

Abstract
Brain function declines with age and is associated with diminishing mitochondrial integrity. The neuronal mitochondrial ultrastructural changes of young (4 months) and old (21 months) F344 rats supplemented with two mitochondrial metabolites, acetyl-L-carnitine (ALCAR, 0.2%[wt/vol] in the drinking water) and R-α-lipoic acid (LA, 0.1%[wt/wt] in the chow), were analysed using qualitative and quantitative electron microscopy techniques. Two independent morphologists blinded to sample identity examined and scored all electron micrographs. Mitochondria were examined in each micrograph, and each structure was scored according to the degree of injury. Controls displayed an age-associated significant decrease in the number of intact mitochondria (P = 0.026) as well as an increase in mitochondria with broken cristae (P < 0.001) in the hippocampus as demonstrated by electron microscopic observations. Neuronal mitochondrial damage was associated with damage in vessel wall cells, especially vascular endothelial cells. Dietary supplementation of young and aged animals increased the proliferation of intact mitochondria and reduced the density of mitochondria associated with vacuoles and lipofuscin. Feeding old rats ALCAR and LA significantly reduced the number of severely damaged mitochondria (P = 0.02) and increased the number of intact mitochondria (P < 0.001) in the hippocampus. These results suggest that feeding ALCAR with LA may ameliorate age-associated mitochondrial ultrastructural decay and are consistent with previous studies showing improved brain function.
Keywords: neuronal mitochondria, morphometry, electron microscopy, R-alpha-lipoic acid (LA), acetyl-L-carnitine (ALCAR), mitochondrial oxidation
Introduction
Mitochondrial dysfunction may be a principal underlying event in aging, including age-associated brain degeneration [1]. Mitochondria provide energy for basic metabolic processes and their decay with age impairs cellular metabolism and leads to a decline of cellular function. Mitochondrial membrane potential, respiratory control ratios and cellular oxygen consumption decline with age and correlate with increased oxidant production [1–3]. Mutations in genes that encode mitochondrial proteins could compromise mitochondria by altering components of the electron transport chain [4], resulting in inefficient electron transport and increased superoxide production [4–6]. The resultant oxidative damage to mitochondria may compromise their ability to meet the steady energy demands of the brain. Oxidized proteins accumulate with age [7, 8] and these may also cause mitochondrial inefficiencies, leading to unwanted oxidant formation. Mitochondrial complexes III and IV significantly increase their Km and decrease Vmax with age [9]. Chronic, accumulated oxidants most likely also cause increased damage and consume critical metabolites such as ubiquinone or small molecular weight antioxidants. The significant loss of cardiolipin in aging may be in part because of greater oxidative damage and/or reduced biosynthesis. Loss of cardiolipin, coupled with oxidation of critical thiol groups in key proteins, may adversely affect transport of substrates and cytochrome c oxidase activity [10] that are necessary for normal mitochondrial function. These changes more than likely hinder the ability of mitochondria to maintain their membrane potential. Mitochondrial dysfunction, which may be due to the accumulated damage that accompanies normal aging but is amplified by disease-specific factors, is probably a key step in the development, maturation and progression of neurodegenerative disease including Alzheimer's disease (AD) [11–14]. Damaged mitochondria are associated with decreased ATP production and increased reactive oxygen species production, both of which characterize AD [12, 14, 15]. Indeed, alterations of biochemical components and structural degradation of mitochondria are extensively reported in AD [14–17] and animal models that mimic human AD [13].
Multiple cellular and systemic changes occur with age. Mitochondrial membrane potential, cardiolipin level, respiratory control ratio and cellular O2 uptake are lower, and levels of oxidants/O2, neuronal RNA oxidation and mutagenic aldehydes from lipid peroxidation are higher in old rats (compared with young rats) [2, 13, 18–24]. Ambulatory activity and cognition decline with age [20, 22]. We previously reported that feeding acetyl-L-carnitine (ALCAR) with R-a-lipoic acid (LA) to old rats for a few weeks restores mitochondrial function, lowers oxidant production, neuronal RNA oxidation and mutagenic aldehydes, and increases rat ambulatory activity and cognition [20, 23, 24].
Accumulating evidence strongly suggests age-associated mitochondrial decline is present not only in neurons but also in all other brain cells (i.e. glia, endothelial cells and pericytes) in human AD [25, 26] and in animal models that mimic AD pathology [13, 17, 27]. For the present study, we employed qualitative and quantitative electron microscopic techniques to examine the age-associated effects on mitochondrial morphology in hippocampal neurons and the effect of long-term (3 months) feeding of ALCAR + LA to young and old rats [15, 17].
Materials and methods
Animals and experimental procedure
Male Fisher 344 rats were obtained from the NIH and reared in the Shock Center at the University of Texas Health Science Center at San Antonio on a 12-hr light/dark cycle. Upon arrival, the animals were transferred immediately into a barrier facility where they were individually housed in plastic cages with wire mesh floors in the Hazleton-Enviro Rack System (Hazleton System, Inc., Aberdeen, MD, USA) to maintain the specific pathogen-free conditions. All protocols utilized were reviewed and approved by the Institutional Animal Care and Use committee at the University of Texas Health Science Center at San Antonio. The animals were fed an AIN93M diet (Diets, Bethlehem, PA: USA) and acidified drinking water, pH 2.5. The following groups were used: (n = 10/group): 4-month-old young males and 21-month-old aging males: both fed either a normal diet or a normal diet with dietary supplementation [0.2% (wt/vol) ALCAR + 0.1% (wt/wt) LA]. The LA was fortified in AIN93M diet containing 0.15% dexlipotam (R-lipoic acid tris salt from Viatris, Germany); the ALCAR was given in drinking water containing 0.2% ALCAR hydrochloride salt (pH was adjusted to 2.5 as the control water). All groups were fed ad libitum for a period of 3 months. The food intake of each rat was measured every 3 days; body weight and food consumption were monitored and measured at 2-week intervals.
Perfusion fixation and tissue collection
After 3 months of feeding, the animals were perfused transcardially with 4% paraformaldehyde for at least 30–40 min to enable effective in situ hybridization as previously described [17]. The brains were harvested into a fresh portion of the same fixative for another 24 hrs for further processing of the tissue for electron microscopy. The perfusion procedure was performed as previously described [17].
Electron microscopy
After several subsequent washes with 0.1 M PBS, the left halves of the brains were trimmed by the Vibratome to produce coronal sections. They were post-fixed with 2.5% glutaraldehyde in 0.1 M PBS for 1 hr washed again with 0.1 M PBS, and then exposed to 1% osmium tetraoxide in cacodylate buffer, pH 7.3, for 1 hr and then processed for standard transmission electron microscopy as described earlier [13–15, 17, 28]. Finally the tissues were dehydrated and embedded in Spurr's embedding media. Randomly selected ultrathin sections of the CA1 and CA3 areas were stained with uranyl acetate and lead citrate and examined using a transmission electron microscope (Jeol 100CX or Jeol 1200CX).
