex, but not in Guam controls. For the α-synucleinopathies—Parkinson's disease, Parkinson's disease dementia, and dementia with Lewy bodies—concomitant TDP-43 pathology has been reported [4, 44, 57, 71]. Recently, it was also shown that in Huntington's disease TDP-43 frequently co-localized with huntingtin in dystrophic neurites and various intracellular inclusions, but not in intranuclear inclusions [88]. Further, the concept of pathological deposits of TDP-43 and disease has to be widened to include sporadic or familial myopathies (i.e., inclusion body myositis, oculopharyngeal muscular dystrophy, distal myopathies with rimmed vacuoles, polymyositis with mitochondrial pathology, polymyositis) [53, 98, 106]. Taken together, the co-occurrence of TDP-43 pathology in tauopathies, α-synucleinopathies and other neurological diseases is of a variable degree and frequency compared to ALS and FLTD-U. Also, the co-localization of TDP-43 with other pathological proteins is present only in a subset of inclusions. In summary, the pathogenetic and clinical significance of the variable degree of co-existence in a given brain and the rare co-localization in a given inclusion of pathological TDP-43 with other hallmark proteins remains to be established.
Diseases with minor or no TDP-43 pathology
About 10% of ALS cases are familial with the most common genetic abnormality being mutations in superoxide dismutase-1 (SOD1). Soon after the discovery of TPD-43 as the main pathological protein in FTLD-U and ALS, it was reported by independent groups that the ubiquitinated inclusion pathology of the SOD-1-positive familial ALS does not reflect the much more common sporadic disease cases and is different from other familial cases, as the SOD-1 positive familial cases largely lack TDP-43 immunoreactivity [60, 97], not excluding the possibility that this may rarely occur [81, 96]. Thus, motor neuron degeneration in these apparently similar entities may result from different mechanisms [60]. In the few cases of primary lateral sclerosis and FTLD with upper motor neuron disease (i.e., FTLD-primary lateral sclerosis) reported so far, TDP-43 pathology in the pyramidal motor system is not a consistent feature [25, 49]. Recent publications have reported FTLD-U cases that are negative for TDP-43 [42, 62, 82]. These have been described to be heterogeneous in terms of their clinical and pathological nature, and comprise sporadic early-onset FTD with predominant behavioral and personality abnormalities. The term “atypical FTLD-U” has been suggested to define this entity, reflecting the idea of a new clinico-pathological subtype of FTLD-U occurring in 7–20% of cases with a diagnosis of FTLD-U or dementia lacking distinctive histopathology [62, 82]. Additionally, there are other rare TDP-43 negative entities, such as FTD linked to chromosome 3 due to mutations in the charged multivesicular body protein 2B gene [18, 89]. Data on the remaining neurodegenerative conditions are limited, but the few multiple system atrophy cases, assessed with only restricted CNS examinations, have been reported to be negative for pathological TDP-43 [19]. Prion diseases, basophilic inclusion body disease/motor neuron disease with basophilic inclusions, neuronal intermediate filament inclusion disease, tangle only dementia, hereditary diffuse leucoencephalopathy with spheroids have also been reported to be negative for pathological TDP-43, as is the case in a recent small report on schizophrenia [18, 19, 31, 48, 65]. Further, cardiovascular (anoxic or ischemic) brain injury and brain neoplastic conditions have not been associated with significant TDP-43 pathology [56]. Finally, the frequency or degree of TDP-43 pathology in normal or “elderly” individuals has not been definitely determined yet, but minor, or occasionally a higher, sub-clinical degree of pathology might occur [33, 34, 71] similar to α-synuclein, tau, or amyloid-beta deposition in aged individuals. Global gene expression analysis using total RNA harvested from the frontal cortex showed that controls and FTLD-U differ in over 100 networks [68]. However, the significance of TDP-43 pathology in normal controls remains to be established.
Whole CNS evolution of pathology
Many studies show that the evolution of neurodegenerative pathology throughout the CNS follows a pattern that is typical for each of the various disease groups, as has been defined for pathological aggregations of α-synuclein or tau (and others), and respective grading or staging schemes have been established to account for this temporal and spatial development of neurodegeneration. In the case of the TDP-43 proteinopathies, data on whole brain pathology with clinical correlations have become available [32, 33, 76, 77]. It appears that brain areas afflicted early on or that are associated with a rapid disease course include basal ganglia, medulla, amygdala, hippocampal formation, and spinal cord and primary motor cortex. On the other hand, the cerebellum and the occipital cortex appear to be affected to a lesser/minor degree or later in the disease course. In fact, the distribution of the pathology in ALS with/without dementia has been split into two different types by cluster analysis with one of them being the more severe and FTLD-U-like “dementia” morphological type involving the hippocampal formation, neocortex and basal ganglia, and the other one being more restricted [77]. An additional hint for a concept of temporal and spatial evolution of TDP-43 pathology throughout the brain might emerge from studying diseases in which TDP-43 is present as an additional feature, “secondary” to other disease defining pathologies, such as in Alzheimer's disease in which a progression of TDP-43 pathology was suggested, with higher order association cortices being affected following the involvement of medial temporal lobe structures such as amygdala or hippocampus [45].
