

Abstract
To investigate the mitochondrial decay and oxidative damage resulting from aging, the activities/kinetics of the mitochondrial complexes were examined in the brains of young and old rats as well as in old rats fed R-α-lipoic acid plus acetyl-L-carnitine (LA/ALC). The brain mitochondria of old rats, compared with young rats, had significantly decreased endogenous antioxidants and superoxide dismutase activity; more oxidative damage to lipids and proteins; and decreased activities of complex I, IV and V. Complex I showed a decrease in binding affinity (increase in Km) for substrates. Feeding LA/ALC to old rats partially restored age-associated mitochondrial dysfunction to the levels of the young rats. These results indicate that oxidative mitochondrial decay plays an important role in brain aging and that a combination of nutrients targeting mitochondria, such as LA/ALC, could ameliorate mitochondrial decay through preventing mitochondrial oxidative damage.
Keywords: Binding affinity (Km), Brain mitochondria, Mitochondrial complex activity, Enzyme kinetics, Oxidative damage
Introduction
Increasing evidence demonstrates that aging is closely associated with mitochondrial degeneration [1]. Mitochondria are the primary energy generating organelles in the cell. The final step of electron transport energy generating process involves adding four electrons to oxygen to form water. Approximately 1–2% of the oxygen accepts a single electron to form reactive oxidant by-products. These oxidants attack mitochondrial membrane proteins, lipids and nucleic acids, resulting in lower efficiency in electron transfer. In turn, the damaged electron respiratory chain increases the production of oxidants, leading to a cycle of increasing oxidant production and mitochondrial damage with age [2].
The oxidative modification of proteins is implicated in the etiology or progression of a number of the degenerative diseases of aging [3]. One consequence of protein oxidation is the deformation of enzymes causing loss of binding affinity (increased Km) for coenzymes and substrates with age [4]. Oxidative damage of enzymes contributes to the mitochondrial degeneration of aging [5]. Mitochondrial complex I, III, and IV lose activity and function (a significant increase in Km and decrease in Vmax) with aging, which is accompanied by an increase in oxidants [6]. In old rats (vs. young rats), mitochondrial membrane potential, cardiolipin level, respiratory control ratio and cellular O2 uptake are lower; oxidants/O2, neuron RNA/DNA oxidation, and mutagenic aldehydes from lipid peroxidation are higher [7, 8].
Mitochondrial cofactors may improve mitochondrial function. α-Lipoic acid (LA) is a disulfide compound that is found naturally in mitochondria as a coenzyme for pyruvate dehydrogenase and α-ketoglutarate dehydrogenase [9]. LA in its reduced form is a powerful antioxidant [9]. LA is a potent inducer of about 200 phase 2 antioxidant enzymes, thus raising cellular antioxidant defenses [10]. Acetyl-L-carnitine (ALC) is an acetyl derivative of L-carnitine, which facilitates the entry and exit of fatty acids from the mitochondria. ALC fed high doses improves mitochondrial function in old rats [11]. Feeding old rats a combination of LA and ALC restored mitochondrial integrity, function; lowers oxidant production, neuronal RNA/DNA oxidation, and mutagenic aldehydes and improves rat ambulatory activity and cognition [8, 11]. Using electron microscopy to observe the ultrastructural changes, we have demonstrated that feeding LA and ALC ameliorated age-associated mitochondrial ultrastructural decay [12]. More recently, it was further shown that the combination of LA and ALC reduced cognitive dysfunction and maintained cognition in aged beagle dogs [13]. A meta-analysis of 21 double blind clinical trials of ALC in the treatment of mild cognitive impairment and mild Alzheimer’s disease showed significant, but modest, efficacy of ALC [14]. A meta-analysis of four clinical trials of LA for treatment of neuropathic deficits in diabetes showed modest efficacy [15]. A recent trial of LA/ALC in human hypertension showed efficacy [16].
Our previous study suggested that the substrate binding affinity and the activity of acetyl-L-carnitine transferase in the brains of old rats was decreased and that impaired mitochondrial function was possibly caused by increased oxidative damage and could be ameliorated by dietary supplementation with LA and ALC [8]. This work suggested that changes in the kinetics of key enzymes in the mitochondria are important biomarkers to evaluate mitochondrial dysfunction in brain aging [4, 17] and that mitochondrial nutrients may target mitochondria to repair enzyme dysfunction [18].
The effects of LA/ALC on the brain mitochondrial electron respiratory chain, especially, the complex activities/kinetics, complex expression, and mitochondrial oxidative stress, have not been well studied. In the present study, we examined the effects of feeding the combination of ALC and LA to old rats on the activity/kinetics and expression of mitochondrial complexes (complex I: NADH: ubiquinone oxidoreductase; complex II: Succinate-ubiquinol oxidoreductase, complex III: ubiquinol-cytochrome c oxidoreductase; complex IV: cytochrome c oxidase; and complex V: ATP synthase), and mitochondrial oxidative stress biomarkers with biochemical assays in the brain of young and old rats.
Materials and Methods
Materials
ALC (hydrochloride salt) was purchased from Biosint USA, Inc. (Larchmont, NY), and R-α-lipoic acid (tris salt) was a gift from Dr. K. Wessel, Viatris, Germany. All other chemicals/kits were reagent grade or the highest quality available from Sigma or otherwise as specified. The antibodies for complex II (succinate-ubiquinone oxidoreductase 70-kDa subunit), III (ubiquinol-cytochrome c oxidoreductase core II 50-kDa), IV (ubiquinol-cytochrome c oxidase 48-kDa), and V (ATP synthase, beta subunit, 56 kDa) were all mouse monoclonal antibodies and purchased from Molecular Probes (Molecular Probes, Eugene, OR); rabbit polyclonal complex I antibody (NADH ubiquinol oxidoreductase 39-kDa subunit), was a gift from Dr. R. Betarbet (Emory University, GA).