Classification of mitochondrial damage and mitochondrial quantification
For the double-blind analysis, all specimens were randomized and micrographs were taken of at least 50 neurons of each group in the rat hip-pocampal tissue on a plane containing the nucleolus at a magnification of 5000x and additionally at 20,000x so that a montage, including the entire cytoplasm, could be made. Randomized photomicrographs were examined with a stereomicroscope at 10–20x. Two independent morphologists: blinded to sample identity, classified and counted mitochondria and the counts were averaged. Quantitative analysis was performed using 5 rats from the YC, YT, OC and OT groups.
We have used two of our previously described criteria to classify mitochondrial damage [17, 28, 29]. The first, a 5-category classification system used to determine structural features of mitochondrial damage, has been applied previously by our groups [29] and includes intact (‘normal’) mitochondria, electron-dense mitochondria, hypertrophic (‘giant’) mitochondria, partially damaged mitochondria and completely damaged mitochondria or mitochondria-derived lysosomal structures (approximately 0, 25, 50, 75 and 100%, respectively, loss of identifiable cristae). The second, a 4-category system is consistent with our analysis of human AD biopsy samples and includes intact, broken cristae, vacuoles associated with lipofuscin and lipofuscin near vacuolar remnants [15]. The total was the sum of intact mitochondria and mitochondria with broken cristae. Each structure was outlined and the area was determined (NIH Image J program) and compared to the total cytoplasmic area excluding the nucleus [15].
Statistical analysis
The statistical analysis for the 5-category data was performed using oneway anova with Newman-Keuls’post hoc tests and the 4-category data were analysed using Student's t-test as well as two-way anova (Ftest). SPSS for Windows was used for the analyses.
Results
Our previous investigations demonstrated that profound structural changes in neurons affected by age and/or disease occurred in the cell body [13, 15, 30]. Therefore, in this study, we focused on mitochondria in neuronal cell bodies in the hippocampal region of the test rats. Neurons from young rats treated with ALCAR + LA dietary supplementation (YT) showed slight but significant (P = 0.02) improvement of mitochondrial integrity (Fig. 1) in comparison to young untreated rats (YC). In contrast, neurons from old control (OC) animals showed a range of mitochondrial abnormalities: the presence of giant mitochondria, clusters of mitochondria with electron-dense matrices, mitochondria with partially and/or completely damaged cristae, and the presence of membrane disruptions (see Figs 1B, 2A-D). Electron-dense mitochondria were characterized by decreased size and were usually seen close to the perinuclear region of the cell body. Single myelin-like structures with osmiphilic granules in the mitochondrial matrix were occasionally present. A double membrane surrounding lysosome-like structures indicates that they derive from damaged mitochondria (Figs 2–4) [15, 17, 26, 28].
1.
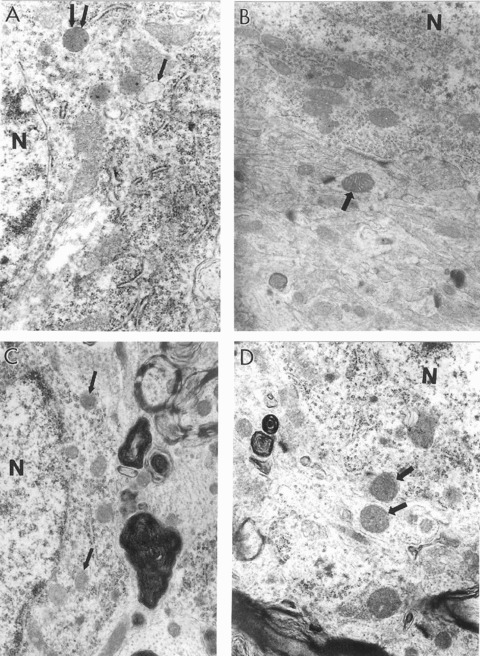
Ultrastructural characteristics of hippocampal neurons in young and old rats with and without 0.2% ALCAR and 0.1% LA diet supplementation. (A) The cell body from a young control rat hippocampus shows the presence of a large number of normal mitochondria. Some of the mitochondria however show minimal changes in their matrix (single arrows indicate the presence of intramitochondrial oedema; double arrows indicate electron dense mitochondria). (B) Mitochondria from the neu-ronal cell body of YT rat show completely intact morphology (arrow indicates normal healthy mitochondrion) (×15,000). (C) The main features of neuronal abnormality in aged control rats appear to be the presence of a range of mitochondria lesions. Some mitochondria however still show intact morphology (single arrows). A giant lipofuscin deposit was close to this neuronal cell body (×15,000). (D) Hippocampal neurons from the OT group are characterized by the absence of mitochondria degeneration. Single arrows indicate normal mitochondria. Lipofuscin was also absent in the neuronal cell body as well as in other cellular compartments in this group of experiments (×15.000). N, nucleus.
2.

Features of hippocampal neurons from old control rats. (A) A neuron under lower magnification. The neuronal cell body shows different degrees of mitochondrial abnormality (×5000). (B) A giant mitochondrion (arrow) is present in the bottom central portion of this neuron seen under high magnification. Many mitochondria are at the stage of transforming to mitochondrial-derived lysosomal structures (×15,000). (C) Another neuron under the lower magnification. A large lipofuscin deposit characterizes the cell body (X5000). (D) Neuronal cell body under high magnification. Giant mitochondria (arrows) and mitochondria transforming to mitochondrial-derived lysosomes (that appear to be part of lipofuscin formation) are consistent features in this neuron (x15,000). N, nucleus.
Hippocampal neuronal cell bodies of YC animals occasionally had mitochondria characterized by transitional, minimal changes in their ultrastructure (i.e. intra-mitochondrial oedema or an electron-dense matrix) (Fig. 1), indicating a spectrum of degeneration from normal to vacuolar lipofuscin. However, a majority of neurons showed mitochondria with intact morphology without any visible alterations. Hippocampal neurons of old rats are always characterized by a range of mitochondrial alterations, though some mitochondria still show intact morphology. The presence of mitochondria with varying degrees of ultrastructural damage appeared to be a permanent feature of the neurons of older animals. The formation of lipofuscin and/or lysosomal structures, characterized by the presence of an osmiphilic electron-dense matrix, often occupied much of the cytoplasmic matrix of OC hippocampi.
One key observation in our study is that the ALCAR + LA supplementation diet greatly reduced the mitochondrial damage, but also reduced formation of lipofuscin and/or myelin-like structures in neurons (Fig. 2C and D). In OT groups, lipofuscin granules were seen only occasionally in the neuronal cell bodies in any of the animals included in our present study (Fig. 6).
6.