Conclusions
The TDP-43 diseases represent a novel class of neurodegenerative disorders akin to α-synucleinopathies and tauopathies, and thus can be categorized according to the predominant accumulated pathological protein into two groups: the major TDP-43 diseases and those with TDP-43 aggregations being present in addition to other, disease defining, pathological proteins. Notwithstanding this, the definite pathogenetic role of TDP-43 inclusions in disease is not yet established [70, 80, 86]. Many neurodegenerative diseases other than ALS and FTLD-U can show variable degrees of TDP-43 pathology. As to the major or primary TDP-43 proteinopathies, the widespread distribution of pathological TDP-43 establishes the diffuse involvement of the CNS. The idea of a predominantly frontotemporal degeneration pattern in FTLD-U and a primarily pyramidal tract degeneration pattern in MND should be refined in favor of a broad clinico-pathological spectrum disorder involving multiple systems with differences being present at group level, but not necessarily in a given case, denoting that they may share similar disease mechanisms linked to pathological TDP-43 [14, 61, 75]. However, despite initial data suggesting that certain brain areas are particularly vulnerable to TDP-43 pathology, including basal ganglia, brainstem, mesolimbic and the corticospinal system, a staging or grading scheme of the evolutionary pattern(s) of pathological TDP-43 throughout the CNS has not yet been developed, and hence represents a major task for future research.
Acknowledgments
The authors would like to thank T. Schuck and J. Robinson for their expert technical assistance and our collaborators within and beyond the Center for Neurodegenerative Disease Research (CNDR) who contributed to the studies reviewed here from CNDR. Further, they thank their patients and families who made this research possible. This work was funded by the National Institutes of Health (AG10124, AG17586).
References
- 1.Abhyankar MM, Urekar C, Reddi PP. A novel CpG-free vertebrate insulator silences the testis-specific SP-10 gene in somatic tissues: role for TDP-43 in insulator function. J Biol Chem. 2007;282:36143–36154. doi: 10.1074/jbc.M705811200. [DOI] [PubMed] [Google Scholar]
- 2.Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R, Graff-Radford NR, Hutton ML, Dickson DW. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann Neurol. 2007;61:435–445. doi: 10.1002/ana.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 4.Arai T, Mackenzie IR, Hasegawa M, Nonoka T, Niizato K, Tsuchiya K, Iritani S, Onaya M, Akiyama H. Phosphorylated TDP-43 in Alzheimer's disease and dementia with Lewy bodies. Acta Neuropathol. 2009;117:125–136. doi: 10.1007/s00401-008-0480-1. [DOI] [PubMed] [Google Scholar]
- 5.Armstrong RA, Lantos PL, Cairns NJ. Overlap between neurodegenerative disorders. Neuropathology. 2005;25:111–124. doi: 10.1111/j.1440-1789.2005.00605.x. [DOI] [PubMed] [Google Scholar]
- 6.Ayala YM, Misteli T, Baralle FE. TDP-43 regulates retinoblastoma protein phosphorylation through the repression of cyclin-dependent kinase 6 expression. Proc Natl Acad Sci USA. 2008;105:3785–3789. doi: 10.1073/pnas.0800546105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ayala YM, Zago P, D'Ambrogio A, Xu YF, Petrucelli L, Buratti E, Baralle FE. Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci. 2008;121:3778–3785. doi: 10.1242/jcs.038950. [DOI] [PubMed] [Google Scholar]
- 8.Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 9.Banks GT, Kuta A, Isaacs AM, Fisher EM. TDP-43 is a culprit in human neurodegeneration, and not just an innocent bystander. Mamm Genome. 2008;19:299–305. doi: 10.1007/s00335-008-9117-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bigio EH. TAR DNA-binding protein-43 in amyotrophic lateral sclerosis, frontotemporal lobar degeneration, and Alzheimer disease. Acta Neuropathol. 2008;116:135–140. doi: 10.1007/s00401-008-0405-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bigio EH. Update on recent molecular and genetic advances in frontotemporal lobar degeneration. J Neuropathol Exp Neurol. 2008;67:635–648. doi: 10.1097/NEN.0b013e31817d751c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bose JK, Wang IF, Hung L, Tarn WY, Shen CK. TDP-43 overexpression enhances exon 7 inclusion during the survival of motor neuron pre-mRNA splicing. J Biol Chem. 2008;283:28852–28859. doi: 10.1074/jbc.M805376200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brandmeir NJ, Geser F, Kwong LK, Zimmerman E, Qian J, Lee VM, Trojanowski JQ. Severe subcortical TDP-43 pathology in sporadic frontotemporal lobar degeneration with motor neuron disease. Acta Neuropathol. 2008;115:123–131. doi: 10.1007/s00401-007-0315-5. [DOI] [PubMed] [Google Scholar]
