

Abstract
Foxg1 is a transcription factor that is critical for forebrain development. Foxg1+/Cre mice were used to test the hypotheses 1) that the subventricular zone (SZ) generates supragranular neurons, 2) that Foxg1-regulated activities define the output from the SZ, and 3) that Foxg1 is involved in the suppression of p21-initiated cell-cycle exit. Foxg1+/Cre mice have thinner neocortices than wild-type controls, specifically in the supragranular layers, as detected by Brn2 immunostaining. Cell proliferation in the ventricular zone (VZ) and SZ was examined to investigate the reduction in upper layer neurons. The number of cycling VZ cells was similar in Foxg1+/+ and Foxg1+/Cre brains. Interestingly, cell proliferation in the SZ and intermediate progenitor cell (IPC) production (noted by Tbr2-immunostaining) was reduced in Foxg1+/Cre brains. These decreases coincided with increased expression of the cell-cycle inhibitor p21 in the VZ and SZ. Furthermore, colocalization of p21 with markers of cell proliferation and IPCs indicated that p21 was temporally expressed to influence the proliferative fate of IPCs. Thus, the present data are consistent with the above hypotheses, particularly, that during corticogenesis, Foxg1-regulated activities enable the expansion of the IPC population likely through suppression of p21-dependent cell-cycle exit.
Keywords: cell cycle, Ki-67, p27, progenitor cells, subventricular zone, ventricular zone
Introduction
Two proliferative zones, the ventricular zone (VZ) and the subventricular zone (SZ), are active during the period of neocortical neuronogenesis. It appears that the SZ, unlike the VZ, does not contain a fixed neural progenitor population (Haubensak et al. 2004; Englund et al. 2005). Rather, the majority of the SZ is comprised of an intermediate progenitor cell (IPC) population of translocated VZ progenitors that undergo a final cell division in the SZ before becoming postmitotic neurons (Miyata et al. 2004; Noctor et al. 2004).
Foxg1 is a transcription factor that is strongly expressed in the developing retina, optic stalks, superior colliculus, and telencephalon and has purported roles in 1) cortical arealization, 2) expansion of the cortical progenitor pool, and 3) regulation of progenitor cell-cycle length (Tao and Lai 1992; Xuan et al. 1995; Dou et al. 1999; Hanashima et al. 2002, 2004; Martynoga et al. 2005; Muzio and Mallamaci 2005). Loss of Foxg1 severely compromises the growth of the telencephalon; however, mice that are haploinsufficient for Foxg1 exhibit subtler developmental defects in the forebrain. Specifically, adult Foxg1+/Cre, Foxg1+/LacZ, and Foxg1+/TTA mice (in which one Foxg1 allele is replaced with Cre recombinase [Cre], LacZ β-D-galactosidase [LacZ], or tetracycline transactivator gene [TTA], respectively) have smaller cortical volumes, and Foxg1+/Cre mice can display a specific reduction in the thickness of layer II/III (Shen et al. 2006; Eagleson et al. 2007).
Interestingly, it has been proposed that a large proportion of layer II/III neurons are born (i.e., undergo their final cell cycle) in the SZ (Miller 1989; 1992; Tarabykin et al. 2001; Nieto et al. 2004; Noctor et al. 2004; Zimmer et al. 2004; Englund et al. 2005; Ferrere et al. 2006; Martinez-Cerdeno et al. 2006). The implication would be that the IPC population in the SZ is affected in Foxg1+/Cre mice. Thus, the microencephaly in Foxg1+/− mice may result from specific defects in progenitor cell-cycle regulation and exit in the VZ and/or SZ. Further, the Foxg1+/Cre mice, which do not exhibit the severe cortical arealization defects apparent in the null mice (Hebert and McConnell 2000), are potentially a better model for determining how Foxg1 influences cortical cell number.
The present study examined the prenatal origins of cortical deficits in Foxg1+/Cre mice. Evaluation of both VZ and SZ cell proliferation at different stages of corticogenesis reveals a significant decrease in the size of the SZ in the Foxg1+/Cre cortex due to decreased production of IPCs and, late in corticogenesis, an increase in VZ cell-cycle length. Loss of IPCs coincides with increased expression of p21, a cyclin-dependent kinase inhibitor (CKI), that potently inhibits cell-cycle progression in the VZ and the transcription of which is directly inhibited by Foxg1 (Seoane et al. 2004; Siegenthaler and Miller 2005). Collectively, this evidence suggests that Foxg1 promotes cortical growth in part by enabling the expansion of the IPC pool in the developing cortex by inhibiting expression of the cell-cycle inhibitor p21.
Materials and Methods
Foxg1-Deficient Mice
Foxg1+/Cre mice were obtained from Pat Levitt (Vanderbilt University, Nashville TN). These animals were derived from Foxg1+/Cre mice generated on a mixed genetic background (Hebert and McConnell 2000). The mice were backcrossed on a C57BL/6J background (Eagleson et al. 2007). Animals were maintained in facility at the Syracuse Veterans Affairs Medical Center (VAMC) that is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care and experimental protocols were approved 1) by the Committee on Humane Use of Animals at Upstate Medical University and 2) by the Institutional Animal Care and Use Committee at the Syracuse VAMC.
Females were placed with breeding males at 5:00 PM. The next day, at 9:00 AM, females were examined, and if a sperm-positive vaginal plug was observed, that time was designated as embryonic day (E) 0.5. Following various bromodeoxyuridine (BrdU) injection paradigms (see below), pregnant females were anesthetized and the fetuses were harvested on E13.5, E15.5, E16.5, or E17.5.
Adult and fetal mice were genotyped using primers designed to amplify both the Foxg1 wild-type and Cre-inserted allele. The 3 primers used were 5′-GCC GCC CCC CGA CGC CTG GGT GAT G-3′, 5′-TGG TGG TGG TGA TGA TGG TGA TGC TGG-3′, and 5′-ATA ATC GCG AAC ATC TTC AGG TTC TGC GGG-3′ (Muzio and Mallamaci 2005). The polymerase chain reaction (PCR) products were 186 bp (wild-type allele) and 220 bp (null allele).
Immunoblotting
The veracity of the Foxg1 haploinsufficiency and knockout was determined with immunoblots for the expression of Foxg1. Two pregnant Foxg1+/Cre mice were anesthetized on E15.5 with a cocktail of ketamine (1.0 mg/kg) and xylazine (1.0 mg/kg), and fetuses were delivered by Cesarean section. Brains were rapidly extracted. Cortices were isolated, placed in lysis buffer (1.0% Nonidet P-40, 0.50% deoxycholic acid, 0.010% sodium dodecyl sulfate [SDS], Complete Mini-Protease inhibitor cocktail tablets [1 tablet per 10 ml buffer; Roche, Indianapolis, IN]) in 100 mM phosphate buffered saline (pH 7.4; PBS), and sonicated and spun at 14 000 rpm for 10 min. The protein concentration of the supernatant was determined using a Bio-Rad Protein Assay (Bio-Rad, Hercules, CA).
An aliquot of the supernatant containing 40 μg/ml protein was combined with electrophoresis sample buffer (300 mM Tris–HCl, 50% glycerol, 5.0% SDS, 0.025% bromophenol blue, 250 mM β-mercaptoethanol). Samples were loaded on a 7.5% SDS–polyacrylamide gel, separated by electrophoresis, and transferred to nitrocellulose membranes. Nonspecific immunoreactivity on the membranes were blocked with a 1 h rinse in 5.0% nonfat dehydrated milk in PBS with 0.10% Tween 20 (TPBS). Blots were probed overnight at 4 °C with rabbit polyclonal anti-Foxg1 antibody (1:500; generously provided by Yoshiki Sasai, RIKEN Institute, Kobe, Japan) in 5.0% bovine serum albumin (BSA) in TPBS. Blots were probed with horseradish peroxidase–linked anti-rabbit antibody (1:3000) in 5.0% BSA in TPBS for 1 h. A chemiluminescent detection reagent (Amersham, Piscataway, NJ) was used to visualize immunotagged protein bands. Samples of littermates taken from 2 litters were analyzed for each genotype.
Membranes were stripped of the immunoreaction and processed for actin expression as a check for consistent loading of the samples. After stripping, blots were immunostained with a mouse antiactin monoclonal antibody (1:4000, Sigma, St. Louis, MO) in TPBS for 90 min at room temperature. Immunodetection of the bound primary (anti-actin) antibody was performed as described above.
Immunohistochemistry
Pregnant dams were anesthetized, and fetuses were delivered (see above). Brains were rapidly extracted and fixed overnight at 4 °C in buffered 4.0% paraformaldehyde. The fixed brains were processed for cryosectioning, frozen, and cut into 12 μm sections. Prior to incubation with primary antibodies, all sections were washed in 3.0% H2O2 for 5 min, steamed in 10 mM citric acid (pH 6.0) for 15 min, and cooled to room temperature in PBS. Nonspecific immunoreactivity was blocked by washing sections in a solution of 1.0% BSA and 0.75% Triton X-100 for 45 min.