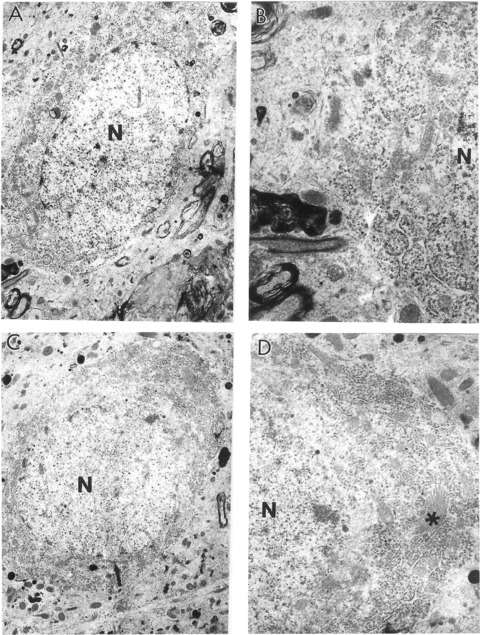
The characteristics of hippocampal neurons from aged rats treated with 0.2% ALCAR and 0.1% LA for 3 months. (A) A neuron under low magnification (×6000). (B) The cell body of this neuron under higher magnification. Only some mitochondria show partial damage to their cristae (×15,000). (C) Other neurons from this rat. The main feature of this neuron is the absence of lipofuscin in the neuronal cell body (×5000). (D) Top portion of this neuronal cell body under higher magnification. The single asterisk indicates the Golgi apparatus. No abnormalities were seen in the Golgi structure. Additional features of this neuron appear to be well developed granular and agranular endoplasmic reticu-lum and the presence of free ribosomes in the matrix of the cell body (×10,000).N, nucleus.
An age-associated characteristic of the mitochondrial abnormality seen in old rat brain was the presence of enlarged (giant) mitochondria (Fig. 3C and D). Neuronal cell bodies from OT rats showed fewer giant mitochondria than age-matched controls. Another abnormality seen in OC rat hippocampal neurons was the presence of a large number of mitochondria with a highly electron-dense matrix (Figs 4 and 5). However, OT rat hippocampal neurons lacked mitochondrial ultrastructural abnormalities, and most of the mitochondria appeared to be intact or with minimal damage. The ultrastructural pattern of the mitochondrial morphology was similar to that of the young control and ALCAR + LA dietary supplementation groups (Fig. 6).
3.

Features of mitochondrial abnormalities in aged OC rat hippocampi. (A) Normal mitochondria (arrows) in neuron under low magnification (×6000). (B) The cell body of this neuron under higher magnification. The cell body contains different populations of mitochondria including normal mitochondria (arrows). Microtubules coexist with normal but not damaged mitochondria (×15,000). (C) Another neuron from the hippocampal area under the low power. Double arrows indicate giant mitochondria (×5000). (D) Cell body of this neuron under higher magnification. Double asterisks indicate giant mitochondria (×15,000). N, nucleus.
4.
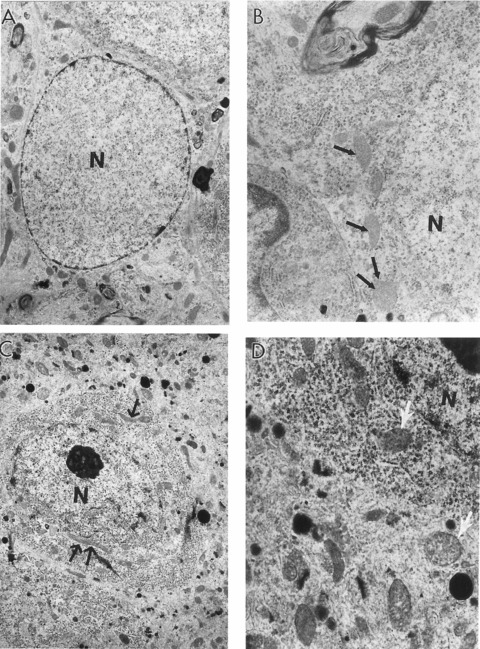
The features of aged rat hippocampal astrocytes and neurons. (A) Astrocytes under low magnification (×6000). (B) High magnification image of the cell bodies of this astrocytes reveals mitochondria with electron-dense matrices (indicated by single arrow) (×10,000). (C) A neuron under low magnification. The single and double arrows indicate different sized giant mitochondria (X5000). (D) The cell body of this neuron shows the presence of oedema in the mitochondrial matrix (single arrows) (×15,000). N, nucleus.
5.
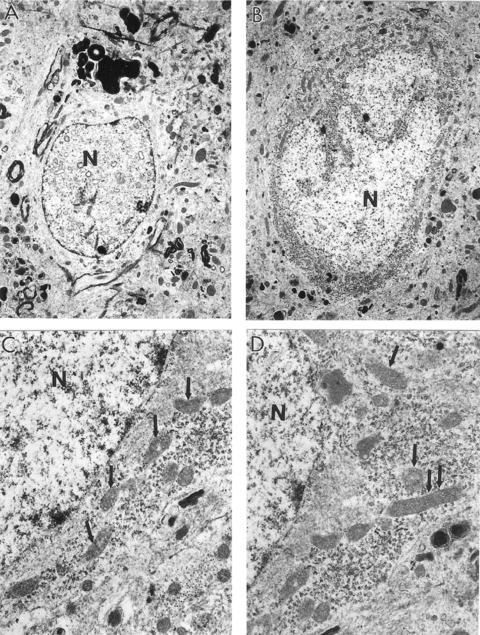
The heterogeneous morphology of mitochondria in aged OC rat hippocampal neurons. (A) The presence of vacuolar structures in the matrix of nuclei appears to be a feature of non-reversibly damaged neurons. The density of the cell body is notably less than that of non-damaged neurons (×5000). (B) Neurons under low magnification. Mitochondrial damage was found adjacent to areas of lipofuscin formation (right bottom portion of this neuron) (×5000). (C and D) Cell body under the higher magnification. Aged, neuronal mitochondria are characterized by increased electron density of their matrices. Oedema appeared to be a consistent feature of these neurons (Fig. C). The cell body contains a range of mitochondrial abnormalities. Hippocampal neurons display mitochondrial density transformations (single and double arrows indicate electron dense and giant mitochondria respectively) (×15,000, C and D).
The presence of vacuolar structures in the matrix of nuclei appears to be afeature of non-reversibly damaged neurons (Fig. 5A). Very often the mitochondrial damage colocalized with areas of lipofuscin formation (Fig. 5B). Aged neuronal mitochondria were characterized by increased electron density of their matrices. Oedema appeared to be a consistent feature of these neurons (Fig. 5C). The cell body contains a range of mitochondrial abnormalities (Fig. 5C and D).
The characteristics of hippocampal neurons from aged rats treated with 0.2% ALCAR and 0.1% LA treatment indicate that only some mitochondria show partial damage to their cristae (Fig. 6A and B). A feature of these neurons is the absence of lipofuscin in the neuronal cell bodies (Fig. 6C). No abnormalities were seen in the Golgi structure. Additional features of this neuron appear to be well developed granular and agranular endoplasmic reticulum and the presence of free ribosomes in the matrix of the cell body (Fig. 6D).