- 14.Brownell B, Oppenheimer DR, Hughes JT. The central nervous system in motor neurone disease. J Neurol Neurosurg Psychiatry. 1970;33:338–357. doi: 10.1136/jnnp.33.3.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buratti E, Baralle FE. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front Biosci. 2008;13:867–878. doi: 10.2741/2727. [DOI] [PubMed] [Google Scholar]
- 16.Buratti E, Brindisi A, Pagani F, Baralle FE. Nuclear factor TDP-43 binds to the polymorphic TG repeats in CFTR intron 8 and causes skipping of exon 9: a functional link with disease penetrance. Am J Hum Genet. 2004;74:1322–1325. doi: 10.1086/420978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buratti E, Dork T, Zuccato E, Pagani F, Romano M, Baralle FE. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 2001;20:1774–1784. doi: 10.1093/emboj/20.7.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ, White CL, III, Schneider JA, Grinberg LT, Halliday G, Duyckaerts C, Lowe JS, Holm IE, Tolnay M, Okamoto K, Yokoo H, Murayama S, Woulfe J, Munoz DG, Dickson DW, Ince PG, Trojanowski JQ, Mann DM. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114:5–22. doi: 10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ, Foong C, White CL, III, Schneider JA, Kretzschmar HA, Carter D, Taylor-Reinwald L, Paulsmeyer K, Strider J, Gitcho M, Goate AM, Morris JC, Mishra M, Kwong LK, Stieber A, Xu Y, Forman MS, Trojanowski JQ, Lee VM, Mackenzie IR. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol. 2007;171:227–240. doi: 10.2353/ajpath.2007.070182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen-Plotkin AS, Geser F, Plotkin JB, Clark CM, Kwong LK, Yuan W, Grossman M, Van Deerlin VM, Trojanowski JQ, Lee VM. Variations in the progranulin gene affect global gene expression in frontotemporal lobar degeneration. Hum Mol Genet. 2008;17:1349–1362. doi: 10.1093/hmg/ddn023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cruts M, Gijselinck I, van der ZJ, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den BM, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- 22.Daoud H, Valdmanis PN, Kabashi E, Dion P, Dupre N, Camu W, Meininger V, Rouleau GA. Contribution of TARDBP mutations to sporadic amyotrophic lateral sclerosis. J Med Genet. 2008;46:112–114. doi: 10.1136/jmg.2008.062463. [DOI] [PubMed] [Google Scholar]
- 23.Davidson Y, Kelley T, Mackenzie IR, Pickering-Brown S, Du PD, Neary D, Snowden JS, Mann DM. Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol. 2007;113:521–533. doi: 10.1007/s00401-006-0189-y. [DOI] [PubMed] [Google Scholar]
- 24.Dickson DW. TDP-43 immunoreactivity in neurodegenerative disorders: disease versus mechanism specificity. Acta Neuropathol. 2008;115:147–149. doi: 10.1007/s00401-007-0323-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dickson DW, Josephs KA, Amador-Ortiz C. TDP-43 in differential diagnosis of motor neuron disorders. Acta Neuropathol. 2007;114:71–79. doi: 10.1007/s00401-007-0234-5. [DOI] [PubMed] [Google Scholar]
- 26.Elman LB, McCluskey L, Grossman M. Motor neuron disease and frontotemporal lobar degeneration: a tale of two disorders linked to TDP-43. Neurosignals. 2008;16:85–90. doi: 10.1159/000109762. [DOI] [PubMed] [Google Scholar]
- 27.Farrer MJ, Hulihan MM, Kachergus JM, Dachsel JC, Stoessl AJ, Grantier LL, Calne S, Calne DB, Lechevalier B, Chapon F, Tsuboi Y, Yamada T, Gutmann L, Elibol B, Bhatia KP, Wider C, Vilarino-Guell C, Ross OA, Brown LA, Castanedes-Casey M, Dickson DW, Wszolek ZK. DCTN1 mutations in Perry syndrome. Nat Genet. 2009;41:163–165. doi: 10.1038/ng.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Foulds P, McAuley E, Gibbons L, Davidson Y, Pickering-Brown SM, Neary D, Snowden JS, Allsop D, Mann DM. TDP-43 protein in plasma may index TDP-43 brain pathology in Alzheimer's disease and frontotemporal lobar degeneration. Acta Neuropathol. 2008;116:141–146. doi: 10.1007/s00401-008-0389-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Freeman SH, Spires-Jones T, Hyman BT, Growdon JH, Frosch MP. TAR-DNA binding protein 43 in Pick disease. J Neuropathol Exp Neurol. 2008;67:62–67. doi: 10.1097/nen.0b013e3181609361. [DOI] [PubMed] [Google Scholar]