Brn2 and Tbr1 expression were detected in the cortices of 6-day-old pups immunohistochemically. After the blocking step, sections were incubated in a solution with a primary antibody against Brn2 (rabbit polyclonal, diluted 1:200 in PBS; Santa Cruz Biotechnology, Santa Cruz, CA) or Tbr1 (rabbit polyclonal, 1:400; Chemicon, Temecula, CA) diluted in 10% goat serum and 0.75% Triton X-100 in PBS (GSTPBS) for 90 min. Following several washes in PBS, sections were incubated for 1 h in biotinylated goat anti-rabbit secondary antibody (Vector Laboratories, Burlingame, CA) and diluted 1:200 in GSTPBS. Sections were rinsed prior to incubation in an avidin-containing complex (Elite Kit; Vector Laboratories). Immunoreactivity was visualized with 0.29% 3,3′-diaminobenzidine (Vector Laboratories) in the presence of 0.019% H2O2.
Combined iododeoxyuridine (IdU)/BrdU/Ki-67 labeling was examined in wild-type and Foxg1 heterozygous fetuses. Sections were incubated with primary antibodies to BrdU/IdU (mouse monoclonal, 1:100; BD Biosciences, San Diego, CA), BrdU (rat polyclonal, 1:50 in GSTPBS; Novus Biologicals, Littleton, CO), and Ki-67 (rabbit monoclonal, 1:100 in GSTPBS; LabVision, Fremont, CA). For Tbr2/BrdU immunolabeling, sections were incubated with primary antibodies to BrdU (mouse monoclonal, 1:100 in GSTPBS; BD Biosciences) and Tbr2 (rabbit polyclonal, 1:100 in GSTPBS; Chemicon). Note that Tbr2 is a marker of IPCs (Englund et al. 2005). For multiple antibody labeling involving p21, the anti-p21 antibody (mouse monoclonal, 1:1000 in GSTPBS; BD Biosciences) was used in conjunction with the Tyramide System Amplification Kit #10 (Molecular Probes, Eugene, OR) as per the manufacturer's instructions. Following immunolabeling of p21, sections were incubated with primary antibodies to 1) proliferating cell nuclear antigen (PCNA; mouse monoclonal, 1:200 in GSTPBS; BD Biosciences) and Tbr2 or 2) the CKI p27 (mouse monoclonal, 1:400 in GSTPBS; BD Biosciences) and Tbr2. For Tuj1 immunolabeling, sections were incubated with primary antibody to Tuj1 (mouse monoclonal, 1:1000 in GSTPBS; Covance, Berkeley, CA). Some sections were immunostained with an anti-active caspase 3 antibody (rabbit polyclonal, 1:200 in GSTPBS; Santa Cruz Biotechnology). For Pax6 immunolabeling, sections were incubated with primary antibody to Pax6 (rabbit polyclonal, 1:500 in GSTPBS; Covance), followed by incubation with the anti-PCNA antibody. Processing with primary antibodies was followed by incubation with the appropriate fluorescein-conjugated secondary antibody (1:200 in GSTPBS; Jackson Immunologicals, West Grove, PA). All immunofluorescence was visualized using a Bio-Rad MRC-1024 Confocal Microscope and the associated Bio-Rad LaserSharp 2000 software.
Quantitative Analyses
The depth of various aspects of the developing cortex was measured. Distance was always calculated along a radial dimension (a line perpendicular to the pial surface). Distances that were measured include the depth of the cortical plate on E13.5, E15.5, and E17.5 in wild-type and heterozygous fetuses and the depth of the band that was immunolabeled for Brn2 or Tbr1. At least 3 animals were examined for each mean measure.
Various aspects of the cell-cycle kinetics were determined using a dual thymidine marker approach (Martynoga et al. 2005). On the designated embryonic age, the pregnant dam was injected with IdU at a time designated as time 0 (in hours) (t0). This was followed 1.5 h later by an injection of BrdU. Half an hour after the injection of BrdU, the pregnant dam was anesthetized and fetual were harvested. Such studies were performed with fetuses on E13.5, E15.5, or E17.5.
The length of the S-phase (TS) and the total length of the cell cycle (TC) were determined based on the relative numbers of cells that incorporated one or both of the thymidine analogs (Martynoga et al. 2005). Briefly, the density (the number of immunolabeled cells in a 62 500 μm2 box in the middorsal telencephalon) was determined for Ki-67+ cells (proliferating cells), IdU+/BrdU+ cells (those remaining in the S-phase at the end of the experiment), and IdU+/BrdU− cells (cells that were in S at the time of the IdU injection, but left S prior to the BrdU injection). The length of S-phase, TS, was calculated as the interval between injections (1.5 h) divided by the quotient of the density of IdU+/BrdU− cells and IdU+/BrdU+ cells. The total length of cell cycle was determined by dividing the TS by the quotient of the density of IdU+/BrdU+ cells and Ki-67+ cells.
For the analysis of cell proliferation in the VZ and SZ, the density of Ki-67+ cells was determined in the same counting space used in the cell-cycle kinetics analysis. The VZ and SZ were delineated by the presence of IdU+/BrdU+ at the border between the VZ and SZ. For each embryonic age, wild-type and heterozygous siblings from a minimum of 3 litters were used.
The amount of IPC production was estimated using a BrdU pulse–chase paradigm. On either E12.5 or E15.5, pregnant dams were injected with BrdU at t0 and their fetuses collected 16 (t16) or 24 (t24) h later, respectively. Sections of the cortices of wild-type and Foxg1+/Cre mice at each gestational age were processed for BrdU and Tbr2 immunoreactivity. An estimate of the frequency of BrdU+ cells that became Tbr2+ (as a measure of IPC production) for each genotype was calculated as the density of Tbr2+/BrdU+ cells divided by the density of BrdU+ cells. For each embryonic age, a pair of wild-type and heterozygous littermates from each of 4 separate litters was used.
Following p21 immunostaining, the density of p21+ cells at E13.5 and E16.5 was determined. For each gestational age, sections from wild-type and heterozygous littermates from 4 litters were used.
Quantitative data were examined for statistically significant differences using a 2-tiered approach. Initially, analyses of variance were used to determine the overall effects of genotype and age. In instances in which statistical significance was detected, a post-hoc Holms–Sidak test was performed for pairwise comparisons.
Results
Characterization of Cortex in Fetal and Neonatal Foxg1+/Cre Mice
To confirm that mice heterozygous for Foxg1 have reduced Foxg1 protein expression, cortices from Foxg1+/+, Foxg1+/Cre, and Foxg1Cre/Cre fetuses were collected for analyses of protein expression. The genotype of the fetuses was determined using PCR primers designed to amplify a portion of the wild-type Foxg1 allele (186 bp PCR product) or a portion of the null Foxg1 allele (220 bp PCR product) (Muzio and Mallamaci 2005) (Fig. 1A). Immunoblot analysis of all 3 genotypes using an antibody specific for Foxg1 revealed that cortices in Foxg1+/Cre mice exhibited reduced (−33 ± 2%) protein expression as compared with wild-type mice. The specificity of the antibody was further confirmed by the total absence of Foxg1 protein in the Foxg1-null animal (Foxg1Cre/Cre). Immunofluorescence using the same Foxg1 antibody indicated that there was global reduction in Foxg1 protein expression in the telencephalons of Foxg1+/Cre mice (Fig. 1B).
Figure 1.

Expression of Foxg1. (A) A representative gel of PCR products from amplification of genomic DNA from Foxg1+/+, Foxg1+/Cre, and Foxg1Cre/Cre mice. The 186- and 220-bp bands result from amplification of the Foxg1 wild-type and null allele, respectively. Foxg1 immunoblots showed decreased Foxg1 protein in the Foxg1+/Cre cortex and the absence of Foxg1 protein in the Foxg1Cre/Cre. (B) Immunohistochemical preparations show differential expression of Foxg1 in forebrains of 15.5-day-old wild-type and heterozygous mice. Scale bars are 500 μm.
To begin to assess the developmental origins of the microencephaly observed in Foxg1+/Cre adult mice, fetal brains were collected at 3 disparate stages of corticogenesis (E13.5, E15.5, and E17.5) and labeled with a marker for young, postmitotic neurons, Tuj1. Early in corticogenesis (E13.5), there was no obvious difference in the thickness of the cerebral wall or in the complement of Tuj1+ cells (Fig. 2; Table 1). By E15.5, however, the cortical plate was significantly (P < 0.01) thinner (−28%) in Foxg1+/Cre animals. The discrepancy in radial growth between the 2 genotypes (−26%) persisted on E17.5.
Figure 2.