Mitochondrial damage was not limited to hippocampal neurons. Mitochondrial alterations were seen in other brain tissue cells. The aged rat hippocampal astrocytes also reveal mitochondria with electron-dense matrices (see Fig. 4A and B). Capillary vessels from YC rat hippocampi exhibited little ultrastructural damage (Fig. 7) in comparison to those of OC rats. OC animals displayed a range of ultrastructural lesions including the presence of giant lipofuscin granules and/or mitochondria-derived autophagic structures in the cytoplasmic matrix of perivascular and vascular endothelial cells. Contrary to this observation, vascular endothelial cells in OT rat hippocampus showed few changes in their ultrastructure. Perivascular cells occasionally displayed lipofuscin granules in the cytoplasmic matrix, but the number (P = 0.02) and density (P = 0.008) of lipofuscin granules were significantly less than those of OC animals (see below).
7.
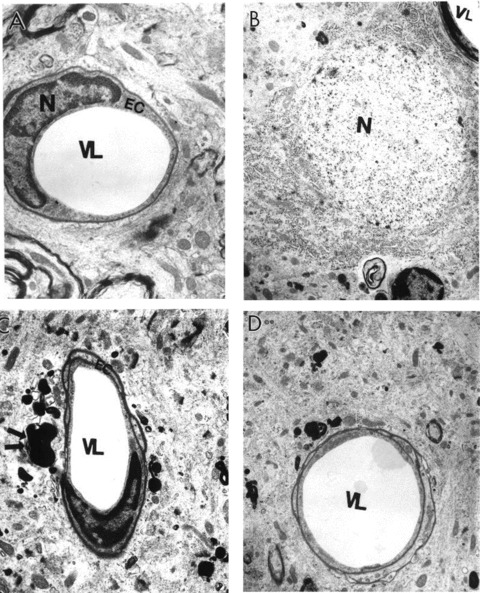
Ultrastructural characteristics of microvessels in rat hippocampus with and without ALCAR + LA dietary supplementation. (A) Capillary vessels from young control non-treated hippocampal tissues had an absence of abnormalities in their ultrastructure. Mitochondria were intact and showed no abnormalities in the vascular endothelium or perivascular cellular compartments (×8000). (B) Young treated rat hippocampal microvessels were similar to young control non-treated animals, characterized by intact morphology (×6000). (C) Old non-treated rat hippocampal capillary vessel endothelium showed degenerative changes in their ultrastructure, which appeared to be membrane disruption and nuclear contraction. A giant lipofuscin formation derived from a completely damaged mitochondrion in the matrix of a perivascular cell was also seen (double arrow) (×6000). (D) A hippocampal capillary from old ALCAR + LA treated rat. Vascular endothelial and perivascular cells showed no damage in their ultrastructure. Occasionally, a single small lipofuscin formation was present in the matrix of perivascular cells (×6000). EC, endothelial cell; N, nucleus; VL, vessel lumen.
Quantification of the percentage of the different types of mitochondria is summarized in Figure 8. In general, mitochondrial morphology within each neuron was heterogeneous in all tested groups. ALCAR + LA treatment significantly ameliorated the age-associated decrease in the percentage of intact mitochondria in old rat brains (P = 0.009). Increased age-associated abnormal mitochondria were present in aged non-treated experimental animals (Fig. 8). The preventive improvement by the ALCAR + LA supplementation was seen in aged animals in stark contrast to OC groups (Fig. 8). Neurons of old rats treated with ALCAR + LA (OT), compared to OC animals, showed a significant decrease in the prevalence of damaged mitochondria (P < 0.001) and increased normal (intact) mitochondria (P = 0.02). The proportion of severely damaged mitochondria in old non-treated rats is significantly higher than that in young rats (P = 0.001); a significant improvement followed ALCAR + LA supplementation (P = 0.012).
8.

(A) The percentage of intact mitochondria in hippocampal neurons. Abbreviations: YC, young control non-treated rats; YT, young rats treated with ALCAR + LA dietary supplementation; OC, old control rats non-treated; OT, old rats treated with ALCAR + LA. (B) The effect of ALCAR + LA dietary supplementation on the distribution of the different types of mitochondria. Significance differences are indicated: *P < 0.05, treated groups compared to respective control; #P< 0.05, significant decrease compared to young control; ##P < 0.01, increase compared to young control.
We also quantitatively examined mitochondria and vacuolar lipofuscin from specimens obtained from YC, YT, OC and OT animals (Fig. 9). Morphometric analysis showed that the number of intact mitochondria is significantly decreased in old non-treated groups (P < 0.001). Significantly larger mitochondria with broken cristae were present in old control animals compared to old treated animals (P = 0.003) but no significant difference was determined in the size of mitochondria associated with vacuolar lipofuscin. Treatment significantly reduced the number of vacuole and lipofuscin formations in old rat hippocampal neurons (P = 0.015 and 0.02, respectively). Quantification of the area associated with different types of mitochondria revealed that ALCAR + LA treatment improved the percentage of area occupied by intact mitochondria in old (P = 0.028) as well as young treated groups (P = 0.009) compared to OC and YC, respectively. The percentage of area occupied by vacuoles was not significantly different in the experimental groups but the lipofuscin area was significantly lower in OT compared to OC (P = 0.008) (Fig. 9C).
9.
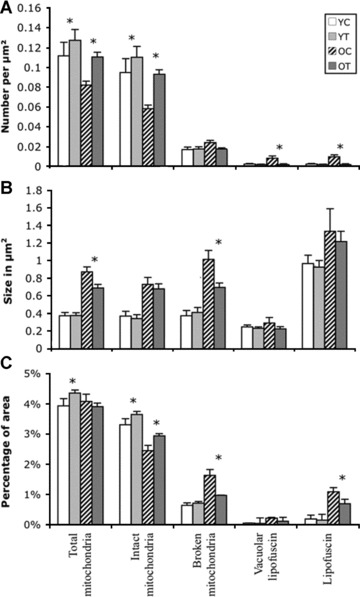
Morphometric quantification of (A) number, (B) size and (C) percentage coverage of cytoplasmic area by intact mitochondria, mitochondria with broken cristae, total mitochondria (intact plus mitochondria with broken cristae), vacuole associated lipofuscin, and lipofuscin alone in cases of young control (YC), young treated (YT), old control (OC) and old treated (OT). (A) Number of intact mitochondria is significantly lower in OC (P = 0.001). No significant change was seen in the number of mitochondria with broken cristae; however, the number of vacuoles associated with lipofuscin and lipofuscin alone were significantly higher in OC, which was significantly decreased with dietary supplementation. (B) A significant decrease in the average size of mitochondria with broken cristae was seen in old non-treated groups compared to treated animals. No significant difference was seen in the average size of intact mitochondria, lipofuscin-associated vacuoles or lipofuscin. (C) Treatment with ALCAR + LA supplementation significantly increased the percentage of area of intact mitochondria in old and young animals. However, a significant difference was seen in the percentage of area covered by mitochondria with broken cristae and lipofuscin only in OC compared to OT groups.