- 30.Fujishiro H, Uchikado H, Arai T, Hasegawa M, Akiyama H, Yokota O, Tsuchiya K, Togo T, Iseki E, Hirayasu Y. Accumulation of phosphorylated TDP-43 in brains of patients with argyrophilic grain disease. Acta Neuropathol. 2008;117:151–158. doi: 10.1007/s00401-008-0463-2. [DOI] [PubMed] [Google Scholar]
- 31.Fujita K, Ito H, Nakano S, Kinoshita Y, Wate R, Kusaka H. Immunohistochemical identification of messenger RNA-related proteins in basophilic inclusions of adult-onset atypical motor neuron disease. Acta Neuropathol. 2008;116:439–445. doi: 10.1007/s00401-008-0415-x. [DOI] [PubMed] [Google Scholar]
- 32.Geser F, Martinez-Lage M, Robinson J, Uryu K, Neumann M, Brandmeir NJ, Xie SX, Kwong L, Elman L, McCluskey L, Clark CM, Malunda J, Miller B, Zimmerman E, Qian J, Van Deerlin VM, Grossman M, Lee VMY, Trojanowski JQ. The clinical and pathological continuum of multisystem TDP-43 proteinopathies. Arch Neurol. 2009 doi: 10.1001/archneurol.2008.558. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Geser F, Brandmeir NJ, Kwong LK, Martinez-Lage M, Elman L, McCluskey L, Xie SX, Lee VM, Trojanowski JQ. Evidence of multisystem disorder in whole-brain map of pathological TDP-43 in amyotrophic lateral sclerosis. Arch Neurol. 2008;65:636–641. doi: 10.1001/archneur.65.5.636. [DOI] [PubMed] [Google Scholar]
- 34.Geser F, Winton MJ, Kwong LK, Xu Y, Xie SX, Igaz LM, Garruto RM, Perl DP, Galasko D, Lee VM, Trojanowski JQ. Pathological TDP-43 in parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam. Acta Neuropathol (Berl) 2007;115:133–145. doi: 10.1007/s00401-007-0257-y. [DOI] [PubMed] [Google Scholar]
- 35.Gijselinck I, Sleegers K, Engelborghs S, Robberecht W, Martin JJ, Vandenberghe R, Sciot R, Dermaut B, Goossens D, van der ZJ, De Pooter T, Del Favero J, Santens P, De Jonghe P, De Deyn PP, Van Broeckhoven C, Cruts M. Neuronal inclusion protein TDP-43 has no primary genetic role in FTD and ALS. Neurobiol Aging. 2007 doi: 10.1016/j.neurobiolaging.2007.11.002. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 36.Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D, Hatanpaa KJ, White CL, III, Bigio EH, Caselli R, Baker M, Al Lozi MT, Morris JC, Pestronk A, Rademakers R, Goate AM, Cairns NJ. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008;63:535–538. doi: 10.1002/ana.21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, Shiekhattar R. The microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432:235–240. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- 38.Grossman M, Wood EM, Moore P, Neumann M, Kwong L, Forman MS, Clark CM, McCluskey LF, Miller BL, Lee VM, Trojanowski JQ. TDP-43 pathologic lesions and clinical phenotype in frontotemporal lobar degeneration with ubiquitin-positive inclusions. Arch Neurol. 2007;64:1449–1454. doi: 10.1001/archneur.64.10.1449. [DOI] [PubMed] [Google Scholar]
- 39.Guerreiro RJ, Schymick JC, Crews C, Singleton A, Hardy J, Traynor BJ. TDP-43 is not a common cause of sporadic amyotrophic lateral sclerosis. PLoS ONE. 2008;3:e2450. doi: 10.1371/journal.pone.0002450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hasegawa M, Arai T, Akiyama H, Nonaka T, Mori H, Hashimoto T, Yamazaki M, Oyanagi K. TDP-43 is deposited in the Guam parkinsonism-dementia complex brains. Brain. 2007;130:1386–1394. doi: 10.1093/brain/awm065. [DOI] [PubMed] [Google Scholar]
- 41.Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y, Beach TG, Buratti E, Baralle F, Morita M, Nakano I, Oda T, Tsuchiya K, Akiyama H. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol. 2008;64:60–70. doi: 10.1002/ana.21425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hatanpaa KJ, Bigio EH, Cairns NJ, Womack KB, Weintraub S, Morris JC, Foong C, Xiao G, Hladik C, Mantanona TY, White CL., III TAR DNA-binding protein 43 immunohistochemistry reveals extensive neuritic pathology in FTLD-U: a midwest-southwest consortium for FTLD study. J Neuropathol Exp Neurol. 2008;67:271–279. doi: 10.1097/NEN.0b013e31816a12a6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Higashi S, Iseki E, Yamamoto R, Minegishi M, Hino H, Fujisawa K, Togo T, Katsuse O, Uchikado H, Furukawa Y, Kosaka K, Arai H. Appearance pattern of TDP-43 in Japanese frontotemporal lobar degeneration with ubiquitin-positive inclusions. Neurosci Lett. 2007;419:213–218. doi: 10.1016/j.neulet.2007.04.051. [DOI] [PubMed] [Google Scholar]