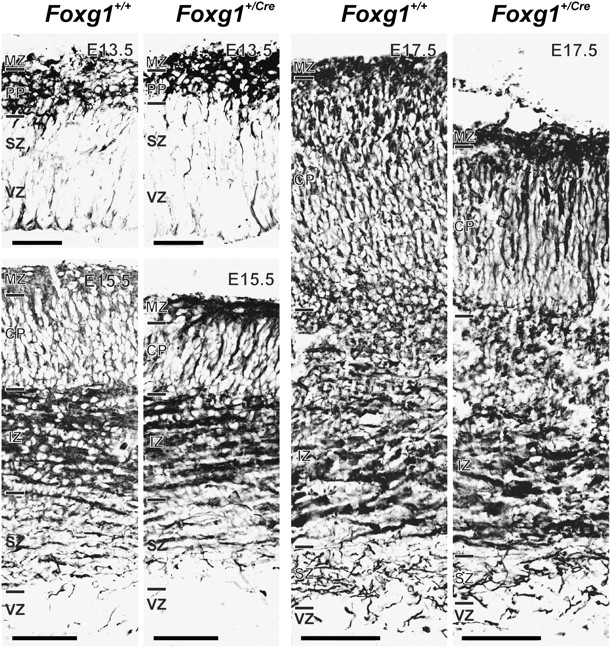
Cortical growth defects in the Foxg1+/Cre fetal brain. The full depth of the cerebral wall is shown for Foxg1+/+ and Foxg1+/Cre fetuses on E13.5, E15.5, and E17.5. Samples were immunostained for Tuj1 expression. Disparity in thickness of the cortical plate (CP)was evident on E15.5 and E17.5 (see Table 1). IZ, intermediate zone; MZ, marginal zone; PP, preplate; SZ, subventricular zone; VZ, ventricular zone. Scale bars are 100 μm (G13.5) and 200 μm (E15.5 and E17.5).
Table 1.
Depth of the cortical plate in wild-type and Foxg1 heterozygous fetuses
Statistically significant (P < 0.01) differences relative to the wild-type controls. Each value is the mean of 3 fetuses ± standard errors of the means. All depths are described in terms of micrometers.
In 6-day-old mice, immunolabeling for Brn2 (a marker for layer II/III) was attenuated in the Foxg1+/Cre mice relative to wild-type controls (Fig. 3). The depth of the Brn2-positive zone in Foxg1+/Cre mice was 73% less than it was in wild-type mice (Table 2). In contrast, there was no apparent difference in Tbr1 immunostaining (a marker of deep layer neurons) in wild-type and heterozygous mice. Thus, the thinner cortex in Foxg1+/Cre mice was attributable to a decrease in the thickness of superficial laminae.
Figure 3.
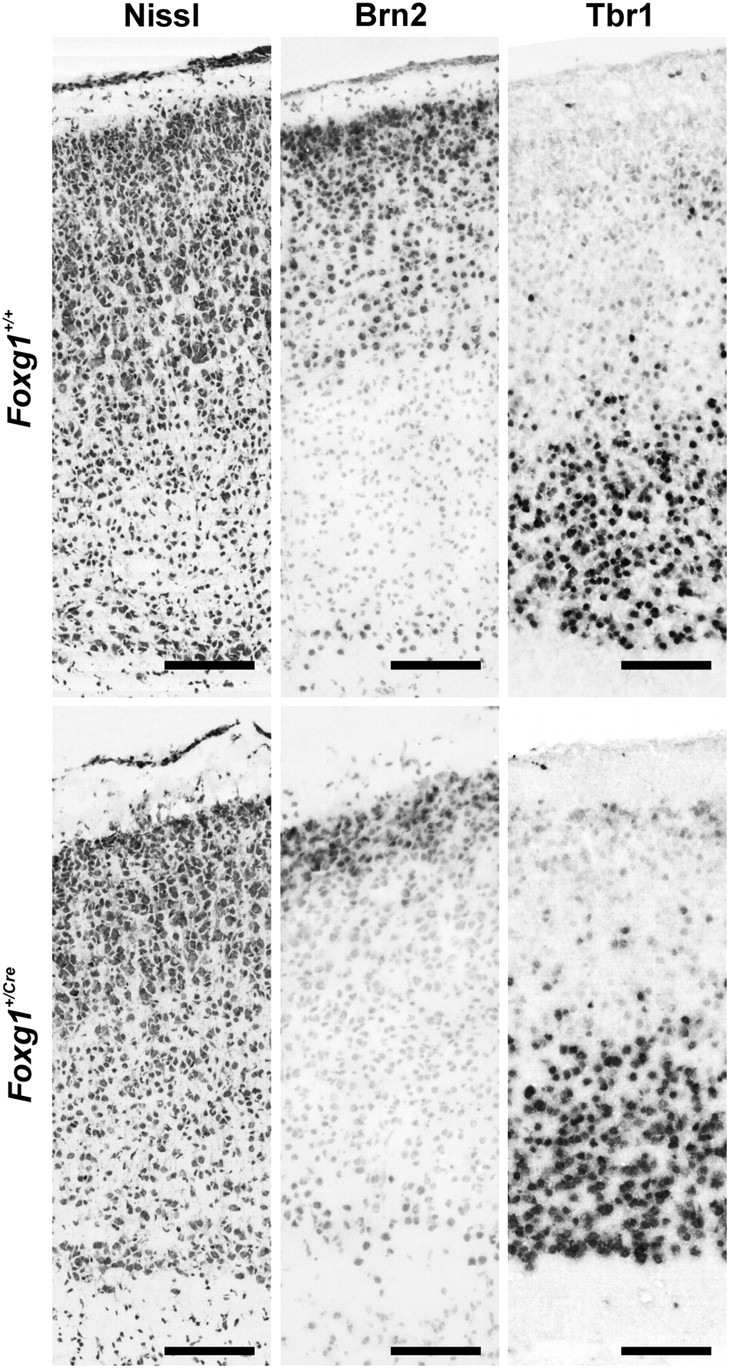
Brn2 and Tbr1 expression in cortex of Foxg1+/+ and Foxg1+/Cre mice on postnatal day 6. The organization of cerebral cortex in the Foxg1+/+ and Foxg1+/Cre mice is depicted in the cresyl violet–stained sections (left). Brn2 and Tbr1 (middle and right, respectively) were expressed in separate compartments of cortex. Brn2 expression in superficial cortex was reduced in Foxg1+/Cre mice, whereas Tbr1 expression was similar in the deep laminae of both genotypes of mice (see Table 2). Scale bars are 100 μm.
Table 2.
Depth of immunoreactive compartments in 6-day-old wild-type and Foxg1 heterozygous mice
| Compartment | Foxg1+/+ | Foxg1+/Cre |
| Brn2 | 276 ± 32 | 75 ± 10a |
| Tbr1 | 228 ± 20 | 238 ± 25 |
Statistically significant (P < 0.01) differences relative to the wild-type controls.
Note: Sections were immunolabeled with an anti-Brn2 or anti-Tbr1 antibody, and the radial depth of the immunolabeled band in superficial or deep cortex, respectively, was measured.
Foxg1 Haploinsufficiency Alters Cell Proliferation
On E13.5, E15.5, and E17.5, Ki-67+ cells filled the VZ and were common in the SZ (Fig. 4A and Supplementary Fig. 1). The number of cycling (Ki-67+) cells in the 2 telencephalic proliferative zones was determined. A significant (F2,15 = 22.930; P < 0.001) effect of age was evident in the VZ in that the frequency of cycling cells decreased with age by greater than 25% (Fig. 4B). There was no difference between the genotypes in the number of proliferating cells in the VZ at any age examined. To further support this finding, cells were immunostained for Pax6 expression (a VZ cell marker; Englund et al. 2005) at E13.5 and E16.5 (data not shown). The density of Pax6-expressing cells in wild-type and Foxg1+/Cre mice was quantified. No differences in the number of Pax6-positive cells were detected between genotypes at either age.
Figure 4.