Animals and Diet
Fischer 344 male rats were obtained from the National Institute on Aging and divided into 3 groups: young control (Young, 7 rats), Old control (Old, 13 rats), Old treated (LA/ALC, 10 rats). The young and old rats were 4.7 and 22 months old at the start of the experiment, and were acclimatized at the Northwest Animal Facilities on the University of California at Berkeley campus for at least 2 weeks before treatment. Control animals were fed AIN93 M diet from Dyets (Bethlehem, PA) and MilliQ water (pH 5.2). The rats in the experimental groups were fed a combination 0.2% (wt/vol) ALC in MilliQ water (pH was adjusted to 5.2 with 1 N NaOH), 0.1% (wt/wt) LA in AIN93 M diet for 4.5 months. The food consumption was determined by weighing the diet and measuring the volume of water weekly; the average daily consumption was then calculated. The weight gain during the course of the experiment was also measured. We did not find any significant differences in diet, water consumption, or weight gain between the un-supplemented old rats and the old supplemented rats, consistent with previous report [7]. During the period of feeding, 10 old rats (6 in Old control, 4 in Old treated) were euthanized due to poor health. Therefore, 20 rats (7 Young control; 7 Old control, and 6 Old treated) were terminated and used in this study.
Assays for Activities of Mitochondrial Complex I, II, III, IV and V
Brain mitochondria (P2 pellet) was prepared by Paula’s method [19]. The yield of mitochondria were 1.25 ± 0.11, 1.27 ± 0.15, 1.30 ± 0.13 mg/100 mg brain tissue for Young, Old and Old + LA/ALC group, respectively. Complex I, II, III, IV, and V were determined as described [20–22]. Briefly, Complex I activity was assayed by monitoring the decrease of NADH at 340 nm. Final concentration of mitochondria protein was 30 μg/ml. Reaction was started by adding 200 μmol/1 NADH and scanned at 340 nm for 3 min. Rotenone (3 μmol/1) was added into the reaction system as blank control. Complex II was assayed with mitochondria (final concentration 30 μg/ml) and the reaction was started with 10 mmol/1 succinate and scanned at 600 nm for 2 min. Complex III activity was measured in the mixture containing 250 mmol/1 sucrose, 1 mmol/1 EDTA, 50 mmol/1 KPi, pH value adjusted to 6.5 to reduce auto-oxidation of reduced CoQ1, 2 mmol/1 KCN, 50 μmol/1 cytochrome C, 0.1% BSA, and the reaction was initiated by 20 μg/ml brain mitochondria and 50 μmol/1 reduced CoQ1, recording the increase of absorption at 550 nm for 2 min. Complex IV was measured by monitoring the decrease of reduced cytochrome C at 550 nm. Complex V (ATP synthase) was assayed in an assay mixture consisting of 50 mM Hepes, pH 8.0, 5 mM MgSO4, 0.35 mM NADH, 250 mM sucrose, 2.5 mM phosphoenolpyruvate, 50 μg pyruvate kinase, 50 μg lactate dehydrogenase, 2 μg anti-mycin A, 40 μM rotenone, 2 mM potassium cyanide, 15 μg of brain mitochondrial protein in the absence or presence of 3 μg oligomycin. The reaction was initiated by the addition of 2.5 mM ATP and ATP synthase activity was determined as the oligomycin-sensitive activity.
Enzyme kinetics of Mitochondrial Complex I and IV
The complex I assay medium contain 250 mmol/1 sucrose, 1 mmol/1 EDTA, 50 mmol/1 Tris-Cl, pH 7.4, 30 μg/ml mitochondria, 10 μmol/1 CoQ1, 3 mg/ml BSA, 50 μmol/1 NADH, 2 mmol/1 KCN and 2 μmol/1 antimycin, reaction started by mitochondria, and scanning 3 min at the 340 nm [20]. The kinetics were determined over a range of CoQ1 2 to 40 μmol/1 with a constant concentration of 50 μmol/1 NADH, or over a range of NADH 10 to 200 μmol/1 with a constant concentration of 10 μmol/1 CoQ1. The results were plotted with the double-reciprocal plot of reciprocal rate 1/V against reciprocal substrate concentration 1/S. Results were also calculated by the direct linear plot with the equation of Km = VmS/V–S. On complex I kinetic assay, the medium contain 10 μg/ml mitochondria, the assay was determined over a range of 2 to 40 μmol/1 reduced cytochrome C.
Western Blot of Mitochondrial Complex I, II, III, IV andV
Brain mitochondrial protein (10 μg) was resolved by 10% SDS-PAGE, transferred to PDF membrane, and blocked with 5% non-fat milk. The PDF membranes were incubated with primary monoclonal antibodies complex I, II, III, IV, V (1:1000, Molecular Probes) overnight at 4°C. Membranes were washed and incubated with anti-mouse or anti-rabbit (1:8000) IgG labeled with horseradish peroxidase (Vector Laboratories) for 1 h and visualized using an enhanced chemiluminescence Western blotting detection system (GE healthcare, UK). As the mitochondria protein loading control, polyacrylamide resolving gels (10%, w/v) loaded same amount of samples were stained with Coomassie Brilliant Blue R250.