- 44.Higashi S, Iseki E, Yamamoto R, Minegishi M, Hino H, Fujisawa K, Togo T, Katsuse O, Uchikado H, Furukawa Y, Kosaka K, Arai H. Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer's disease and dementia with Lewy bodies. Brain Res. 2007;1184:284–294. doi: 10.1016/j.brainres.2007.09.048. [DOI] [PubMed] [Google Scholar]
- 45.Hu WT, Josephs KA, Knopman DS, Boeve BF, Dickson DW, Petersen RC, Parisi JE. Temporal lobar predominance of TDP-43 neuronal cytoplasmic inclusions in Alzheimer disease. Acta Neuropathol. 2008;116:215–220. doi: 10.1007/s00401-008-0400-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Igaz LM, Kwong LK, Xu Y, Truax AC, Uryu K, Neumann M, Clark CM, Elman LB, Miller BL, Grossman M, McCluskey LF, Trojanowski JQ, Lee VM. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am J Pathol. 2008;173:182–194. doi: 10.2353/ajpath.2008.080003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Inukai Y, Nonaka T, Arai T, Yoshida M, Hashizume Y, Beach TG, Buratti E, Baralle FE, Akiyama H, Hisanaga S, Hasegawa M. Abnormal phosphorylation of Ser409/410 of TDP-43 in FTLD-U and ALS. FEBS Lett. 2008;582:2899–2904. doi: 10.1016/j.febslet.2008.07.027. [DOI] [PubMed] [Google Scholar]
- 48.Isaacs AM, Powell C, Webb TE, Linehan JM, Collinge J, Brandner S. Lack of TAR-DNA binding protein-43 (TDP-43) pathology in human prion diseases. Neuropathol Appl Neurobiol. 2008;34:446–456. doi: 10.1111/j.1365-2990.2008.00963.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Josephs KA, Dickson DW. Frontotemporal lobar degeneration with upper motor neuron disease/primary lateral sclerosis. Neurology. 2007;69:1800–1801. doi: 10.1212/01.wnl.0000277270.99272.7e. [DOI] [PubMed] [Google Scholar]
- 50.Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande VC, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- 51.Kasai T, Tokuda T, Ishigami N, Sasayama H, Foulds P, Mitchell DJ, Mann DM, Allsop D, Nakagawa M. Increased TDP-43 protein in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neuropathol. 2009;117:55–62. doi: 10.1007/s00401-008-0456-1. [DOI] [PubMed] [Google Scholar]
- 52.Kuhnlein P, Sperfeld AD, Vanmassenhove B, Van D V, Lee VM, Trojanowski JQ, Kretzschmar HA, Ludolph AC, Neumann M. Two German kindreds with familial amyotrophic lateral sclerosis due to TARDBP mutations. Arch Neurol. 2008;65:1185–1189. doi: 10.1001/archneur.65.9.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kusters B, van Hoeve BJ, Schelhaas HJ, Ter Laak H, van Engelen BG, Lammens M. TDP-43 accumulation is common in myopathies with rimmed vacuoles. Acta Neuropathol. 2009;117:209–211. doi: 10.1007/s00401-008-0471-2. [DOI] [PubMed] [Google Scholar]
- 54.Kwong LK, Neumann M, Sampathu DM, Lee VM, Trojanowski JQ. TDP-43 proteinopathy: the neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta Neuropathol. 2007;114:63–70. doi: 10.1007/s00401-007-0226-5. [DOI] [PubMed] [Google Scholar]
- 55.Kwong LK, Uryu K, Trojanowski JQ, Lee VM. TDP-43 proteinopathies: neurodegenerative protein misfolding diseases without amyloidosis. Neurosignals. 2008;16:41–51. doi: 10.1159/000109758. [DOI] [PubMed] [Google Scholar]
- 56.Lee EB, Lee VM, Trojanowski JQ, Neumann M. TDP-43 immunoreactivity in anoxic, ischemic and neoplastic lesions of the central nervous system. Acta Neuropathol. 2008;115:305–311. doi: 10.1007/s00401-007-0331-5. [DOI] [PubMed] [Google Scholar]
- 57.Lin WL, Dickson DW. Ultrastructural localization of TDP-43 in filamentous neuronal inclusions in various neurodegenerative diseases. Acta Neuropathol. 2008;116:205–213. doi: 10.1007/s00401-008-0408-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liscic RM, Grinberg LT, Zidar J, Gitcho MA, Cairns NJ. ALS and FTLD: two faces of TDP-43 proteinopathy. Eur J Neurol. 2008;15:772–780. doi: 10.1111/j.1468-1331.2008.02195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mackenzie IR, Baborie A, Pickering-Brown S, Du PD, Jaros E, Perry RH, Neary D, Snowden JS, Mann DM. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol. 2006;112:539–549. doi: 10.1007/s00401-006-0138-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, Kwong LK, Forman MS, Ravits J, Stewart H, Eisen A, McClusky L, Kretzschmar HA, Monoranu CM, Highley JR, Kirby J, Siddique T, Shaw PJ, Lee VMY, Trojanowski JQ. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007;61:427–434. doi: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- 61.Mackenzie IR, Feldman HH. Ubiquitin immunohistochemistry suggests classic motor neuron disease, motor neuron disease with dementia, and frontotemporal dementia of the motor neuron disease type represent a clinicopathologic spectrum. J Neuropathol Exp Neurol. 2005;64:730–739. doi: 10.1097/01.jnen.0000174335.27708.0a. [DOI] [PubMed] [Google Scholar]