Discussion
This study examines for the first time the qualitative and quantitative pattern of mitochondria in hippocampal neurons affected by age and demonstrates the restoration of mitochondrial ultrastructure by the dietary supplementation of ALCAR + LA. Perhaps even more important is the observation that dietary supplementation stimulated mitochondrial proliferation in young animals, suggesting early interventions are important in fighting age-related decline.
An accumulating body of evidence strongly suggests that mitochondrial decay plays a key role in aging and age-associated neurodegenerative disease, such as AD [14, 17, 26, 28, 31]. Mutant mitochondrial DNA is implicated in impairing cognitive functioning [32] and cellular energy production, disrupting the normal connections between brain cells and increasing the generation of oxidants [33]. Further, beta-amyloid-binding alcohol dehydrogenase is a direct molecular link from amyloid p to mitochondrial toxicity [34]. A recent study of extending mouse lifespan with overexpression of catalase in mitochondria provides strong support to the oxidative mitochondrial decay theory of aging [35] and age-associated diseases, including mice that mimic human AD [17, 26]. However, no detailed quantitative electron microscopic study examines rat brain mitochondrial changes with age.
In the present study, the data indicate a significant decrease in the number and percentage of intact mitochondria and a significant increase in the number and percentage of damaged mitochondria (mitochondria with broken cristae, vacuoles and vacuolar lipofuscin) in aged hippocampal neurons. Neuronal damage is characterized by an increase in the number of vacuolar lipofuscin structures as well as the number of electron-dense and giant mitochondria, lysosomes, lipofuscin deposits, mitochondrial matrix oedema, internal and external membrane breaks and a decrease in the density of microtubules per neurons [36].
It is important to note that the tissues in our present study were fixed with paraformaldehyde. Moreover, previous studies also demonstrate the consistency of optimal tissue preservation by this technique and the consequent EM images [17]. In addition, our unpublished data from these experiments show that mitochondrial DNA overproliferation and/or deletion were dependent on mitochondrial structural damage seen in hippocampal neurons. Vibratome sections from each brain were used for the in situ hybridization study to compare the spectrum of mitochondrial structural and DNA damage and/or prevention by using selective mitochondrial antioxidants, consequently greatly reducing the risk of potential artifacts.
Feeding ALCAR + LA ameliorated the age-associated neuronal damage evident by marked decreases of vacuoles and lipofuscin as well as decreases in partially and completely damaged mitochondria. In our preliminary study using in situ hybridization of mitochondrial DNA, we have found that the main source (almost 100%) of lipofuscin formation appeared to be mitochondria because ‘young’ lipofuscin contains mtDNA positive signals. Well-developed granular and agranular endoplasmic reticulum and free ribosomes (or polysomes) are present in the matrix of cell bodies, which were often comparable to the ultrastructure of hippocampal neurons in young rat brains. In addition, ALCAR + LA dietary supplementation also increased the percentage of area covered by intact mitochondria not only in old groups but also in young treated animals, suggesting a much more significant effect of the ALCAR + LA dietary supplementation can be achieved if treatment is started at an early age.
Our observations support recent meta-analysis of 21 double blind clinical trials of ALCAR in the treatment of mild cognitive impairment and mild AD that showed modest but significant efficacy of ALCAR [37]. A meta-analysis of four clinical trials of LA for treatment of neuropathic deficits in diabetes showed modest efficacy [37]. In addition, immune function decline with age has been extensively documented in human beings [38, 39] and in rodents [40, 41]. The deleterious effects of oxidants on immune cells such as T and B lymphocytes were ameliorated by ALCAR treatment in aged animals [42, 43]. Moreover, Franceschi and colleagues demonstrated that ALCAR treatment increased phyto-haemagglutinin-induced peripheral blood lymphocyte proliferation in young and old subjects [44] and LA, as an antioxidant, may have protective effects on immune cells leading to improved immune system functioning in general.
A recent study by McMackin and coworkers in a double-blind crossover study showed the effect of combined ALCAR + LA on vasodilator function and blood pressure in patients with coronary artery disease [45]. Active treatment increased brachial artery diameter and had a significant effect on individuals with either elevated blood pressure or metabolic syndrome [45]. Recent studies report protective effects of LA on endothelial function and the reduction of markers of inflammation in metabolic syndrome and in patients with diabetic syndrome, albuminuria, and symptomatic diabetic polyneuropathy as well as in aging rats. [27, 46–48]. Metabolic function as a possible key factor in the mitochondrial decay seen in aging and micronutrient deficiency has been discussed [49]. Future studies are needed to determine in more detail the potential protective effect of ALCAR + LA treatment in different experimental models such as stroke and cere-brovascular disease.
We previously reported preliminary qualitative electron microscopic observations of age-associated mitochondrial morphological decay and the positive effects of ALCAR and/or LA in old rat brain [22, 23, 44]. Compared with young rats, old rats showed some disruption and loss of cristae in about half of the mitochondria in the dentate gyms area, indicating structural decay. Animals treated with 0.5% ALCAR and/or 0.2% LA showed preservation of cristae and less structural disruption compared to controls. Formation or accumulation of lipofuscin granules in the cytoplasmic matrix of hippocampal neurons and other AD brain cell types [15, 50, 51] and rodent brain samples demonstrating AD-like pathology [13, 17,52] appears to be a feature of mitochondrial damage associated with age and especially disease. As a major source of reactive oxygen species, mitochondria are particularly vulnerable to oxidative stress [15, 50, 51]. A recent morphometric study found a significant reduction in intact mitochondria in different cellular compartments of AD and AD-like rodent brain [13,15, 17] and other cells (e.g. fibroblasts) obtained from patients with AD [53]. In addition, old rats had more lipofuscin in the cytoplasm of granular cells of the dentate gyrus, and the combined-treatment rats appeared to have less lipofuscin accumulation.
In the present study, we replicated our previous experiment with more animals for quantitative analysis of mitochondria in the hippocampus. The morphological observations clearly show that old rat brain is characterized by age-associated increases in the number of electron-dense and giant mitochondria, mitochondrial oedema, external membrane breaks, lysosomes, lipofuscin formation and decreases in the density of microtubules. Golgi, granular and agranular endoplasmic reticulum and microvessels structures display abnormalities. Treatment with ALCAR + LA for 3 months ameliorated the age-associated neuronal damage and increased proliferation of intact mitochondria in hippocampal neurons in the young animals.