Measurement of Reduced Glutathione (GSH) in Rat Brain Mitochondria
5,5′-Dithiobis-2-nitrobenzoic acid (DTNB) reacts with reduced glutathione to form a yellow product. The optical density, measured at 412 nm, is directly proportional to the glutathione concentration in the sample with GSH Detection Kit (Bioassay Systems, CA).
Assays for Superoxide Dismutase and Catalase in Rat Brain Mitochondria
Superoxide dismutase (SOD; E.G.: 1.15.1.1) activity was assayed using the SOD Detection Kit (Sigma, St. Louis, MO). The kit utilizes Dojindo’s highly water-soluble tetrazolium salt, WST-1 (2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt) that produces a water-soluble formazan dye upon reduction with a superoxide anion. The rate of the reduction with superoxide anion are linearly related to the xanthine oxidase (XO) activity, and is inhibited by SOD, therefore, the inhibition activity of SOD or SOD-like materials can be determined colorimetrically.
Catalase assay was performed utilizing a kit (Biochem, San Carlos, CA).
Measurement of Lipid Peroxidation in Rat Brain Mitochondria
Malonaldehyde (MDA) was used as the index of lipid peroxidation and detected with the MDA Detection Kit (Oxis International, Inc., Foster City, CA).
Detection of Protein Carbonyls in Rat Brain Mitochondria
Protein carbonyls in brain mitochondria were assayed with the Oxyblot protein oxidation detection kit (Cell Biolabs, Inc., San Diego, CA). The carbonyl groups in the protein side chains were derivatized to 2,4-dinitrophenylhydrazone (DNP-hydrazone), then separated by polyacrylamide gel electrophoresis followed by Western blotting. Proteins which have undergone oxidative modification will be identified by appearing as a band in the lane containing the derivatized sample [3]; Another polyacrylamide resolving gels loading the same amount of samples was electrophoresed and stained with Coomassie Brilliant Blue R250 as the loading control (Fig. 5a).
Fig. 5.
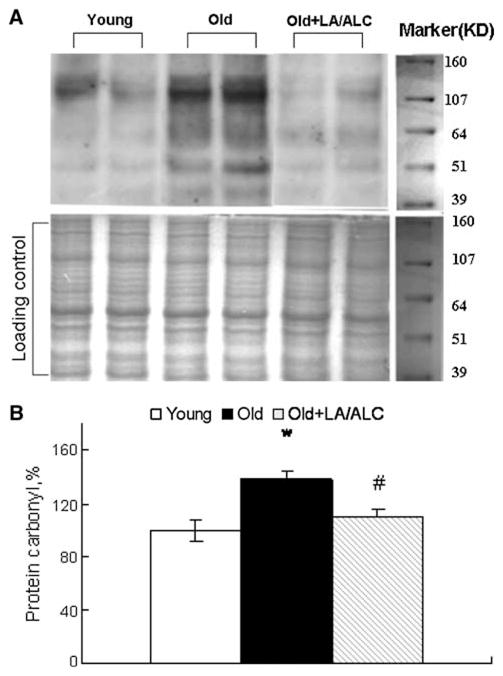
Protein carbonyls in the mitochondria of rat brain. Values were normalized by loading control, setting the protein carbonyls in Young rats as 100%. a Representative blotting of Protein carbonyl and duplicate Gel dyed with Coomassie brilliant blue as loading control, b Quantitative results. Values are mean ± SEM from 7 rats in the group of the Young or the Old, and 6 rats in the group of the Old fed with Old + LA/ALC. *P < 0.05 vs. Young, and #P < 0.05 vs. Old
Statistical Analyses
Data of biochemical assays shown are the mean ± SEM. Statistical comparisons were performed using one way ANOVA and Tukey post-hoc comparison, and P < 0.05 was considered statistically different. SPSS for Windows was used for the analyses.
Results
Effect of LA/ALC on Age-Associated Dysfunction of Brain Mitochondria Complexes
Compared with young rats, activities of complex I, IV and V were significantly decreased about 30% (P = 0.001), 20% (P = 0.041) and 15% (P = 0.011) in the old rat brain, respectively (Fig. 1). LA/ALC treatment significantly restored the complex I activity (P = 0.05).
Fig. 1.