- 62.Mackenzie IR, Foti D, Woulfe J, Hurwitz TA. Atypical frontotemporal lobar degeneration with ubiquitin-positive, TDP-43-negative neuronal inclusions. Brain. 2008;131:1282–1293. doi: 10.1093/brain/awn061. [DOI] [PubMed] [Google Scholar]
- 63.Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, Kovacs GG, Ghetti B, Halliday G, Holm IE, Ince PG, Kamphorst W, Revesz T, Rozemuller AJ, Kumar-Singh S, Akiyama H, Baborie A, Spina S, Dickson DW, Trojanowski JQ, Mann DM. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol. 2009;117:15–18. doi: 10.1007/s00401-008-0460-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mackenzie IR, Rademakers R. The role of transactive response DNA-binding protein-43 in amyotrophic lateral sclerosis and frontotemporal dementia. Curr Opin Neurol. 2008;21:693–700. doi: 10.1097/WCO.0b013e3283168d1d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mateen FJ, Josephs KA. TDP-43 is not present in brain tissue of patients with schizophrenia. Schizophr Res. 2008;108:297–298. doi: 10.1016/j.schres.2008.08.033. [DOI] [PubMed] [Google Scholar]
- 66.McCluskey LF, Elman LB, Martinez-Lage M, Van D V, Yuan W, Clay D, Siderowf A, Trojanowski JQ. Amyotrophic lateral sclerosis-plus syndrome with TAR DNA-binding protein-43 pathology. Arch Neurol. 2009;66:121–124. doi: 10.1001/archneur.66.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mercado PA, Ayala YM, Romano M, Buratti E, Baralle FE. Depletion of TDP 43 overrides the need for exonic and intronic splicing enhancers in the human apoA-II gene. Nucleic Acids Res. 2005;33:6000–6010. doi: 10.1093/nar/gki897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mishra M, Paunesku T, Woloschak GE, Siddique T, Zhu LJ, Lin S, Greco K, Bigio EH. Gene expression analysis of frontotemporal lobar degeneration of the motor neuron disease type with ubiquitinated inclusions. Acta Neuropathol. 2007;114:81–94. doi: 10.1007/s00401-007-0240-7. [DOI] [PubMed] [Google Scholar]
- 69.Mori F, Tanji K, Zhang HX, Nishihira Y, Tan CF, Takahashi H, Wakabayashi K. Maturation process of TDP-43-positive neuronal cytoplasmic inclusions in amyotrophic lateral sclerosis with and without dementia. Acta Neuropathol. 2008;116:193–203. doi: 10.1007/s00401-008-0396-9. [DOI] [PubMed] [Google Scholar]
- 70.Nakamura M, Ito H, Wate R, Nakano S, Hirano A, Kusaka H. Phosphorylated Smad2/3 immunoreactivity in sporadic and familial amyotrophic lateral sclerosis and its mouse model. Acta Neuropathol. 2008;115:327–334. doi: 10.1007/s00401-007-0337-z. [DOI] [PubMed] [Google Scholar]
- 71.Nakashima-Yasuda H, Uryu K, Robinson J, Xie SX, Hurtig H, Duda JE, Arnold SE, Siderowf A, Grossman M, Leverenz JB, Woltjer R, Lopez OL, Hamilton R, Tsuang DW, Galasko D, Masliah E, Kaye J, Clark CM, Montine TJ, Lee VM, Trojanowski JQ. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol. 2007;114:221–229. doi: 10.1007/s00401-007-0261-2. [DOI] [PubMed] [Google Scholar]
- 72.Neumann M, Kwong LK, Lee EB, Kremmer E, Flatley A, Xu Y, Forman MS, Troost D, Kretzschmar HA, Trojanowski JQ, Lee VM. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 2009;117:137–149. doi: 10.1007/s00401-008-0477-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Neumann M, Kwong LK, Sampathu DM, Trojanowski JQ, Lee VM. TDP-43 proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis: protein misfolding diseases without amyloidosis. Arch Neurol. 2007;64:1388–1394. doi: 10.1001/archneur.64.10.1388. [DOI] [PubMed] [Google Scholar]
- 74.Neumann M, Mackenzie IR, Cairns NJ, Boyer PJ, Markesbery WR, Smith CD, Taylor JP, Kretzschmar HA, Kimonis VE, Forman MS. TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropathol Exp Neurol. 2007;66:152–157. doi: 10.1097/nen.0b013e31803020b9. [DOI] [PubMed] [Google Scholar]
- 75.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VMY. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 76.Nishihira Y, Tan CF, Hoshi Y, Iwanaga K, Yamada M, Kawachi I, Tsujihata M, Hozumi I, Morita T, Onodera O, Nishizawa M, Kakita A, Takahashi H. Sporadic amyotrophic lateral sclerosis of long duration is associated with relatively mild TDP-43 pathology. Acta Neuropathol. 2009;117:45–53. doi: 10.1007/s00401-008-0443-6. [DOI] [PubMed] [Google Scholar]