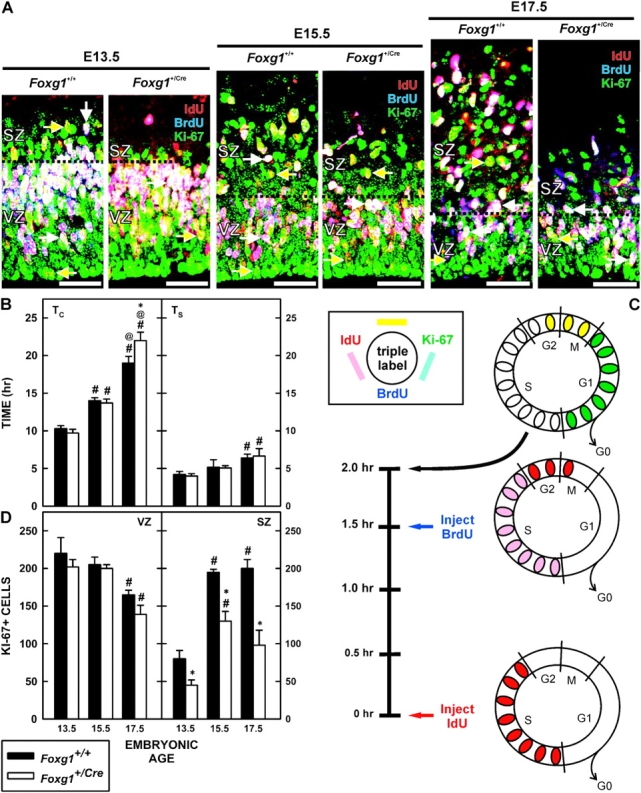
Cell proliferation in Foxg1 haploinsufficient mice. (A) Pregnant dams were injected with IdU and BrdU 1.5 h apart and fetuses were collected 30 min later. Representative images of IdU/BrdU/Ki-67 triple labeling from brains of Foxg1+/+ and Foxg1+/Cre mice on E13.5, E15.5, and E17.5 shows the distributions of BrdU−/IdU+/Ki-67+ (yellow arrows) close to the ventricular surface and BrdU+/IdU+/Ki-67+ (white arrows) in the outer third of the VZ and intermingled in the SZ. Note that the depth of the VZ was similar between the 2 genotypes at all ages, whereas the SZ was clearly reduced in the Foxg1+/Cre fetuses. Scale bars are 100 μm. (B) Total cell-cycle length (TC) (left) and the length of S-phase (TS) (right) were determined for Foxg1+/+ and Foxg1+/Cre mice on E13.5, E15.5, and E17.5. * denotes a significant difference relative to the wild type at the same age, # indicates a significant difference from the mean TC on E13.5 (within genotype), and @ denotes significant differences relative to the expression on E15.5 (within genotype). (C) The illustration describes the experimental paradigm for determining the cell-cycle kinetics (see Quantitative Analyses, Materials and Methods) and the color scheme to immunolabeling in the micrographs. (D) The total number of proliferating (Ki-67+ cells) cells in a 62 500 μm2 field in the VZ (left) and SZ (right) is shown. Separate analyses for Foxg1+/+ and Foxg1+/Cre mice were performed. Notations as for (B).
In the SZ of wild-type mice, the density of Ki-67+ cells rose significantly (2.4-fold; F2,15 = 63.675; P < 0.001) between E13.5 and E15.5 and stabilized between E15.5 and E17.5. In Foxg1+/Cre mice, a similar trend (a 2.8-fold increase) in the SZ population was observed (F2,15 = 63.675; P < 0.001). There were significantly (F2,15 = 60.879; P < 0.001) fewer Ki-67+ cells in the SZ of Foxg1+/Cre fetuses than those of wild-type mice at all embryonic ages (E13.5, 42%; E15.5, 33%; and E17.5, 51%). To determine whether an increase in cell death might contribute to the reduction in number of proliferating cells in the SZ, the expression of active caspase 3 in the proliferative zones on E15.5 and E17.5 was examined. No genotype-related differences in immunolabeling were detected at either age (data not shown).
The total length of the cell cycle was assessed using a BrdU/IdU pulse–chase paradigm (Fig. 4C). BrdU was administered 1.5 h after injecting IdU, and fetuses were harvested 30 min later. Cells triple-labeled for IdU, BrdU, and Ki-67 predominated in the outer third of the VZ (Fig. 4A). In contrast, IdU+/BrdU−/Ki-67+ cells were common in the inner VZ. These were cycling cells that were in the S-phase at the time of the IdU injection, but had exited S prior to exposure to BrdU.
The TC for VZ cells changed significantly (F2,16 = 239.190; P < 0.001) over the period of cortical neuronogenesis (Fig. 4D). In the cortices of wild-type fetuses, the TC for VZ cells nearly doubled between E13.5 (10.3 h) and E17.5 (19.0 h). This increase was statistically significant (F2,16 = 9.795; P < 0.007). A similar temporal 2.3-fold increase (from 9.7 to 22 h) in the TC was observed in Foxg1+/Cre (F2,16 = 8.018; P = 0.018). Furthermore, there was a significant interaction between genotype and age (F2,16 = 7.319; P < 0.005). Specifically, TC was significantly (P < 0.05) longer in the VZs of Foxg+/Cre mice at E17.5 than in the VZs of wild-type mice; TC was not significantly different at the earlier ages examined.
The TS increased significantly (F2,15 = 12.239; P < 0.001) during cortical neuronogenesis (Fig. 4D). Such an increase was evident in wild-type (4.2 to 6.4 h; F2,15 = 5.065; P = 0.038) and heterozygous (4.0 to 6.6 h; F2,15 = 7.043; P = 0.030) mice. The change occurred after E15.5; in the wild-type mice, there was a 24% increase and in the heterozygotes, the increase was 31%. No significant difference between the genotypes was detected.
Generation of IPCs in Foxg1+/cre Fetuses
The difference in Ki-67+ labeling in the SZ of Foxg1+/Cre fetuses prompted an examination of the IPC marker, Tbr2, in wild-type and heterozygous mice. At both E13.5 and E16.5, the number of Tbr2+ cells was significantly (F3,17 = 78.517; P < 0.001) lower (−37.6% at E13.5; −38.8% at E16.5) in the cerebral walls of Foxg1+/Cre mice than in wild-type controls (Fig. 5A,C).
Figure 5.
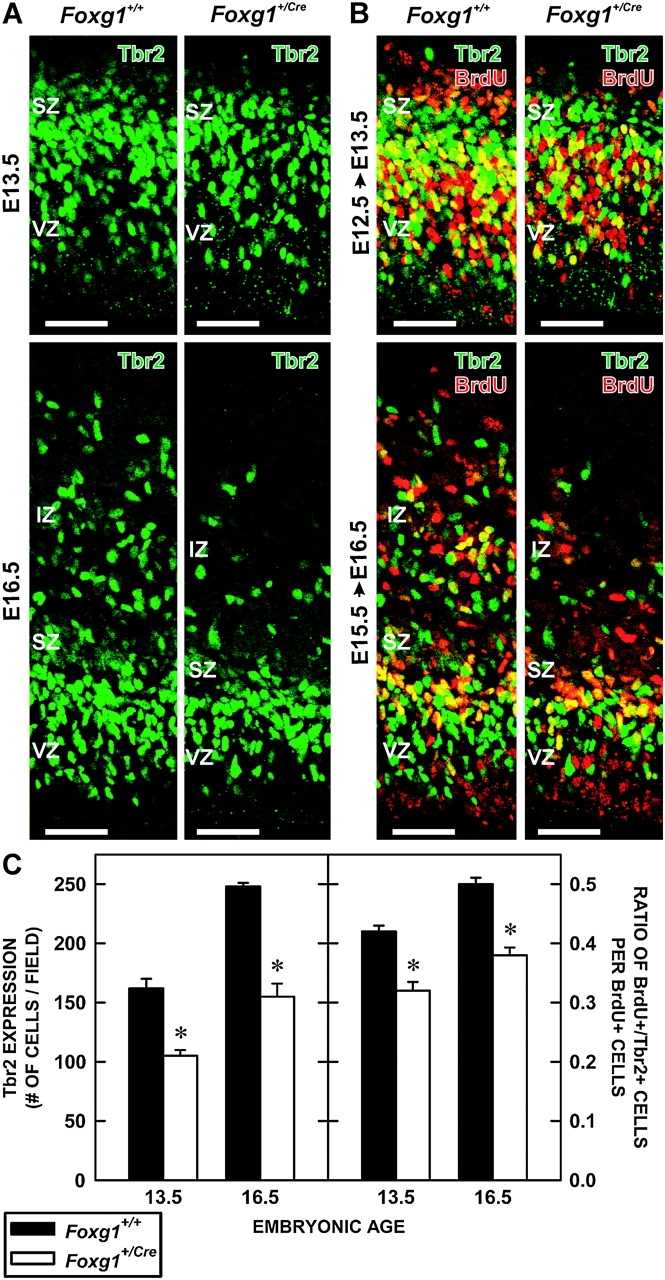
Production of IPCs in Foxg1+/+ and Foxg1+/Cre fetuses. (A) Sections of the cerebral walls of Foxg1+/+ and Foxg1+/Cre fetuses on E13.5 (top) and E16.5 (bottom) were immunolabeled with an antibody to Tbr2, an IPC cell marker. Scale bars are 100 μm. (B) Pregnant dams were injected with BrdU on E12.5 (top) or E15.5 (bottom) and their fetuses collected 16 (E13.5) or 24 h later (E16.5), respectively. Sections were processed for BrdU (red) and Tbr2 (green) immunolabeling. Yellow cells coexpressed BrdU and Tbr2. Scale bars are 100 μm. (C) The 2 graphs depict the total number of Tbr2+ cells in a defined portion of the cerebral wall of Foxg1+/+ and Foxg1+/Cre mice at E13.5 and E16.5 (left) and an estimation of Tbr2-cell production as determined by the number of Tbr2+/BrdU+ cells divided by the total BrdU+ population (right). Asterisks identify statistically significant (P < 0.05) differences between genotypes.