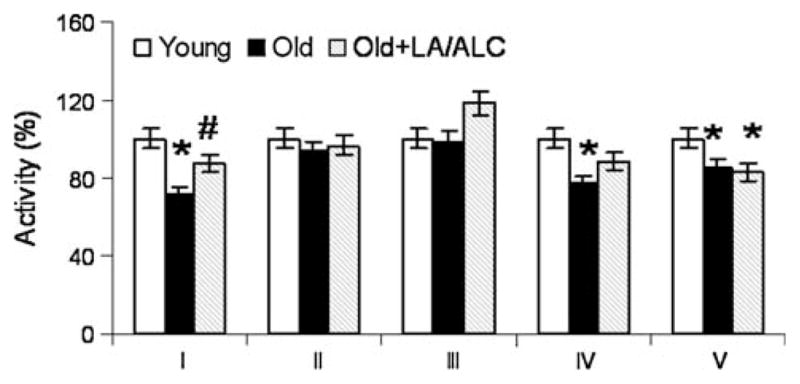
Activities of mitochondrial complexes in the mitochondria of rat brain. Values are mean ± SEM from 7 rats in the group of the Young or the Old, and 6 rats in the group of the Old fed with Old + LA/ALC. *P < 0.05 vs. Young, #P < 0.05 vs. Old
Binding of Complex I to CoQ and NADH decreased with age and this decrease was not reversed by feeding LA/ALC (Fig. 2a–c). To detect changes in kinetics of complex I and IV due to aging and LA/ALC supplementation, we calculated the Vm and the Km of both complexes. The apparent Km of complex I for either CoQ1 or NADH substrate was significantly higher (weaker binding) in old rats than in young ones (Fig. 2a, b): Km for CoQ1 exhibits a 73% increase from 1.94 to 3.36 μmol/1 (P = 0.039, Fig. 2c), and for NADH, a 20% increase from 10.13 to 12.04 μmol/1 (P = 0.044, Fig. 2c). Supplementation with LA/ALC increased the Vm of complex I of old rats to the level of young rats but did not affect the Km (Fig. 2d). For the kinetics of complex IV in old rats, compared with young rats, the Km showed no significant difference in either old rats or old treated (Fig. 3a, b). The Vm in old rats decreased significantly, compared with young rats (P = 0.041) and was almost restored by the supplementation of LA/ALC (no significant difference between Young and Old + LA/ALC, and Old vs. Old + LA/ALC, P = 0.08) (Fig. 3c).
Fig. 2.

Double-reciprocal plots of reaction velocity of complex I versus substrate CoQ1 (a) or NADH (b) in the mitochondria of rat brain, (c) Km for CoQ1 and NADH; (d) Vmax. Values are mean ± SEM from 7 rats in the group of the Young or the Old, and 6 rats in the group of the Old fed with Old + LA/ALC. Km was expressed as μmol/1 and the Vmax was expressed as nmol/min/mg protein. *P < 0.05 vs. Young, and #P < 0.05 vs. Old. The long-dash line, short-dash line and solid line represent the groups of Old, Old + LA/ALC and Young mice, respectively
Fig. 3.
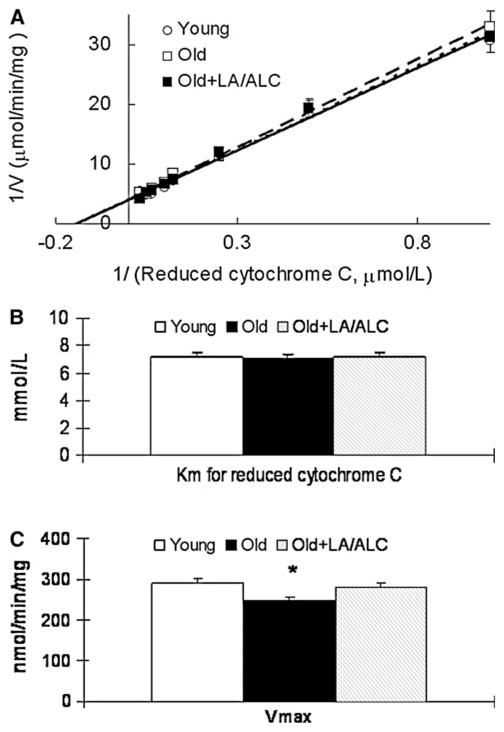
Double-reciprocal plots of reaction velocity of complex I versus substrate reduced cytochrome C (a) in the mitochondria of rat brain, (b) Km for reduced cytochrome C; (c) Vmax. Values are mean ± SEM from 7 rats in the group of the Young or the Old, and 6 rats in the group of the Old fed with Old + LA/ALC. Km was expressed as μmol/1 and the Vmax was expressed as nmol/min/mg protein. *P < 0.05 vs. Young. The long-dash line, short-dash line and solid line represent the groups of Old, Old + LA/ALC and Young mice, respectively
Effect of LA/ALC on Mitochondrial Oxidative Damage
Mitochondrial MDA
MDA level, an index of lipid peroxidation, exhibited a twofold increase in old rats as compared to that of young rats in the brain mitochondria (P = 0.021), and was significantly decreased by the LA/ALC supplementation to old animals (P = 0.044) (Fig. 4).
Fig. 4.
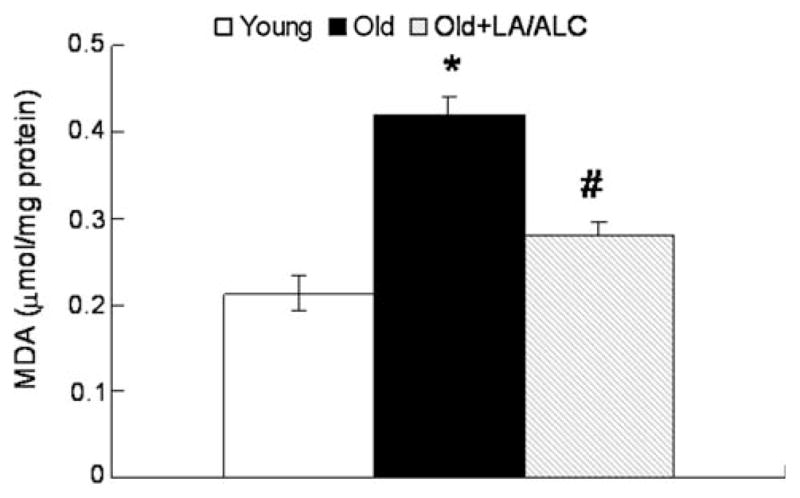
MDA level in the mitochondria of rat brain. Values are mean ± SEM from 7 rats in the group of the Young or the Old, and 6 rats in the group of the Old fed with Old + LA/ALC. *P < 0.05 vs. Young, and #P < 0.05 vs. Old
Mitochondrial Protein Carbonyl
Protein carbonyl, an index of protein oxidation, increased significantly in the brain mitochondria of old rats, compared with that of young rats (P = 0.019), and the increase was significantly reduced by the LA/ALC supplementation to old rats (Old vs. Old + LA/ALC, P = 0.026) (Fig. 5b).