Mitochondria are essential to the functions of neurons because their limited glycolytic capacity makes them highly dependent on aerobic oxidative phosphorylation. We and others demonstrated that oxidative stress is one of the earliest events in susceptible neurons and vascular endothelium in AD [13,14, 17, 25–27, 31] and mitochondrial dysfunction is implicated in the associated increased oxidative stress. Among other mitochondrial abnormalities, we found that the levels of mitochondrial DNA and cytochrome oxidase-1 in susceptible neurons and vascular endothelial cells are significantly increased compared with those of age-matched controls, even though the number of mitochondria per neuron is significantly decreased [15, 27]. The present results confirm our previous qualitative observation in aged rats and provide not only quantitative morphological confirmation of mitochondrial decay, but also suggest that the ameliorating effect of ALCAR + LA on age-associated activity and memory decline [20, 23, 54–58] is a result of repairing mitochondrial structure, thereby restoring mitochondrial function in aged animal brain. A preventive effect of ALCAR + LA supplementation was seen in all brain cellular compartments including microvascular systems, indicating a systemic effect of the treatment.
Our results demonstrate for the first time that ultrastructural morphometric analysis provides criteria for documenting mitochondrial damage that increases with aging. Treatment with ALCAR + LA restores intact mitochondrial morphology normally degraded with age. Moreover, quantitative electron microscopic observation for the first time demonstrated that with ALCAR + LA supplementation neuronal mitochondria from young treated animals show better intact morphology compared to young non-treated controls. Further investigation on the effect of ALCAR + LA supplementation by using electron microscopic quantitative/qualitative study will provide new information for the current model of aging as well as neurodegenerative diseases such as AD where mitochondrial damage and energy failure appear to be a primary step in the development of cognitive impairment or brain pathology such as amyloid beta deposition, a hallmark for AD. In conclusion, old rats showed morphological mitochondrial decay and dietary supplementation with ALCAR + LA restored mitochondrial morphology and prevented decay. Further, a key observation of this study is the proliferation of intact mitochondria in young treated rat brain neurons. These results support the role of mitochondrial decay as an important factor in aging. Further, dietary supplementation with selective mitochondrial antioxidants and metabolites such as ALCAR and LA may be an effective strategy for delaying aging, as well as neurodegeneration such as AD, where mitochondrial damage appears to be a primary target before the development of any amyloid deposition or cognitive impairment.
Abbreviations
ALCAR, acetyl-L-carnitine; AD, Alzheimer's disease; LA, R-α-lipoic acid; OC, old control; OT, old treated; YC, young control; YT, young treated; CD, completely destroyed; PD, partially damaged.
Acknowledgments
This work was supported by The Ellison Medical Foundation Grant SS-0422–99, the National Institute on Aging AG17140 (to B.N.A.), Alzheimer's Association IIRG (G.P.), and NIGMS MBRS-RISE GM 60655 (J.C.S.). We thank Sigma-Tau for acetyl carnitine and Viatris for R-lipoic acid.
References
- 1.de Grey ADJ. The mitochondrial free radical theory of aging. Georgetown, TX: R.G. Landers Company; 1999. [Google Scholar]
- 2.Hagen TM, Yowe DL, Bartholomew JC, Wehr CM, Do KL, Park JY, Ames BN. Mitochondrial decay in hepatocytes from old rats: membrane potential declines, heterogeneity and oxidants increase. Proc NatlAcad Sci USA. 1997;94:3064–9. doi: 10.1073/pnas.94.7.3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci USA. 1994;91:10771–8. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sohal RS, Mockett RJ, Orr WC. Mechanisms of aging: an appraisal of the oxidative stress hypothesis. Free Radic BiolMed. 2002;33:575–86. doi: 10.1016/s0891-5849(02)00886-9. [DOI] [PubMed] [Google Scholar]
- 6.Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273:59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stadtman ER. Protein oxidation and aging. Science. 1992;257:1220–4. doi: 10.1126/science.1355616. [DOI] [PubMed] [Google Scholar]
- 8.Stadtman ER, Levine RL. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids. 2003;25:207–18. doi: 10.1007/s00726-003-0011-2. [DOI] [PubMed] [Google Scholar]
- 9.Feuers RJ. The effects of dietary restriction on mitochondrial dysfunction in aging. Ann N Y Acad Sci. 1998;854:192–201. doi: 10.1111/j.1749-6632.1998.tb09902.x. [DOI] [PubMed] [Google Scholar]
- 10.Paradies G, Ruggiero FM, Petrosillo G, Gadaleta MN, Quagliariello E. Effect of aging and acetyl-L-carnitine on the activity of cytochrome oxidase and adenine nucleotide translocase in rat heart mitochondria. FEBS Lett. 1994;350:213–5. doi: 10.1016/0014-5793(94)00763-2. [DOI] [PubMed] [Google Scholar]
- 11.Castellani R, Hirai K, Aliev G, Drew KL, Nunomura A, Takeda A, Cash AD, Obrenovich ME, Perry G, Smith MA. Role of mitochondrial dysfunction in Alzheimer's disease. JNeurosci Res. 2002;70:357–60. doi: 10.1002/jnr.10389. [DOI] [PubMed] [Google Scholar]
- 12.Gibson GE, Sheu KF, Blass JP. Abnormalities of mitochondrial enzymes in Alzheimer disease. J Neural Transm. 1998;105:855–70. doi: 10.1007/s007020050099. [DOI] [PubMed] [Google Scholar]
- 13.Obrenovich ME, Smith MA, Siedlak SL, Chen SG, de la Torre JC, Perry G, Aliev G. Overexpression of GRK2 in Alzheimer disease and in a chronic hypoperfusion rat model is an early marker of brain mitochondrial lesions. Neurotox Res. 2006;10:43–56. doi: 10.1007/BF03033333. [DOI] [PubMed] [Google Scholar]