To assess the production of Tbr2+ IPCs in wild-type and heterozygous fetuses, a cohort of proliferating cells in the VZ and SZ was labeled with BrdU on E12.5 or E15.5 and examined for Tbr2 coexpression 16 or 24 h later, respectively. The interval between the injection and collection times allowed for 1) VZ cells destined to become IPCs in the SZ time to express Tbr2 and 2) Tbr2+ cells in the SZ that were in S-phase at the time of the BrdU injection time to downregulate Tbr2+ and exit the cell cycle. Using this method, an estimate of the number of IPCs generated from the VZ was made. This minimized contamination from Tbr2+ cells in the S-phase that were already in the SZ at the time of injection.
Numerous BrdU+/Tbr2+ cells were present at the interface of the VZ and SZ and most BrdU+ cells in the SZ were Tbr2− at both E13.5 and E16.5 (Fig. 5B). This indicated that the injection/collection paradigm successfully targeted cells transiting from the VZ to the SZ and that IPCs in S at the time of injection had exited the cell cycle (i.e., downregulated Tbr2 expression). Thus, the frequency of Tbr2 expression by BrdU-labeled cells in both genotypes reflected the production of IPCs by the VZ.
The proportion of Tbr2+/BrdU+ cells was affected by the genotype of the fetus at both time points (Fig. 5C). This proportion was significantly (F2,15 = 85.828; P < 0.001) lower in the Foxg1+/Cre fetuses than in controls at both E13.5 (−28.6%) and E16.5 (−23.8%).
p21 Expression in the VZ and SZ of Foxg1+/Cre Mice
Excess expression of p21, a protein that induces cell-cycle exit in cortical VZ (Siegenthaler and Miller 2005), is evident in the fetal cortices of Foxg1Cre/Cre mice (Seoane et al. 2004). Thus, the expression of p21 in Foxg1+/Cre fetuses was examined on E13.5 and E16.5 (Fig. 6A). p21+ cells were apparent in the VZ and SZ of wild-type mice at both fetal ages, and the density of p21+ cells increased 3.4-fold over time (Fig. 6B). The number of p21+ cells was significantly (F2,15 = 25.281; P < 0.001) greater (3.3-fold) in Foxg1+/Cre fetuses than wild-type mice at both E13.5 and E16.5. In both genotypes, most p21+ cells were in the VZ.
Figure 6.
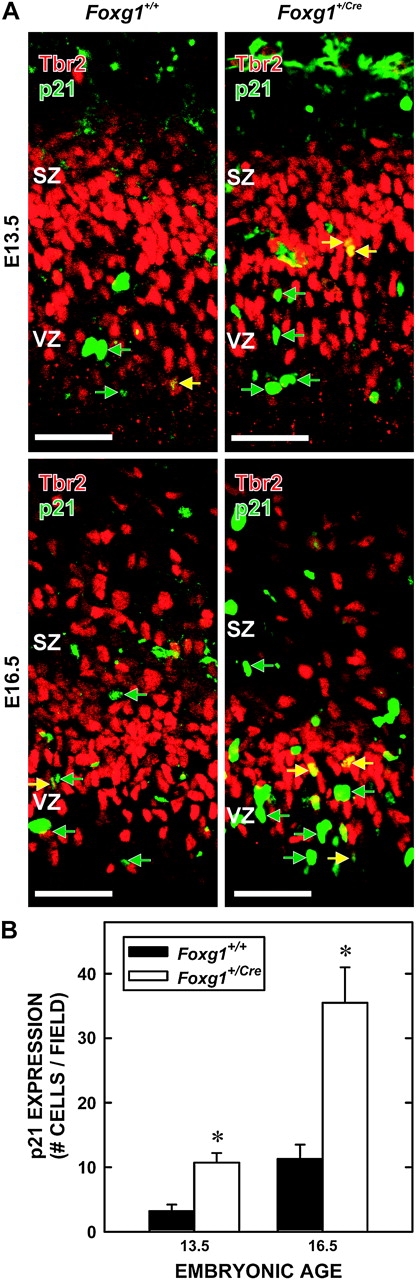
p21 expression among Tbr2+ cells. (A) p21+ cells were evident in the VZ and SZ (green arrows) of brains of Foxg1+/+ (left) and Foxg1+/Cre (right) fetuses on E13.5 (top) and E16.5 (bottom). In both genotypes, p21+/Tbr2+ cells (yellow arrows) were present. Scale bars are 100 μm. (B) Quantification of p21+ cell number in Foxg1+/+ and Foxg1+/Cre at E13.5 and E16.5 reveals a temporal increase in p21+ cells in both genotypes, though significantly more so in the Foxg1+/Cre cortex. Asterisks identify statistically significant differences between genotypes (P < 0.05).
To confirm that the p21+ was expressed by proliferating cells in the VZ and SZ, sections of wild-type and Foxg1+/Cre cortices were triple immunolabeled for possible coexpression of p21, Tbr2, and PCNA. In wild-type and Foxg1+/Cre cortex, most p21+ cells in the VZ and SZ coexpressed PCNA and triple-labeled cells were apparent in both proliferative zones (Fig. 7A). Notably, p21+/PCNA+/Tbr2− cells were largely limited to the VZ in both genotypes. This pattern of labeling suggested that the upregulation of p21 influenced the cell-cycle fate of VZ progenitors both prior to and after the decision to become a Tbr2+ IPC. Note also that density of Tbr2 population was lower in the Foxg1+/Cre fetuses (Supplementary Fig. 2).
Figure 7.
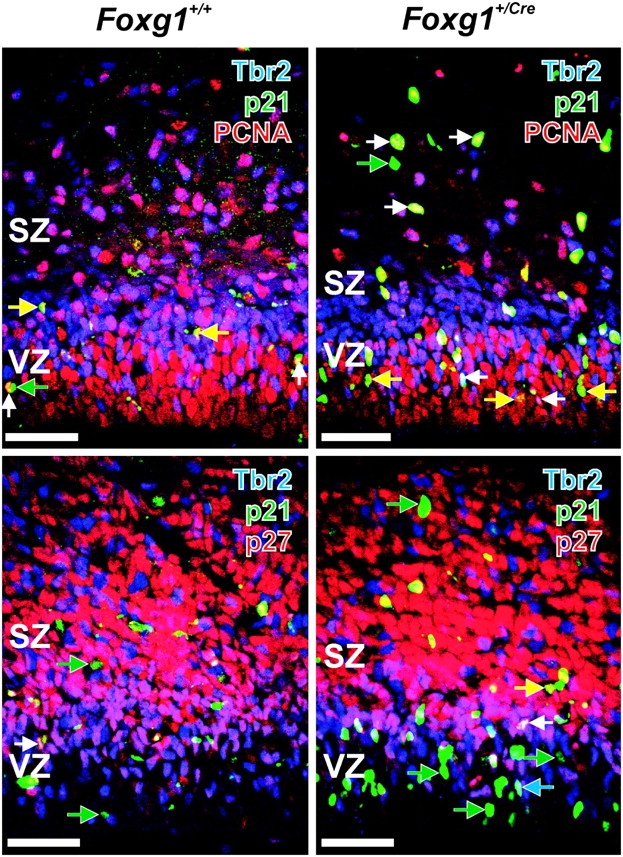
Expression of Tbr2 and p21 in proliferating and postproliferative cells. (Top) Sections of brains from 16.5-day-old Foxg1+/+ (left) and Foxg1+/Cre (right) fetuses were immunolabeled with antibodies to Tbr2 (blue), p21 (green), and PCNA (red), a marker of proliferating cells. Tbr2+/p21+/PCNA+ cells are indicated by white arrows, Tbr2−/p21+/PCNA+ cells are indicated by yellow arrows, and green arrows indicate Tbr2−/p21+/PCNA− cells. (Bottom) Most p21+ cells, both Tbr2- (green arrow) and Tbr2+ (blue arrows) did not coexpress p27, though examples of p21–p27 coimmunoreactivity were present (white and yellow arrows). Scale bars are 100 μm.
p27 is a CKI expressed in cortical progenitors (Delalle et al. 1999; Nguyen, et al. 2006a; Nguyen, et al. 2006b). It regulates cell-cycle exit in the cortical proliferative zones (Goto et al. 2004) and can be coexpressed with p21 by VZ cells (Siegenthaler and Miller 2005). The overall expression of p27 was similar in the 2 genotypes (Fig. 7B). Note that 1) most p21+ VZ cells in Foxg1+/Cre mice did not express p27 (Supplementary Fig. 3), 2) in wild-type and heterozygous fetuses, many Tbr2+ cells at the VZ/SZ transition coexpressed p27, and 3) almost none of the Tbr2+ cells in the mid- and upper SZ coexpressed p27. The lack of overlap between p21 and p27 in IPCs in the VZ indicated that the 2 CKIs have disparate roles in regulating IPC fate in the developing cortex.