Effect of LA/ALC on Mitochondrial Antioxidant Defense Systems
Mitochondrial Antioxidant GSH
Compared with young rats, GSH was significantly decreased from 1.48 to 1.12 μg/g mitochondria in old rats (P = 0.046) and completely restored by the LA/ALC supplementation in brain mitochondria (1.64 μg/g in Old + LA/ALC vs. 1.12 μg/g in Old, P = 0.04) (Fig. 6).
Fig. 6.
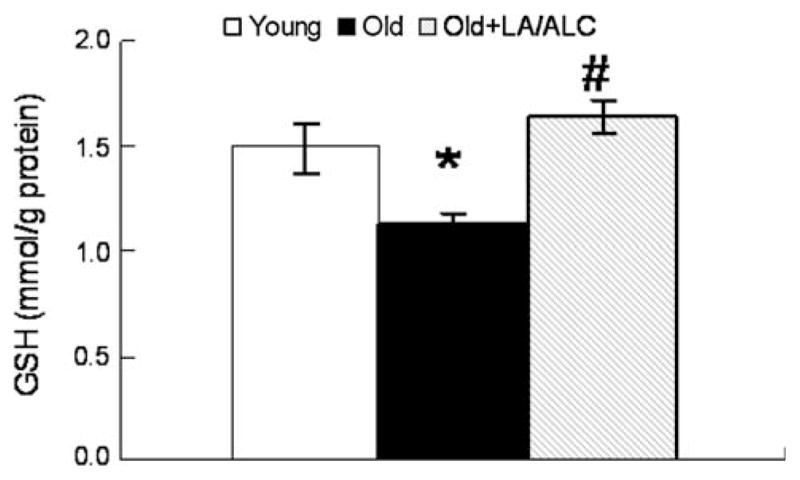
GSH level in the mitochondria of rat brain. Values are mean ± SEM from 7 rats in the group of the Young or the Old, and 6 rats in the group of the Old fed with Old + LA/ALC. *P < 0.05 vs. young, and #P < 0.05 vs. Old
Activity of SOD and Catalase
SOD activity in old rats showed a 25% decrease, compared with that in young rats (P = 0.036), and the decrease was significantly restored by the LA/ALC supplementation (Old vs. Old + LA/ALC, P = 0.039) (Fig. 7). However, catalase activity showed no difference in the three groups (data not shown).
Fig. 7.
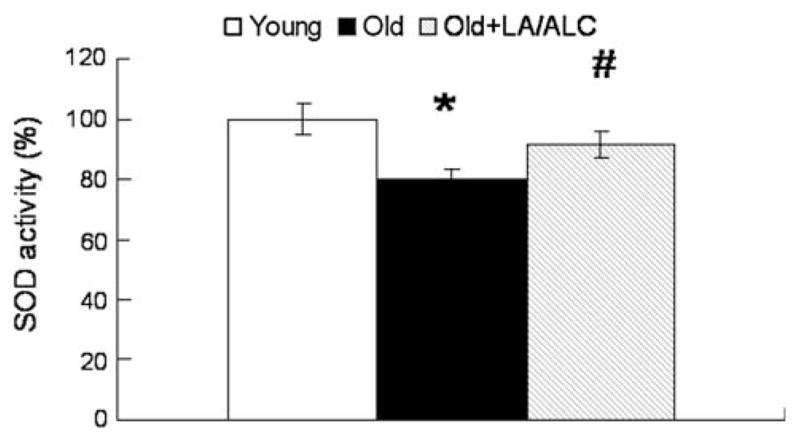
SOD activity in the mitochondria of rat brain. Values are mean ± SEM from 7 rats in the group of the Young or the Old, and 6 rats in the group of the Old fed with Old + LA/ALC. *P < 0.05 vs. Young, and #P < 0.05 vs. Old
Protein Expressions of Mitochondrial Complexes
To test whether the age-associated loss of enzyme activity resulting from protein levels of the enzymes and whether the improving effect of nutrients is due to enhancement of enzyme protein expression, we measured the protein levels of complex I, II, III, IV and V in brain mitochondria using a Western blotting assay. The results are shown in Table 1. We found no significant changes in mitochondrial complex protein levels in the brain between the young and the old animals; however, the LA/ALC supplementation appears to increase the protein level of complex V.
Table 1.