- 14.Zhu X, Smith MA, Perry G, Aliev G. Mitochondrial failures in Alzheimer's disease. Am J Alzheimer's Dis Other Demen. 2004;19:345–52. doi: 10.1177/153331750401900611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA. Mitochondrial abnormalities in Alzheimer's disease. J Neurosci. 2001;21:3017–23. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aliev G, Smith MA, de la Torre JC, Perry G. Mitochondria as a primary target for vascular hypoperfusion and oxidative stress in Alzheimer's disease. Mitochondrion. 2004;4:649–63. doi: 10.1016/j.mito.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 17.Aliyev A, Chen SG, Seyidova D, Smith MA, Perry G, de la Torre J, Aliev G. Mitochondria DNA deletions in atherosclerotic hypoperfused brain microvessels as a primary target for the development of Alzheimer's disease. J Neurol Sci. 2005;229–230:285–92. doi: 10.1016/j.jns.2004.11.040. [DOI] [PubMed] [Google Scholar]
- 18.Hagen TM, Ingersoll RT, Lykkesfeldt J, Liu J, Wehr CM, Vinarsky V, Bartholomew JC, Ames AB. (R)-alpha-lipoic acid-supplemented old rats have improved mitochondrial function, decreased oxidative damage, and increased metabolic rate. FASEB J. 1999;13:411–8. doi: 10.1096/fasebj.13.2.411. [DOI] [PubMed] [Google Scholar]
- 19.Hagen TM, Ingersoll RT, Wehr CM, Lykkesfeldt J, Vinarsky V, Bartholomew JC, Song MH, Ames BN. Acetyl-L-carnitine fed to old rats partially restores mitochondrial function and ambulatory activity. Proc Natl Acad Sci USA. 1998;95:9562–6. doi: 10.1073/pnas.95.16.9562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hagen TM, Liu J, Lykkesfeldt J, Wehr CM, Ingersoll RT, Vinarsky V, Bartholomew JC, Ames BN. Feeding acetyl-L-carnitine and lipoic acid to old rats significantly improves metabolic function while decreasing oxidative stress. Proc Natl Acad Sci USA. 2002;99:1870–5. doi: 10.1073/pnas.261708898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hagen TM, Vinarsky V, Wehr CM, Ames BN. (R)-alpha-lipoic acid reverses the age-associated increase in susceptibility of hepatocytes to tert-butylhydroperoxide both in vitro and in vivo. Antioxid Redox Signal. 2000;2:473–83. doi: 10.1089/15230860050192251. [DOI] [PubMed] [Google Scholar]
- 22.Liu J, Atamna H, Kuratsune H, Ames BN. Delaying brain mitochondrial decay and aging with mitochondrial antioxidants and metabolites. Ann N Y Acad Sci. 2002;959:133–66. doi: 10.1111/j.1749-6632.2002.tb02090.x. [DOI] [PubMed] [Google Scholar]
- 23.Liu J, Head E, Gharib AM, Yuan W, Ingersoll RT, Hagen TM, Cotman CW, Ames BN. Memory loss in old rats is associated with brain mitochondrial decay and RNA/DNA oxidation: partial reversal by feeding acetyl-L-carnitine and/or R-alpha-lipoic acid. Proc NatlAcad Sci USA. 2002;99:2356–61. doi: 10.1073/pnas.261709299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu J, Killilea DW, Ames BN. Age-associated mitochondrial oxidative decay: improvement of carnitine acetyltransferase substrate-binding affinity and activity in brain by feeding old rats acetyl-L-carnitine and/or R-alpha-lipoic acid. Proc Natl Acad Sci USA. 2002;99:1876–81. doi: 10.1073/pnas.261709098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aliev G, Smith MA, Seyidova D, Neal ML, Lamb BT, Nunomura A, Gasimov EK, Vinters HV, Perry G, LaManna JC, Friedland RP. The role of oxidative stress in the pathophysiology of cerebrovascular lesions in Alzheimer's disease. Brain Pathol. 2002;12:21–35. doi: 10.1111/j.1750-3639.2002.tb00419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aliev G, Seyidova D, Lamb BT, Obrenovich ME, Siedlak SL, Vinters HV, Friedland RP, LaManna JC, Smith MA, Perry G. Mitochondria and vascular lesions as a central target for the development of Alzheimer's disease and Alzheimer disease-like pathology in transgenic mice. Neurol Res. 2003;25:665–74. doi: 10.1179/016164103101201977. [DOI] [PubMed] [Google Scholar]
- 27.Aliev G, Seyidova D, Neal ML, Shi J, Lamb BT, Siedlak SL, Vinters HV, Head E, Perry G, LaManna JC, Friedland RP, Cotman CW. Atherosclerotic lesions and mitochondria DNA deletions in brain microvessels as a central target for the development of human AD and AD-like pathology in aged transgenic mice. Ann N Y Acad Sci. 2002;977:45–64. doi: 10.1111/j.1749-6632.2002.tb04798.x. [DOI] [PubMed] [Google Scholar]
- 28.Aliyev A, Seyidova D, Rzayev N, Obrenovich ME, Lamb BT, Chen SG, Smith MA, Perry G, de la Torre JC, Aliev G. Is nitric oxide a key target in the pathogenesis of brain lesions during the development of Alzheimer's disease. Neurol Res. 2004;26:547–53. doi: 10.1179/01610425017613. [DOI] [PubMed] [Google Scholar]
- 29.Aliev G, Cirillo R, Salvatico E, Paro M, Prosdocimi M. Changes in vessel ultra-structure during ischemia and reperfusion of rabbit hindlimb: implications for therapeutic intervention. Microvasc Res. 1993;46:65–76. doi: 10.1006/mvre.1993.1035. [DOI] [PubMed] [Google Scholar]
- 30.Aliev G, Smith MA, Obrenovich ME, Perry G. Role of vascular hypoperfusion-induced oxidative stress and mitochondrial failure in the pathogenesis of Alzheimer disease. Neurotox Res. 2003;5:385–90. doi: 10.1007/BF03033159. [DOI] [PubMed] [Google Scholar]
- 31.Zhu X, Perry G, Moreira PI, Aliev G, Cash AD, Hirai K, Smith VA. Mitochondrial abnormalities and oxidative imbalance in Alzheimer disease. J Alzheimer's Dis. 2006;9:147–53. doi: 10.3233/jad-2006-9207. [DOI] [PubMed] [Google Scholar]
- 32.Roubertoux PL, Sluyter F, Carlier M, Marcet B, Maarouf-Veray F, Cherif C, Marican C, Arrechi P, Godin F, Jamon M, Verrier B, Cohen-Salmon C. Mitochondrial DNA modifies cognition in interaction with the nuclear genome and age in mice. Nat Genet. 2003;35:65–9. doi: 10.1038/ng1230. [DOI] [PubMed] [Google Scholar]
- 33.Coskun PE, Beal MF, Wallace DC. Alzheimer's brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc Natl Acad Sci USA. 2004;101:10726–31. doi: 10.1073/pnas.0403649101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science. 2004;304:448–52. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 35.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–11. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 36.Cash AD, Aliev G, Siedlak SL, Nunomura A, Fujioka H, Zhu X, Raina AK, Vinters HV, Tabaton M, Johnson AB, Paula-Barbosa M, Avila J, Jones PK, Castellani RJ, Smith MA, Perry G. Microtubule reduction in Alzheimer's disease and aging is independent of tau filament formation. AmJPathol. 2003;162:1623–7. doi: 10.1016/s0002-9440(10)64296-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Montgomery SA, Thai LJ, Amrein R. Meta-analysis of double blind randomized controlled clinical trials of acetyl-L-carni-tine versus placebo in the treatment of mild cognitive impairment and mild Alzheimer's disease. Int Clin Psychopharmacol. 2003;18:61–71. doi: 10.1097/00004850-200303000-00001. [DOI] [PubMed] [Google Scholar]