Discussion
Cortical development in Foxg1+/Cre mice is compromised. During early development, the cortical anlages in Foxg1+/+ and Foxg1+/Cre fetuses appear similar, but as corticogenesis proceeds, a thinner cortical plate becomes evident in Foxg1+/Cre mice. The emerging defect is embodied in a targeted reduction in supragranular cortex. This corroborates the data of Eagleson et al. (2007). The thinner cortical plate in Foxg1+/Cre mice derives from a loss of neuron-generating IPCs during mid- and late corticogenesis and, to a lesser extent, from a lengthening of the cell cycle in late corticogenesis. The smaller IPC population is linked with an increased expression of p21 in the VZ and SZ. Collectively, these data implicate Foxg1 as an important player in the expansion of the pool of cortical IPCs via regulation of specific cell-cycle proteins.
Methodological Considerations
Some qualifications regarding the role of Foxg1 are necessary. These regard the genetic manipulations—the eliminated (Foxg1) and inserted (Cre) genes.
Consensus is emerging that Foxg1+/− mice exhibit microencephaly, regardless of their genetic background or mode of generation (e.g., Shen et al. 2006; Eagleson et al. 2007). On the other hand, only Foxg1+/Cre mice backcrossed on a C56BL/6 background exhibit cortical thinning in the radial dimension. Unfortunately, no detailed examination of these heterozygous fetuses has been published, and published analyses of cortical size in the radial dimension are limited (Shen et al. 2006).
Cre homozygous mice can have problems in cell proliferation (Forni et al. 2006). Such effects are only evident in Cre+/+ mice. Their brains are microencephalic and can be hydrocephalic. In heterozygous mice (such as those used in the present study), however, there is no evidence of gross anatomical changes in the brain, a change in the frequency of cycling cells in the SZ, or in the frequency of late-generated neurons labeled by an injection of BrdU on E19.5. Indeed, the gross and microscopic appearance of the brains of heterozygous mice is indistinguishable from wild-type mice.
Thus, the implications are 1) that backcrossing reveals the effects of Foxg1 insufficiency and 2) that one copy of Cre does not interfere with the proliferation of neuronal progenitors and action of Foxg1. Nevertheless, it cannot be dismissed that reduced amounts of Foxg1 and Cre in congenic mice conspire together to cause the cortical defects described in the present study.
Cell-Cycle Length
As corticogenesis progresses in normal mice (E10.5–E17.5; Caviness 1982) and rats (E11–E21; Miller 1985; 1988a; 1988b; Al-Ghoul and Miller 1989), the TC increases (Miller and Kuhn 1995; Takahashi et al. 1999; Tarui et al. 2005). There is also a lengthening of the TC in Foxg1 haploinsufficient and null mice relative to wild-type controls (Hanashima et al. 2002, 2004; Martynoga et al. 2005; present study). In the VZ of Foxg1−/− mice, there is a considerable increase in the TC early in corticogenesis (E13.5), which at least partially contributes to the reduction in cell proliferation in the neuroepithelium (Xuan et al. 1995; Hanashima et al. 2002, 2004; Martynoga et al. 2005).
The present study shows that the TC is longer in Foxg1+/Cre mice than in wild-type littermates, but only late in corticogenesis (e.g., E17.5). The late increase in the TC may contribute to the decreased number of neurons in superficial cortex of Foxg1+/Cre brains. The disparity between Foxg1+/+ and Foxg1+/Cre littermates in the thickness of the cerebral wall is evident days before the genotype-induced differences in the TC emerge. Therefore, the increase in the TC must be part of a broader dysregulation of neuronal generation in the heterozygous animals. Further, the increase in cell-cycle length is an unlikely cause for the observed reduction in IPC production in Foxg1+/Cre brains as the increase in the TC occurs later in cortical development.
Foxg1-mediated activities apparently affect cell proliferation during a specific phase of the cell cycle. Given that the lengths of G2+M and S are not affected in Foxg1+/Cre mice (Martynoga et al. 2005; present study), the observed increase in TC can largely be ascribed to G1. The increase in G1 is possibly due to the temporal increase in p21 expression in the heterozygotes. Indeed, there was more than a 3-fold increase in p21 expression between E13.5 and E16.5 in the Foxg1+/Cre fetal cortices. Increased expression of p21 can lengthen as well as block G1 progression through inhibition of protein complexes critical for the transition from G1 to S (Sherr and Roberts 1999).
Sites of Cortical Neuronogenesis
There is controversy about the contribution of the VZ and SZ to the generation of neocortical neurons. Some argue that the VZ is the sole or primary site of neuronal origin and that the SZ generates glia and glial precursors (e.g., Caviness et al. 1995, 2003; Gomes et al. 2003; Samuelsen et al. 2003). Others contend that the SZ is the predominant site of neuronal generation and that the VZ is merely a self-replacing population (fetal cortical neurons: Martens et al. 2000; adult telencephalic neurons: Doetsch et al. 1997, 1999; Alvarez-Buylla and Garcia-Verdugo 2002). A third concept is that the VZ and SZ produce cortical neurons, specifically neurons in deep and superficial laminae, respectively (Miller 1989, 1992, 1996; Miller and Nowakowski 1991; Tarabykin et al. 2001; Nieto et al. 2004; Zimmer et al. 2004; Englund et al. 2005; Ferrere et al. 2006; Martinez-Cerdeno et al. 2006).
Data from the present study provide independent in vivo support for the concept that the VZ and SZ generate cortical neurons. In Foxg1+/Cre mice, there is a targeted decrease in the number of cycling SZ cells that becomes evident during late-cortical neuronogenesis. This is coupled with a specific reduction in the size of layer II/III. Thus, as with the paired changes in the SZ and alterations in supragranular neurons following fetal ethanol exposure (Miller 1988a, 1989), these correlative data link SZ cells and layer II/III neurons. After all, layer II/III neurons are among the last cortical neurons to be generated (Angevine and Sidman 1961; Brückner et al. 1976; Caviness 1982; Miller 1988b). This linkage is further supported by the contributions of an increase in the TC and p21 expression occurring during late cortical neuronogenesis to the reduction in supragranular cortex.
The interplay of activity in the VZ and SZ is highly choreographed. Most radial glia have cell bodies in the VZ, where they generate daughter cells that either continue to proliferate in the VZ, transition to the SZ as proliferative IPCs, or become postmitotic neurons (Malatesta et al. 2000; Noctor et al. 2001, 2004; Tarabykin et al. 2001; Miyata et al. 2004; Nieto et al. 2004; Zimmer et al. 2004; Englund et al. 2005). Most IPCs in the SZ divide symmetrically to produce a pair of neurons that migrate to the cortical plate (Miyata et al. 2004; Noctor et al. 2004). This second, neuron-generating division in the SZ is an important feature of corticogenesis as it substantially increases the number of neurons generated by a single stem cell. Thus, IPCs provide a major source of cortical neurons during mid- and late neuronogenesis when layer II/III neurons are generated.
The population of IPCs is reduced in the Foxg1 heterozygotes throughout cortical neuronogenesis. This occurs despite no change in the number of Ki-67+ neuronal progenitors in the VZ. Preservation of the size of the VZ (and presumably the total number of VZ cells) in the heterozygotes indicates that the numbers of neuronal progenitors remaining in the VZ or moving out of the VZ are not altered. What is affected is the fate of the transitioning cells (i.e., transient, proliferating IPCs vs. young, postmitotic neurons). The observed reduction in IPC production in the Foxg1+/Cre mice suggests that more cells in their final cell cycle within the Foxg1+/Cre VZ do not transit to the SZ as IPCs. Instead, the cells undergo terminal differentiation and migrate to the cortical plate. The selective reduction in upper layer, Brn2+ cells in Foxg1+/Cre brains supports this contention. The reduced population of Brn2+ cells is evidence of the loss of the neuron-generating potential of cortical IPCs in the Foxg1+/Cre fetal brains. On the other hand, it does not support the only other alternative fate for these cells, cell-cycle reentry by the VZ cells, and the potential for an increase in the number of upper layer neurons.
Role of CKIs
p21 is linked to cell-cycle exit during cortical development (Siegenthaler and Miller 2005). Reduction in the population of IPCs in the cortices of Foxg1+/Cre mice coincides with an increase in the overall expression of p21 and in IPCs in cortical proliferative zones compared with wild-type controls. These increases are potentiated during the latter part of cortical neuronogenesis. Importantly, increased p21 expression is evident in proliferating VZ cells both before and after cells commit to becoming IPCs (i.e., expression of Tbr2). Thus, inappropriate p21 expression in the Foxg1+/Cre mice has the potential to induce cell-cycle exit both in determined IPC cells and in VZ progenitors, which under normal circumstances would become IPCs.
Dysregulation of p21 in the Foxg1+/Cre mice is potentially due to a reduction in inhibition of transforming growth factor (TGF) β signaling relative to Foxg1+/Cre mice. TGFβ1 stimulates p21 expression in VZ cells, Foxg1 can potently inhibit this TGFβ-dependent regulation, and Foxg1-null mice exhibit increased telencephalic p21 expression (Dou et al. 2000; Rodriguez et al. 2001; Seoane et al. 2004; Siegenthaler and Miller 2005, 2007). Thus, the loss of a single Foxg1 allele and subsequent increase in p21 expression (present study) implicate that Foxg1 controls the expansion of the IPC population in the SZ as it transitions from the VZ by suppressing TGFβ signaling.