Relative expression of complexes in isolated brain mitochondria
| Group | Complex I | II | III | IV | V |
|---|---|---|---|---|---|
| Young | 100 ± 12.2 | 100 ± 10.3 | 100 ± 4.6 | 100 ± 7.4 | 100 ± 1.8 |
| Old | 104.5 ± 8.3 | 112.8 ± 10.4 | 107.3 ± 4.3 | 104.5 ± 5.2 | 99.3 ± 5.3 |
| Old + LA/ALC | 107.7 ± 5.5 | 103.6 ± 12.5 | 121.8 ± 8.3 | 112.8 ± 5.2 | 120.1 ± 3.3* |
Values are mean ± SEM from 7 rats in the group of the Young or the Old, and 6 rats in the group of the Old fed with LA/ALC. The expression of mitochondria complexes was assessed by Western blotting in isolated brain mitochondria. The photometric intensity of bands was normalized by loading control gel stained by Coomassie Brilliant Blue, setting the expression of complexes in Young rats as 100
P < 0.05 vs. Old
Discussion
The mitochondrial enzymes, complexes (I–V) are the key components in the process of ATP production and simultaneous oxidant by-product generation. Therefore, these enzymes are of particular importance in triggering mitochondrial decay and oxidative damage. Mitochondrial complex dysfunction has a close association with aging and aging-related disease [5, 23]. To understand the underlying mechanisms of brain aging and the ameliorating effects of LA/ALC on retarding brain aging, our objective in the current study was to focus on the activity/kinetics of mitochondrial complexes and oxidative damage to brain mitochondria.
The activity of complex I, IV and V decreased in the brain of the old rats, and the LA/ALC treatment led to a recovery of activity of complex I and IV (Figs. 1, 2). Further investigation of the kinetics of complex I and IV showed that the Km of the complex I for CoQ1 increased from 1.94 in young rats to 3.36 μmol/1 in old rats. These results suggest that mitochondrial complex I efficiency is significantly reduced in old rats. According to the Michaelis–Menten equation, if the level of coenzyme Q keeps around 2 μmol/1 in brain mitochondria, the complex I of old rats would lose about 30% activity assuming no change in Vm in old or young rats. That is, even if the other components of electron transport chain remain normal, the higher Km of complex I would cause a 30% loss of efficiency of brain mitochondrial electron transfer in the old rats. The LA/ALC supplementation could not restore the lost binding affinity of complex I, suggesting much higher level of substrates are needed to maintain routine mitochondrial activity in the old rats. Supplementation with high levels of mitochondria substrates and B vitamins, which can raise coenzyme levels, is a promising therapeutic approach to promote more efficient enzyme activity in old individuals [4, 17]. Supplementation with LA and ALC partially restored activity of complex I and IV in old rats. These results provide evidence for the close link between brain mitochondrial function and cognition as previously reported for LA and/or ALC [7, 13]. The improvement of complex I/IV activity of old rats would stimulate electron transport and reduce oxidant generation due to electron leakage [24, 25].
The expression of mitochondrial complexes (Table 1) did not show an age-associated change in any of the protein levels of mitochondrial complexes. These results suggest that protein level may remain constant while enzyme function gets impaired with age. It is also suggested that protein levels alone do not account for enzyme activity losses as a function of age in rats nor fully account for LA/ALC induced increases in enzyme activity. This characteristic is further supported by our results on protein carbonyls (Fig. 5) and the increase in MDA (Fig. 4) in the aging mitochondria. The higher level of carbonyls in mitochondria in old rats indicates that the vulnerable enzymes in complex I and IV are increasingly inactivated with age due to protein oxidation while their expression is unchanged. MDA is considered to be an index of lipid peroxidation, though it is known that amino acid, protein, and DNA oxidation can also generate MDA [26]. MDA may cause mitochondrial dysfunction [27]. Increased oxidants generation may lower antioxidant defenses and oxidatively modify enzymes. Our results on the endogenous antioxidant GSH (Fig. 6) and antioxidant enzyme SOD (Fig. 7) clearly suggest that brain aging is accompanied with decreased antioxidant defenses in brain mitochondria and that LA/ALC supplementation enhances the antioxidant defenses in the mitochondria of old rats.
The mechanisms of the combination of LA/ALC on brain mitochondria are being clarified and could involve several pathways [4, 18]. First, LA has been shown to be a potent inducer of GSH synthesis, including GSH in mitochondria, and about 200 other phase-2 antioxidant defense enzymes [10, 28]. Intracellular glutathione is critical for neuronal function [29]. ALC, too, at much higher doses induces phase-2 enzymes [30], perhaps indirectly. Second, LA can be reduced in mitochondria to some extent to dihydrolipoic acid, which is a potent antioxidant, to supply dihydrolipoic acid as a cofactor for two mitochondrial enzymes pyruvate dehydrogenase and α-ketoglutarate dehydrogenase. Third, ALC, beside its effects on mitochondrial fatty acid transportation [31] improves the substrate binding affinity and the activity of carnitine acetyltransferase [8]. There is also a report showing ALC acts as an antioxidant, perhaps indirectly [32]. The improvement of carnitine acetyltransferase by feeding ALC may promote the production of substrates like NADH, improving antioxidant status and accelerating electron transfer to produce more ATP. Fourth, LA has a synergistic effect with ALC in improving mitochondrial function. ALC can contribute by providing more ATP for the senescent cell; however, at the same time it could lead to more oxidant generation in the process of electron transfer from complex I to complex IV. Fifth, LA and ALC synergistically ameliorate mitochondrial oxidative stress and recover complex activity in old rats potentially through the mechanism of enhancing mitochondrial biogenesis, which has been shown in neurons [33], adipocytes [34], and beta cells [35].
In conclusion, brain mitochondria decay with age and this decay is associated with an increase in oxidative damage to mitochondria, a decrease in activity and substrate binding affinity of complex I, and decreased activity of complex IV/V. LA/ALC feeding partially or completely restored mitochondrial function to the level of young rat. These results suggest that mitochondrial decay is a key contributor to aging and that feeding LA/ALC, could ameliorate the mitochondrial decay and oxidative damage by improving the mitochondrial redox homeostasis in the brain. These results also suggest that amelioration of mitochondrial decay with dietary supplementation with antioxidants/nutrients targeted to mitochondria might be an effective strategy for delaying brain aging.