- 77.Nishihira Y, Tan CF, Onodera O, Toyoshima Y, Yamada M, Morita T, Nishizawa M, Kakita A, Takahashi H. Sporadic amyotrophic lateral sclerosis: two pathological patterns shown by analysis of distribution of TDP-43-immunoreactive neuronal and glial cytoplasmic inclusions. Acta Neuropathol. 2008;116:169–182. doi: 10.1007/s00401-008-0385-z. [DOI] [PubMed] [Google Scholar]
- 78.Nishihira Y, Tan CF, Toyoshima Y, Yonemochi Y, Kondo H, Nakajima T, Takahashi H. Sporadic amyotrophic lateral sclerosis: widespread multisystem degeneration with TDP-43 pathology in a patient after long-term survival on a respirator. Neuropathology. 2009 doi: 10.1111/j.1440-1789.2008.00999.x. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 79.Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol. 1995;69:3584–3596. doi: 10.1128/jvi.69.6.3584-3596.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pamphlett R, Kum JS. TDP-43 inclusions do not protect motor neurons from sporadic ALS. Acta Neuropathol. 2008;116:221–222. doi: 10.1007/s00401-008-0392-0. [DOI] [PubMed] [Google Scholar]
- 81.Robertson J, Sanelli T, Xiao S, Yang W, Horne P, Hammond R, Pioro EP, Strong MJ. Lack of TDP-43 abnormalities in mutant SOD1 transgenic mice shows disparity with ALS. Neurosci Lett. 2007;420:128–132. doi: 10.1016/j.neulet.2007.03.066. [DOI] [PubMed] [Google Scholar]
- 82.Roeber S, Mackenzie IR, Kretzschmar HA, Neumann M. TDP-43-negative FTLD-U is a significant new clinico-pathological subtype of FTLD. Acta Neuropathol. 2008;116:147–157. doi: 10.1007/s00401-008-0395-x. [DOI] [PubMed] [Google Scholar]
- 83.Rollinson S, Snowden JS, Neary D, Morrison KE, Mann DM, Pickering-Brown SM. TDP-43 gene analysis in frontotemporal lobar degeneration. Neurosci Lett. 2007;419:1–4. doi: 10.1016/j.neulet.2007.03.044. [DOI] [PubMed] [Google Scholar]
- 84.Rutherford NJ, Zhang YJ, Baker M, Gass JM, Finch NA, Xu YF, Stewart H, Kelley BJ, Kuntz K, Crook RJ, Sreedharan J, Vance C, Sorenson E, Lippa C, Bigio EH, Geschwind DH, Knopman DS, Mitsumoto H, Petersen RC, Cashman NR, Hutton M, Shaw CE, Boylan KB, Boeve B, Graff-Radford NR, Wszolek ZK, Caselli RJ, Dickson DW, Mackenzie IR, Petrucelli L, Rademakers R. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008;4:e1000193. doi: 10.1371/journal.pgen.1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sampathu DM, Neumann M, Kwong LK, Chou TT, Micsenyi M, Truax A, Bruce J, Grossman M, Trojanowski JQ, Lee VMY. Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol. 2006;169:1343–1352. doi: 10.2353/ajpath.2006.060438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sanelli T, Xiao S, Horne P, Bilbao J, Zinman L, Robertson J. Evidence that TDP-43 is not the major ubiquitinated target within the pathological inclusions of amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2007;66:1147–1153. doi: 10.1097/nen.0b013e31815c5edd. [DOI] [PubMed] [Google Scholar]
- 87.Schumacher A, Friedrich P, Diehl-Schmid J, Ibach B, Perneczky R, Eisele T, Vukovich R, Foerstl H, Riemenschneider M. No association of TDP-43 with sporadic frontotemporal dementia. Neurobiol Aging. 2009;30:157–159. doi: 10.1016/j.neurobiolaging.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 88.Schwab C, Arai T, Hasegawa M, Yu S, McGeer PL. Colocalization of transactivation-responsive DNA-binding protein 43 and huntingtin in inclusions of Huntington disease. J Neuropathol Exp Neurol. 2008;67:1159–1165. doi: 10.1097/NEN.0b013e31818e8951. [DOI] [PubMed] [Google Scholar]
- 89.Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, Nielsen JE, Hodges JR, Spillantini MG, Thusgaard T, Brandner S, Brun A, Rossor MN, Gade A, Johannsen P, Sorensen SA, Gydesen S, Fisher EM, Collinge J. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 2005;37:806–808. doi: 10.1038/ng1609. [DOI] [PubMed] [Google Scholar]
- 90.Sleegers K, Brouwers N, Maurer-Stroh S, van Es MA, Van Damme P, van Vught PW, van der ZJ, Serneels S, De Pooter T, Van den BM, Cruts M, Schymkowitz J, De Jonghe P, Rousseau F, van den Berg LH, Robberecht W, Van Broeckhoven C. Progranulin genetic variability contributes to amyotrophic lateral sclerosis. Neurology. 2008;71:253–259. doi: 10.1212/01.wnl.0000289191.54852.75. [DOI] [PubMed] [Google Scholar]
- 91.Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al Chalabi A, Miller CC, Nicholson G, Shaw CE. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Steinacker P, Hendrich C, Sperfeld AD, Jesse S, von Arnim CA, Lehnert S, Pabst A, Uttner I, Tumani H, Lee VM, Trojanowski JQ, Kretzschmar HA, Ludolph A, Neumann M, Otto M. TDP-43 in cerebrospinal fluid of patients with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch Neurol. 2008;65:1481–1487. doi: 10.1001/archneur.65.11.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Strong MJ. The syndromes of frontotemporal dysfunction in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2008;9:323–338. doi: 10.1080/17482960802372371. [DOI] [PubMed] [Google Scholar]