Interestingly, the present data do not show that p21 induces cell-cycle exit in “permanent” VZ progenitors. After all, the size of the VZ population is unaffected in the Foxg1+/Cre mice and the temporal increase in p21 expression in both genotypes matches the growth of the IPC population. Thus, these 2 changes appear to be linked. p21 expression in Foxg1+/+ and Foxg1+/Cre cortices may be limited to a subpopulation of VZ cells on the verge of permanently exiting the VZ. There is growing evidence for heterogeneity in the VZ in that selective protein expression and differences in cell-cycle length distinguish cells that exit the cell cycle from cells that remain in the VZ (Iacopetti et al. 1999; Haubensak et al. 2004; Calegari et al. 2005).
The expression of the p27 in IPCs in the cortices of wild-type and Foxg1 haploinsufficient mice is noteworthy. Many IPCs at the VZ/SZ border coexpress p27, but IPCs in the SZ proper do not. Thus, it appears that initiating p27 expression forces IPCs out of the cell cycle, whereas IPCs that do not turn on p27 successfully transit to the SZ where they continue to cycle. Interestingly, overexpression of p27 leads to a thinning of neocortex reminiscent of the Foxg1+/Cre cortex, including a selective loss in supragranular cortex (Tarui et al. 2005). Furthermore, p27-null mice have thicker cortices and specifically an increase in neuronal numbers in layer II/III (Goto et al. 2004).
Summary
The Foxg1 haploinsufficient mouse provides invaluable insight into mechanisms of cortical development. It shows 1) that neurons in layer II/III are generated from IPCs in the SZ, 2) that Foxg1 regulates the expansion of IPCs, and 3) that the expansion of IPCs is likely mediated through the suppression of p21-promoted cell-cycle exit. An important conclusion based on these findings is that the determination of a supragranular fate is a Foxg1-dependent activity.
Supplementary Material
Supplementary figures 1–4 can be found at: http://www.cercor.oxfordjournals.org/
Funding
Funding for this work came from the National Institute of Alcohol Abuse and Alcoholism (AA06916 and AA07568) and the Department of Veterans Affairs.
Supplementary Material
Acknowledgments
Conflict of Interest: None declared.
References
- Al-Ghoul WM, Miller MW. Transient expression of Alz-50-immunoreactivity in developing rat neocortex: a marker for naturally occurring neuronal death? Brain Res. 1989;481:361–367. doi: 10.1016/0006-8993(89)90815-9. [DOI] [PubMed] [Google Scholar]
- Alvarez-Buylla A, Garcia-Verdugo JM. Neurogenesis in adult subventricular zone. J Neurosci. 2002;3:629–634. doi: 10.1523/JNEUROSCI.22-03-00629.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angevine JB, Jr, Sidman RL. Autoradiographic study of cell migration during histiogenesis of cerebral cortex in the mouse. Nature. 1961;192:766–768. doi: 10.1038/192766b0. [DOI] [PubMed] [Google Scholar]
- Brückner G, Marěs V, Biesold D. Neurogenesis in the visual system of the rat. An autoradiographic investigation. J Comp Neurol. 1976;166:245–255. doi: 10.1002/cne.901660208. [DOI] [PubMed] [Google Scholar]
- Calegari F, Haubensak W, Haffner C, Huttner WB. Selective lengthening of the cell cycle in the neurogenic subpopulation of neural progenitor cells during mouse brain development. J Neurosci. 2005;25:6533–6538. doi: 10.1523/JNEUROSCI.0778-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caviness VS., Jr Neocortical histogenesis in normal and reeler mice: a developmental study based upon [3H]thymidine autoradiography. Brain Res. 1982;256:293–302. doi: 10.1016/0165-3806(82)90141-9. [DOI] [PubMed] [Google Scholar]
- Caviness VS, Jr, Goto T, Tarui T, Takahashi T, Bhide PG, Nowakowski RS. Cell output, cell cycle duration and neuronal specification: a model of integrated mechanisms of the neocortical proliferative process. Cereb Cortex. 2003;13:592–598. doi: 10.1093/cercor/13.6.592. [DOI] [PubMed] [Google Scholar]
- Caviness VS, Jr, Takahashi T, Nowakowski RS. Numbers, time and neocortical neuronogenesis: a general developmental and evolutionary model. Trends Neurosci. 1995;18:379–383. doi: 10.1016/0166-2236(95)93933-o. [DOI] [PubMed] [Google Scholar]
- Delalle I, Takahashi T, Nowakowski RS, Tsai LH, Caviness VS., Jr Cyclin E-p27 opposition and regulation of the G1 phase of the cell cycle in the murine neocortical PVE: a quantitative analysis of mRNA in situ hybridization. Cereb Cortex. 1999;9:824–832. doi: 10.1093/cercor/9.8.824. [DOI] [PubMed] [Google Scholar]
- Doetsch F, Garcia-Verdugo JM, Alvarez-Buylla A. Cellular composition and three-dimensional organization of the subventricular germinal zone in the adult mammalian brain. J Neurosci. 1997;13:5046–5061. doi: 10.1523/JNEUROSCI.17-13-05046.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetsch F, Garcia-Verdugo JM, Alvarez-Buylla A. Regeneration of a germinal layer in the adult mammalian brain. Proc Natl Acad Sci USA. 1999;20:11619–11624. doi: 10.1073/pnas.96.20.11619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou C, Lee J, Liu B, Liu F, Massagué J, Xuan S, Lai E. BF-1 interferes with transforming growth factor β signaling by associating with Smad partners. Mol Cell Biol. 2000;20:6201–6211. doi: 10.1128/mcb.20.17.6201-6211.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou CL, Li S, Lai E. Dual role of brain factor-1 in regulating growth and patterning of the cerebral hemispheres. Cereb Cortex. 1999;9:543–550. doi: 10.1093/cercor/9.6.543. [DOI] [PubMed] [Google Scholar]
- Eagleson KL, Schlueter McFadyen-Ketchum LJ, Ahrens ET, Mills PH, Does MD, Nickols J, Levitt P. Disruption of Foxg1 expression by knock-in of Cre recombinase: effects on the development of the mouse telencephalon. Neuroscience. 2007;148:385–399. doi: 10.1016/j.neuroscience.2007.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, Kowalczyk T, Hevner RF. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci. 2005;25:247–251. doi: 10.1523/JNEUROSCI.2899-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrere A, Vitalis T, Gingras H, Gaspar P, Cases O. Expression of Cux-1 and Cux-2 in the developing somatosensory cortex of normal and barrel-defective mice. Anat Rec A Discov Mol Cell Evol Biol. 2006;288:158–165. doi: 10.1002/ar.a.20284. [DOI] [PubMed] [Google Scholar]
- Forni PE, Scuoppo C, Imayoshi I, Taulli R, Dastrù W, Sala V, Betz UA, Muzzi P, Martinuzzi D, Vercelli AE, et al. High levels of Cre expression in neuronal progenitors cause defects in brain development leading to microencephaly and hydrocephaly. J Neurosci. 2006;26:9593–9602. doi: 10.1523/JNEUROSCI.2815-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes WA, Mehler MF, Kessler JA. Transgenic overexpression of BMP4 increases astroglial and decreases oligodendroglia lineage commitment. Dev Biol. 2003;255:164–177. doi: 10.1016/s0012-1606(02)00037-4. [DOI] [PubMed] [Google Scholar]
- Goto T, Mitsuhashi T, Takahashi T. Altered patterns of neuron production in the p27 knockout mouse. Dev Neurosci. 2004;26:208–217. doi: 10.1159/000082138. [DOI] [PubMed] [Google Scholar]
- Hanashima C, Li SC, Shen L, Lai E, Fishell G. Foxg1 suppresses early cortical cell fate. Science. 2004;303:56–59. doi: 10.1126/science.1090674. [DOI] [PubMed] [Google Scholar]
- Hanashima C, Shen L, Li SC, Lai E. Brain factor-1 controls the proliferation and differentiation of neocortical progenitor cells through independent mechanisms. J Neurosci. 2002;22:6526–6536. doi: 10.1523/JNEUROSCI.22-15-06526.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubensak W, Attardo A, Denk W, Huttner WB. Neurons arise in the basal neuroepithelium of the early mammalian telencephalon: a major site of neurogenesis. Proc Natl Acad Sci USA. 2004;101:3196–3201. doi: 10.1073/pnas.0308600100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert JM, McConnell SK. Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev Biol. 2000;222:296–306. doi: 10.1006/dbio.2000.9732. [DOI] [PubMed] [Google Scholar]