Acknowledgments
The authors thank Dr. Afshin Gharib for taking care of the animals. This work was supported by the Ellison Medical Foundation Grants S-0422-99, the National Institute on Aging AG17140, and the National Center for Complementary and Alternative Medicine Research Scientist Award K05 AT001323-4 (B. N. A.), and National Institutes of Health grants NEI EY016101, NIA AG023265, and NCCAM AT01918 (B. N. A. and J. L.) and NIA AG012694 (C. W. C).
Footnotes
Special issue article in honor of Dr. Akitane Mori.
References
- 1.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Judge S, Jang YM, Smith A, Hagen T, Leeuwenburgh C. Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J. 2005;19:419–421. doi: 10.1096/fj.04-2622fje. [DOI] [PubMed] [Google Scholar]
- 3.Leine RL, Garland D, Oliver CN, et al. Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol. 1990;186:464–478. doi: 10.1016/0076-6879(90)86141-H. [DOI] [PubMed] [Google Scholar]
- 4.Ames BN, Suh JH, Liu J. Enzymes lose binding affinity for coenzymes and substrates with age: a strategy for remediation. In: Kaput J, editor. Nutrigenomics: concepts and technologies. Wiley; Hoboken: 2006. pp. 277–291. [Google Scholar]
- 5.Naarro A, Boveris A. Rat brain and lier mitochondria develop oxidative stress and lose enzymatic activities on aging. Am J Physiol Regul Integr Comp Physiol. 2004;287(5):R1244–R1249. doi: 10.1152/ajpregu.00226.2004. [DOI] [PubMed] [Google Scholar]
- 6.Feuers RJ. The effects of dietary restriction on mitochondrial dysfunction in aging. Ann NY Acad Sci. 1998;854:192–201. doi: 10.1111/j.1749–6632.1998.tb09902.x. [DOI] [PubMed] [Google Scholar]
- 7.Liu J, Head E, Gharib AM, et al. Memory loss in old rats is associated with brain mitochondrial decay and RNA/DNA oxidation: partial reversal by feeding acetyl-L-carnitine and/or R-alpha-lipoic acid. Proc Natl Acad Sci USA. 2002;99:2356–2361. doi: 10.1073/pnas.261709299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu J, Killilea DW, Ames BN. Age-associated mitochondrial oxidative decay: improvement of carnitine acetyltransferase substrate-binding affinity and activity in brain by feeding old rats acetyl-L-carnitine and/or R-alpha-lipoic acid. Proc Natl Acad Sci USA. 2002;99:1876–1881. doi: 10.1073/pnas.261709098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Packer L, Witt EH, Tritschler HJ. Alpha-Lipoic acid as a biological antioxidant. Free Radic Biol Med. 1995;19:227–250. doi: 10.1016/0891-5849(95)00017-R. [DOI] [PubMed] [Google Scholar]
- 10.Suh JH, Shenvi SV, Dixon BM, et al. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci USA. 2004;101:3381–3386. doi: 10.1073/pnas.0400282101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hagen TM, Ingersoll RT, Wehr CM, et al. Acetyl-L-carnitine fed to old rats partially restores mitochondrial function and ambulatory activity. Proc Natl Acad Sci USA. 1998;95:9562–9566. doi: 10.1073/pnas.95.16.9562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alie G, Liu J, Shenk JC, et al. Neuronal mitochondrial amelioration by feeding acetyl-L-carnitine and lipoic acid to aged rats. J Cell Mol Med. 2008 doi: 10.1111/j.l582–4934.2008.00324.x. [epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Milgram NW, Araujo JA, Hagen TM, Treadwell B, Ames BN. Acetyl-L-carnitine and alpha-lipoic acid supplementation of aged beagle dogs improves learning in two landmark discrimination tests. FASEB J. 2007;21:3756–3762. doi: 10.1096/fj.07-8531com. [DOI] [PubMed] [Google Scholar]
- 14.Montgomery SA, Thai LJ, Amrein R. Meta-analysis of double blind randomized controlled clinical trials of acetyl-L-carnitine versus placebo in the treatment of mild cognitive impairment and mild Alzheimer’s disease. Int Clin Psychopharmacol. 2003;18:61–71. doi: 10.1097/00004850-200303000-00001. [DOI] [PubMed] [Google Scholar]
- 15.Ziegler D, Luft D. Clinical trials for drugs against diabetic neuropathy: can we combine scientific needs with clinical practicalities? Int Rev Neurobiol. 2002;50:431–463. doi: 10.1016/s0074-7742(02)50085-4. [DOI] [PubMed] [Google Scholar]
- 16.McMackin CJ, Widlansky ME, Hamburg NM, et al. Effect of combined treatment with alpha-Lipoic acid and acetyl-L-carnitine on vascular function and blood pressure in patients with coronary artery disease. J Clin Hypertens (Greenwich) 2007;9:249–255. doi: 10.1111/j.1524-6175.2007.06052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ames BN, Elson-Schwab I, Siler EA. High-dose vitamin therapy stimulates variant enzymes with decreased coenzyme binding affinity (increased K(m)): relevance to genetic disease and polymorphisms. Am J Clin Nutr. 2002;75:616–658. doi: 10.1093/ajcn/75.4.616. [DOI] [PubMed] [Google Scholar]