- 94.Strong MJ, Grace G, Lomen-Hoerth C, Woolley-Levine S, Goldstein LH, Murphy J, Shoesmith C, Rosenfeld J, Leigh P, Bruijn L, Ince P, Figlewicz D. Consensus criteria for the diagnosis of frontotemporal cognitive and behavioural syndromes in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2009 doi: 10.1080/17482960802654364. in press. [DOI] [PubMed] [Google Scholar]
- 95.Strong MJ, Volkening K, Hammond R, Yang W, Strong W, Leystra-Lantz C, Shoesmith C. TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol Cell Neurosci. 2007;35:320–327. doi: 10.1016/j.mcn.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 96.Sumi H, Kato S, Mochimaru Y, Fujimura H, Etoh M, Sakoda S. Nuclear TAR DNA binding protein 43 expression in spinal cord neurons correlates with the clinical course in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2009;68:37–47. doi: 10.1097/NEN.0b013e3181919cb5. [DOI] [PubMed] [Google Scholar]
- 97.Tan CF, Eguchi H, Tagawa A, Onodera O, Iwasaki T, Tsujino A, Nishizawa M, Kakita A, Takahashi H. TDP-43 immunoreactivity in neuronal inclusions in familial amyotrophic lateral sclerosis with or without SOD1 gene mutation. Acta Neuropathol (Berl) 2007;113:535–542. doi: 10.1007/s00401-007-0206-9. [DOI] [PubMed] [Google Scholar]
- 98.Temiz P, Weihl CC, Pestronk A. Inflammatory myopathies with mitochondrial pathology and protein aggregates. J Neurol Sci. 2008;278:25–29. doi: 10.1016/j.jns.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 99.Thorpe JR, Tang H, Atherton J, Cairns NJ. Fine structural analysis of the neuronal inclusions of frontotemporal lobar degeneration with TDP-43 proteinopathy. J Neural Transm. 2008;115:1661–1671. doi: 10.1007/s00702-008-0137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Uryu K, Nakashima-Yasuda H, Forman MS, Kwong LK, Clark CM, Grossman M, Miller BL, Kretzschmar HA, Lee VM, Trojanowski JQ, Neumann M. Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J Neuropathol Exp Neurol. 2008;67:555–564. doi: 10.1097/NEN.0b013e31817713b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB, Clay D, Wood EM, Chen-Plotkin AS, Martinez-Lage M, Steinbart E, McCluskey L, Grossman M, Neumann M, Wu IL, Yang WS, Kalb R, Galasko DR, Montine TJ, Trojanowski JQ, Lee VM, Schellenberg GD, Yu CE. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7:409–416. doi: 10.1016/S1474-4422(08)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang HY, Wang IF, Bose J, Shen CK. Structural diversity and functional implications of the eukaryotic TDP gene family. Genomics. 2004;83:130–139. doi: 10.1016/S0888-7543(03)00214-3. [DOI] [PubMed] [Google Scholar]
- 103.Wang IF, Reddy NM, Shen CK. Higher order arrangement of the eukaryotic nuclear bodies. Proc Natl Acad Sci USA. 2002;99:13583–13588. doi: 10.1073/pnas.212483099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang IF, Wu LS, Chang HY, Shen CK. TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J Neurochem. 2008;105:797–806. doi: 10.1111/j.1471-4159.2007.05190.x. [DOI] [PubMed] [Google Scholar]
- 105.Wang IF, Wu LS, Shen CK. TDP-43: an emerging new player in neurodegenerative diseases. Trends Mol Med. 2008;14:479–485. doi: 10.1016/j.molmed.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 106.Weihl CC, Temiz P, Miller SE, Watts G, Smith C, Forman M, Hanson PI, Kimonis V, Pestronk A. TDP-43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2008;79:1186–1189. doi: 10.1136/jnnp.2007.131334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wider C, Dickson DW, Stoessl AJ, Tsuboi Y, Chapon F, Gutmann L, Lechevalier B, Calne DB, Personett DA, Hulihan M, Kachergus J, Rademakers R, Baker MC, Grantier LL, Sujith OK, Brown L, Calne S, Farrer MJ, Wszolek ZK. Pallidonigral TDP-43 pathology in Perry syndrome. Parkinsonism Relat Disord. 2008 doi: 10.1016/j.parkreldis.2008.07.005. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yokoseki A, Shiga A, Tan CF, Tagawa A, Kaneko H, Koyama A, Eguchi H, Tsujino A, Ikeuchi T, Kakita A, Okamoto K, Nishizawa M, Takahashi H, Onodera O. TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann Neurol. 2008;63:538–542. doi: 10.1002/ana.21392. [DOI] [PubMed] [Google Scholar]