- Iacopetti P, Michelini M, Stuckman I, Oback B, Aaku-Saraste E, Huttner WB. Expression of the antiproliferative gene Tis21 at the onset of neurogenesis identifies single neuroepithelial cells that switch from proliferative to neuron-generating division. Proc Natl Acad Sci USA. 1999;96:4639–4644. doi: 10.1073/pnas.96.8.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malatesta P, Hartfuss E, Gotz M. Isolation of radial glial cells by fluorescent-activated cell sorting reveals a neuronal lineage. Development. 2000;127:5253–5263. doi: 10.1242/dev.127.24.5253. [DOI] [PubMed] [Google Scholar]
- Martens DJ, Tropepe V, van Der Kooy D. Separate proliferation kinetics of fibroblast growth factor-responsive and epidermal growth factor-responsive neural stem cells within the embryonic forebrain germinal zone. J Neurosci. 2000;20:1085–1095. doi: 10.1523/JNEUROSCI.20-03-01085.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Cerdeno V, Noctor SC, Kriegstein AR. The role of intermediate progenitor cells in the evolutionary expansion of the cerebral cortex. Cereb Cortex. 2006;(16 Suppl 1):i152–i161. doi: 10.1093/cercor/bhk017. [DOI] [PubMed] [Google Scholar]
- Martynoga B, Morrison H, Price DJ, Mason JO. Foxg1 is required for specification of ventral telencephalon and region-specific regulation of dorsal telencephalic precursor proliferation and apoptosis. Dev Biol. 2005;283:113–127. doi: 10.1016/j.ydbio.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Miller MW. Co-generation of projection and local circuit neurons in neocortex. Dev Brain Res. 1985;23:187–192. doi: 10.1016/0165-3806(85)90040-9. [DOI] [PubMed] [Google Scholar]
- Miller MW. Effect of prenatal exposure to ethanol on the development of cerebral cortex: 1. Neuronal generation. Alcohol Clin Exp Res. 1988a;12:440–449. doi: 10.1111/j.1530-0277.1988.tb00223.x. [DOI] [PubMed] [Google Scholar]
- Miller MW. Development of projection and local circuit neurons in cerebral cortex. In: Peters A, Jones EG, editors. Cerebral Cortex. Vol. 7. Development and Maturation of Cerebral Cortex. New York: Plenum; 1988b. pp. 133–175. [Google Scholar]
- Miller MW. Effects of prenatal exposure to ethanol on neocortical development: II. Cell proliferation in the ventricular and subventricular zones of the rat. J Comp Neurol. 1989;287:326–338. doi: 10.1002/cne.902870305. [DOI] [PubMed] [Google Scholar]
- Miller MW. Effects of prenatal exposure to ethanol on cell proliferation and neuronal migration. In: Miller MW, editor. Development of the central nervous system: effects of alcohol and opiates. New York: Wiley-Liss; 1992. pp. 47–69. [Google Scholar]
- Miller MW. Limited ethanol exposure selectively alters the proliferation of precursor cells in the cerebral cortex. Alcohol Clin Exp Res. 1996;20:139–143. doi: 10.1111/j.1530-0277.1996.tb01056.x. [DOI] [PubMed] [Google Scholar]
- Miller MW, Kuhn PE. Cell cycle kinetics in fetal rat cerebral cortex: effects of prenatal treatment with ethanol assessed by a cumulative labeling technique with flow cytometry. Alcohol Clin Exp Res. 1995;19:233–237. doi: 10.1111/j.1530-0277.1995.tb01497.x. [DOI] [PubMed] [Google Scholar]
- Miller MW, Nowakowski RS. Effect of prenatal exposure t ethanol on the cell cycle kinetics and growth fraction in the proliferative zones of fetal rat cerebral cortex. Alcohol Clin Exp Res. 1991;15:229–232. doi: 10.1111/j.1530-0277.1991.tb01861.x. [DOI] [PubMed] [Google Scholar]
- Miyata T, Kawaguchi A, Saito K, Kawano M, Muto T, Ogawa M. Asymmetric production of surface-dividing and non-surface-dividing cortical progenitor cells. Development. 2004;131:3133–3145. doi: 10.1242/dev.01173. [DOI] [PubMed] [Google Scholar]
- Muzio L, Mallamaci A. Foxg1 confines Cajal-Retzius neuronogenesis and hippocampal morphogenesis to the dorsomedial pallium. J Neurosci. 2005;25:4435–4441. doi: 10.1523/JNEUROSCI.4804-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L, Besson A, Heng JI, Schuurmans C, Teboul L, Parras C, Philpott A, Roberts J, Guillemot F. p27kip1 independently promotes neuronal differentiation and migration in the cerebral cortex. Genes Dev. 2006;20:1511–1524. doi: 10.1101/gad.377106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L, Besson A, Roberts J, Guillemot F. Coupling cell cycle exit, neuronal differentiation and migration in cortical neurogenesis. Cell Cycle. 2006;5:2314–2318. doi: 10.4161/cc.5.20.3381. [DOI] [PubMed] [Google Scholar]
- Nieto M, Monuki ES, Tang H, Imitola J, Haubst N, Khoury SJ, Cunningham J, Gotz M, Walsh CA. Expression of Cux-1 and Cux-2 in the subventricular zone and upper layers II-IV of the cerebral cortex. J Comp Neurol. 2004;479:168–180. doi: 10.1002/cne.20322. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kreigstein AR. Neurons derived from radial glial cells establish radial units in the neocortex. Nature. 2001;409:714–720. doi: 10.1038/35055553. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martinez-Cerdeno V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7:136–144. doi: 10.1038/nn1172. [DOI] [PubMed] [Google Scholar]
- Rodriguez C, Huang LJ, Son JK, McKee A, Xiao Z, Lodish HF. Functional cloning of the proto-oncogene brain factor-1 (BF-1) as a Smad-binding antagonist of transforming growth factor-β signaling. J Biol Chem. 2001;276:30224–30230. doi: 10.1074/jbc.M102759200. [DOI] [PubMed] [Google Scholar]
- Samuelsen GB, Larsen KB, Bogdanovic N, Laursen H, Graem N, Larsen JF, Pakkenberg B. The changing number of cells in the human fetal forebrain and its subdivisions: a stereologic analysis. Cereb Cortex. 2003;13:115–122. doi: 10.1093/cercor/13.2.115. [DOI] [PubMed] [Google Scholar]
- Seoane J, Le HV, Shen L, Anderson SA, Massagué J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell. 2004;117:211–223. doi: 10.1016/s0092-8674(04)00298-3. [DOI] [PubMed] [Google Scholar]
- Shen L, Nam HS, Song P, Moore H, Anderson SA. FoxG1 haploinsufficiency results in impaired neurogenesis in the postnatal hippocampus and contextual memory deficits. Hippocampus. 2006;16:875–890. doi: 10.1002/hipo.20218. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- Siegenthaler JA, Miller MW. Transforming growth factor β1 promotes cell cycle exit through the cyclin-dependent kinase inhibitor p21 in the developing cerebral cortex. J Neurosci. 2005;25:8627–8636. doi: 10.1523/JNEUROSCI.1876-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegenthaler JA, Miller MW. Generation of Cajal-Retzius neurons in the mouse forebrain is regulated by transforming growth factor β1-Fox signaling. Dev Biol. 2007 doi: 10.1016/j.ydbio.2007.09.036. doi:10.1016/j.ydbio.2007.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Goto T, Miyama S, Nowakowski RS, Caviness VS., Jr Sequence of neuron origin and neocortical laminar fate: relation to cell cycle of origin in the developing murine cerebral wall. J Neurosci. 1999;19:10357–10371. doi: 10.1523/JNEUROSCI.19-23-10357.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao W, Lai E. Telencephalon-restricted expression of BF-1, a new member of the HNF-3/fork head gene family, in the developing rat brain. Neuron. 1992;8:957–966. doi: 10.1016/0896-6273(92)90210-5. [DOI] [PubMed] [Google Scholar]
- Tarabykin V, Stoykova A, Usman N, Gruss P. Cortical upper layer neurons derive from the subventricular zone as indicated by Svet1 gene expression. Development. 2001;128:1983–1993. doi: 10.1242/dev.128.11.1983. [DOI] [PubMed] [Google Scholar]
- Tarui T, Takahashi T, Nowakowski RS, Hayes NL, Bhide PG, Caviness VS., Jr Overexpression of p27Kip1, probability of cell cycle exit, and laminar destination of neocortical neurons. Cereb Cortex. 2005;15:1343–1355. doi: 10.1093/cercor/bhi017. [DOI] [PubMed] [Google Scholar]
- Xuan S, Baptista CA, Balas G, Tao W, Soares VC, Lai E. Winged helix transcription factor BF-1 is essential for the development of the cerebral hemispheres. Neuron. 1995;14:1141–1152. doi: 10.1016/0896-6273(95)90262-7. [DOI] [PubMed] [Google Scholar]
- Zimmer C, Tiveron MC, Bodmer R, Cremer H. Dynamics of Cux2 expression suggests that an early pool of SVZ precursors is fated to become upper cortical layer neurons. Cereb Cortex. 2004;14:1408–1420. doi: 10.1093/cercor/bhh102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.