- 18.Liu J. The effects and mechanisms of mitochondrial nutrient alpha-lipoic acid on improving age-associated mitochondrial and cognitive dysfunction: an overview. Neurochem Res. 2008;33:194–203. doi: 10.1007/s11064-007-9403-0. [DOI] [PubMed] [Google Scholar]
- 19.Keeney PM, Xie J, Capaldi RA, Bennett JP., Jr Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci. 2006;26:5256–5264. doi: 10.1523/JNEUROSCI.0984-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lenaz G, Fato R, Baracca A, Genoa ML. Mitochondrial quinone reductases: complex I. Methods Enzymol. 2004;382:3–20. doi: 10.1016/80076-6879(04)82001-9. [DOI] [PubMed] [Google Scholar]
- 21.Sun L, Luo C, Long J, Wei D, Liu J. Acrolein is a mitochondrial toxin: effects on respiratory function and enzyme activities in isolated rat liver mitochondria. Mitochondrion. 2006;6:136–142. doi: 10.1016/j.mito.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 22.Yarian CS, Rebrin I, Sohal RS. Aconitase and ATP synthase are targets of malondialdehyde modification and undergo an age-related decrease in activity in mouse heart mitochondria. Biochem Biophys Res Commun. 2005;330:151–156. doi: 10.1016/j.bbrc.2005.02.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smigrodzki R, Parks J, Parker WD. High frequency of mitochondrial complex I mutations in Parkinson’s disease and aging. Neurobiol Aging. 2004;25:1273–1281. doi: 10.1016/j.neurobiolaging.2004.02.020. [DOI] [PubMed] [Google Scholar]
- 24.Yoon YS, Lee JH, Hwang SC, Choi KS, Yoon G. TGF betal induces prolonged mitochondrial ROS generation through decreased complex I activity with senescent arrest in M1Lu cells. Oncogene. 2005;24:1895–1903. doi: 10.1038/sj.onc.1208262. [DOI] [PubMed] [Google Scholar]
- 25.Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Federici A, Ruggiero FM. Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: involvement of reactive oxygen species and cardiolipin. Circ Res. 2004;94:53–59. doi: 10.1161/01.RES.0000109416.56608.64. [DOI] [PubMed] [Google Scholar]
- 26.Liu J, Yeo HC, Doniger SJ, Ames BN. Assay of aldehydes from lipid peroxidation: gas chromatography-mass spectrometry compared to thiobarbituric acid. Anal Biochem. 1997;245:161–166. doi: 10.1006/abio.1996.9990. [DOI] [PubMed] [Google Scholar]
- 27.Long J, Wang X, Gao H, et al. Malonaldehyde acts as a mitochondrial toxin: inhibitory effects on respiratory function and enzyme activities in isolated rat liver mitochondria. Life Sci. 2006;79:1466–1472. doi: 10.1016/j.lfs.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 28.Volobouea LA, Liu J, Suh JH, Ames BN, Miller SS. (R)-alpha-lipoic acid protects retinal pigment epithelial cells from oxidative damage. Invest Ophthalmol Vis Sci. 2005;46:4302–4310. doi: 10.1167/ios.04-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Packer L, Tritschler HJ, Wessel K. Neuroprotection by the metabolic antioxidant alpha-lipoic acid. Free Radic Biol Med. 1997;22:359–378. doi: 10.1016/S0891-5849(96)00269-9. [DOI] [PubMed] [Google Scholar]
- 30.Calabrese, Ravagna A, Colombrita C, et al. Acetylcarnitine induces heme oxygenase in rat astrocytes and protects against oxidative stress: involvement of the transcription factor Nrf2. J Neurosci Res. 2005;79:509–521. doi: 10.1002/jnr.20386. [DOI] [PubMed] [Google Scholar]
- 31.Liu J, Atamna H, Kuratsune H, Ames BN. Delaying brain mitochondrial decay and aging with mitochondrial antioxidants and metabolites. Ann NY Acad Sci. 2002;959:133–166. doi: 10.1111/j.1749-6632.2002.tb02090.x. [DOI] [PubMed] [Google Scholar]
- 32.Calo LA, Pagnin E, Davis PA, et al. Antioxidant effect of L-carnitine and its short chain esters: relevance for the protection from oxidative stress related cardiovascular damage. Int J Cardiol. 2006;107:54–60. doi: 10.1016/j.ijcard.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 33.Abdul HM, Butterfield DA. Involvement of PI3K/PKG/ERK1/2 signaling pathways in cortical neurons to trigger protection by cotreatment of acetyl-L-carnitine and alpha-lipoic acid against HNE-mediated oxidative stress and neurotoxicity: implications for Alzheimer’s disease. Free Radic Biol Med. 2007;42:371–384. doi: 10.1016/j.freeradbiomed.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen W, Liu K, Tian C, et al. R-alpha-Lipoic acid and acetyl-L-carnitine complementarity promote mitochondrial biogenesis in murine 3T3-L1 adipocytes. Diabetologia. 2008;51:165–174. doi: 10.1007/s00125-007-0852-4. [DOI] [PubMed] [Google Scholar]
- 35.Shen W, Liu K, Tian C, et al. Protective effects of R-alpha-lipoic acid and acetyl-L-carnitine in MIN6 and isolated rat islet cells chronically exposed to oleic acid. J Cell Biochem. 2008;104:1232–1243. doi: 10.1002/jcb.21701. [DOI] [PubMed] [Google Scholar]