- 38.Linton PJ, Dorshkind K. Age-related changes in lymphocyte development and function. Nat Immunol. 2004;5:133–9. doi: 10.1038/ni1033. [DOI] [PubMed] [Google Scholar]
- 39.Murasko DM, Goonewardene IM. T-cell function in aging: mechanisms of decline. Annu Rev Gerontol Geriatr. 1990;10:71–96. doi: 10.1007/978-3-662-38445-9_5. [DOI] [PubMed] [Google Scholar]
- 40.Pahlavani MA, Harris MD. Effect of dehydroepiandrosterone on mitogen-induced lymphocyte proliferation and cytokine production in young and old F344 rats. Immunol Lett. 1995;47:9–14. doi: 10.1016/0165-2478(95)00057-c. [DOI] [PubMed] [Google Scholar]
- 41.Pahlavani MA, Vargas DM, Guo Z, Richardson A. Normal immune function in young and old DNA polymerase-beta deficient mice. Immunol Lett. 2000;72:17–21. doi: 10.1016/s0165-2478(00)00159-0. [DOI] [PubMed] [Google Scholar]
- 42.Pahlavani MA, Harris MD. Effect of in vitro generation of oxygen free radicals on T cell function in young and old rats. Free Radic BiolMed. 1998;25:903–13. doi: 10.1016/s0891-5849(98)00124-5. [DOI] [PubMed] [Google Scholar]
- 43.Angelucci L, Ramacci MT. Hypothalamo-pituitaryadrenocortical function in aging: effects of acetyl-L-carnitine. In: De Simone C, Arrigoni-Martelli E, editors. Stress, immunity and ageing: a role for acetyl-L-carnitine. Amsterdam: Elsevier; 1989. pp. 109–18. [Google Scholar]
- 44.Franceschi C, Cossarizza A, Troiano L, Salati R, Monti D. Immunological parameters in aging: studies on natural immunomodulatory and immunoprotective substances. Int J Clin Pharmacol Res. 1990;10:53–7. [PubMed] [Google Scholar]
- 45.McMackin CJ, Widlansky ME, Hamburg NM, Huang AL, Weller S, Holbrook M, Gokce N, Hagen TM, Keaney JF, Jr, Vita JA. Effect of combined treatment with alpha-Lipoic acid and acetyl-L-carnitine on vascular function and blood pressure in patients with coronary artery disease. J Clin Hypertens (Greenwich) 2007;9:249–55. doi: 10.1111/j.1524-6175.2007.06052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sola S, Mir MQ, Cheema FA, Khan-Merchant N, Menon RG, Parthasarathy S, Khan BV. Irbesartan and lipoic acid improve endothelial function and reduce markers of inflammation in the metabolic syndrome: results of the Irbesartan and Lipoic Acid in Endothelial Dysfunction (ISLAND) study. Circulation. 2005;111:343–8. doi: 10.1161/01.CIR.0000153272.48711.B9. [DOI] [PubMed] [Google Scholar]
- 47.Morcos M, Borcea V, Isermann B, Gehrke S, Ehret T, Henkels M, Schiekofer S, Hofmann M, Amiral J, Tritschler H, Ziegler R, Wahl P, Nawroth PP. Effect of alpha-lipoic acid on the progression of endothelial cell damage and albuminuria in patients with diabetes mellitus: an exploratory study. Diabetes Res Clin Pract. 2001;52:175–83. doi: 10.1016/s0168-8227(01)00223-6. [DOI] [PubMed] [Google Scholar]
- 48.Ziegler D, Hanefeld M, Ruhnau KJ, Hasche H, Lobisch M, Schutte K, Kerum G, Malessa R. Treatment of symptomatic diabetic polyneuropathy with the antioxidant alpha-lipoic acid: a 7-month multi-center randomized controlled trial (ALADIN III Study). ALADIN III Study Group. Alpha-Lipoic Acid in Diabetic Neuropathy. Diabetes Care. 1999;22:1296–301. doi: 10.2337/diacare.22.8.1296. [DOI] [PubMed] [Google Scholar]
- 49.Ames BN. Low micronutrient intake may accelerate the degenerative diseases of aging through allocation of scarce micronutrients by triage. Proc Natl Acad Sci USA. 2006;103:17589–94. doi: 10.1073/pnas.0608757103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moreira PI, Siedlak SL, Wang X, Santos MS, Oliveira CR, Tabaton M, Nunomura A, Szweda LI, Aliev G, Smith MA, Zhu X, Perry G. Increased autophagic degradation of mitochondria in Alzheimer disease. Autophagy. 2007;3:614–15. doi: 10.4161/auto.4872. [DOI] [PubMed] [Google Scholar]
- 51.Moreira PI, Siedlak SL, Wang X, Santos MS, Oliveira CR, Tabaton M, Nunomura A, Szweda LI, Aliev G, Smith MA, Zhu X, Perry G. Autophagocytosis of mitochondria is prominent in Alzheimer disease. J Neuropathol Exp Neurol. 2007;66:525–32. doi: 10.1097/01.jnen.0000240476.73532.b0. [DOI] [PubMed] [Google Scholar]
- 52.de la Torre JC, Aliev G. Inhibition of vascular nitric oxide after rat chronic brain hypoperfusion: spatial memory and immunocytochemical changes. J Cereb Blood Flow Metab. 2005;25:663–72. doi: 10.1038/sj.jcbfm.9600057. [DOI] [PubMed] [Google Scholar]
- 53.Blass JP, Gibson GE. The role of oxidative abnormalities in the pathophysiology of Alzheimer's disease. Rev Neurol. 1991;147:513–25. [PubMed] [Google Scholar]
- 54.Ando S, Tadenuma T, Tanaka Y, Fukui F, Kobayashi S, Ohashi Y, Kawabata T. Enhancement of learning capacity and cholinergic synaptic function by carnitine in aging rats. J Neurosci Res. 2001;66:266–71. doi: 10.1002/jnr.1220. [DOI] [PubMed] [Google Scholar]
- 55.Farr SA, Poon HF, Dogrukol-Ak D, Drake J, Banks WA, Eyerman E, Butterfield DA, Morley JE. The antioxidants alpha-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. J Neurochem. 2003;84:1173–83. doi: 10.1046/j.1471-4159.2003.01580.x. [DOI] [PubMed] [Google Scholar]
- 56.Ghirardi O, Giuliani A, Caprioli A, Ramacci MT, Angelucci L. Spatial memory in aged rats: population heterogeneity and effect of levocarnitine acetyl. J Neurosci Res. 1992;31:375–9. doi: 10.1002/jnr.490310220. [DOI] [PubMed] [Google Scholar]
- 57.Stoll S, Hartmann H, Cohen SA, Muller WE. The potent free radical scavenger alpha-lipoic acid improves memory in aged mice: putative relationship to NMDA receptor deficits. Pharmacol Biochem Behav. 1993;46:799–805. doi: 10.1016/0091-3057(93)90204-7. [DOI] [PubMed] [Google Scholar]
- 58.Taglialatela G, Caprioli A, Giuliani A, Ghirardi O. Spatial memory and NGF levels in aged rats: natural variability and effects of acetyl-L-carnitine treatment. Exp Gerontol. 1996;31:577–87. doi: 10.1016/0531-5565(96)00052-6. [DOI] [PubMed] [Google Scholar]