

Abstract
Mitochondria play important roles as the powerhouse of the cell. After cerebral ischemia, mitochondria overproduce reactive oxygen species (ROS), which have been thoroughly studied with the use of superoxide dismutase transgenic or knockout animals. ROS directly damage lipids, proteins, and nucleic acids in the cell. Moreover, ROS activate various molecular signaling pathways. Apoptosis-related signals return to mitochondria, then mitochondria induce cell death through the release of pro-apoptotic proteins such as cytochrome c or apoptosis-inducing factor. Although the mechanisms of cell death after cerebral ischemia remain unclear, mitochondria obviously play a role by activating signaling pathways through ROS production and by regulating mitochondria-dependent apoptosis pathways.
Keywords: Mitochondria, Cerebral ischemia, SOD1, Reactive oxygen species, Neuronal death, PIDD
1. Introduction
Mitochondria are the powerhouse of the cell. Their primary physiological function is to generate adenosine triphosphate through oxidative phosphorylation via the electron transport chain, which contains five multi-subunit enzyme complexes, I to V. Reactive oxygen species (ROS) are generated in complex I and complex III during mitochondrial respiration [1]. Therefore, oxygen metabolism can be a potential threat to tissues and cells.
Numerous studies have shown the roles ROS play in the pathophysiology of neurological disorders, including ischemia, trauma, and degenerative diseases. ROS cause macromolecular damage such as lipid peroxidation, protein oxidation, and DNA oxidation, all of which can lead to cell injury and death [2,3]. In addition, ROS can act as intracellular messengers to transduce signals of various pathways, including cell death pathways [4,5], similar to the way in which reactive nitrogen species transduce signals in endothelial cells or neurons [6,7].
Besides triggering molecular signals by overproduction of ROS, mitochondria regulate apoptotic pathways by sequestering Ca2+, storing and releasing pro-apoptotic proteins such as cytochrome c and apoptosis-inducing factor (AIF), and probably by opening the permeability transition pore [8,9]. In this review, we discuss the roles of ROS generated in mitochondria and mitochondria-dependent apoptotic pathways in several in vivo models of cerebral ischemia.
2. The roles of ROS generated by mitochondria
2.1. Generation and clearance of ROS under normal physiological conditions
Because mitochondria generate superoxide anions (O2-) and hydrogen peroxide (H2O2) during mitochondrial respiration under normal physiological conditions [1], oxygen metabolism poses a potential threat to cells. It is, nevertheless, essential for cell survival. Pro-oxidant enzymes, such as nitric oxide synthases (NOS), cyclooxygenases, xanthine dehydrogenase, xanthine oxidase, NADPH oxidase, myeloperoxidase, and monoamine oxidase, generate the ROS O2-, H2O2, nitric oxide, and lipid peroxides.
To detoxify such ROS, cells develop ROS clearance systems. Superoxide dismutase (SOD), glutathione peroxidase (GSHPx), and catalase contribute to scavenging these ROS. SOD has three isoforms: copper/zinc SOD (SOD1), manganese SOD (SOD2), and extracellular SOD (SOD3) (Table 1). All three SOD isoforms dismutate O2- to H2O2 and molecular oxygen. Then, GSHPx scavenges H2O2 to water at the expense of glutathione. Catalase also dismutates H2O2 to water [2]. Other small molecular non-enzymatic antioxidants such as vitamin E and vitamin C are also involved in the detoxification of free radicals [10].
Table 1.
Mammalian superoxide dismutases
| □ | SOD1 (CuZnSOD) |
SOD2 (MnSOD) |
SOD3 (ECSOD) |
|---|---|---|---|
| Location | Cytosol | Mitochondria | Extracellular space |
| Molecular weight | 32,000 | 88,000 | 120,000 |
| Structure | Dimer | Tetramer | Tetramer |
| Metals, g-atoms/subunit | Cu 1, Zn 1 | Mn 1 | Cu 1, Zn 1 |
| Phenotype of transgenic mouse (+/+) | Normal | Normal | Normal |
| Phenotype of knockout mutant (−/−) | Normal | Neonatal lethality | Normal |
| 21 (human) | 6 (human) | 4 (human) | |
| 16 (mouse) | 17 (mouse) | 5 (mouse) | |
| Chromosome | 11 (rat) | 1 (rat) | 14 (rat) |
CuZn, copper, zinc; Mn, manganese; EC, extracellular.
Oxidative stress is defined as the pathogenic outcome of ROS overproduction beyond the capacity of ROS clearance in cells. After cerebral ischemia, the balance between ROS production and clearance shifts to the production side, resulting in induction of oxidative stress-induced signaling and cell injury.
2.2. Reperfusion injury and ROS
Reperfusion injury is brain damage caused by the return of blood flow, resulting in progression of vasogenic edema, hemorrhagic transformation, and an increase in stroke volume. ROS involvement in reperfusion injury has been described since the early 1980s [11,12]. Numerous subsequent reports have presented the relationship between reperfusion injury and ROS. In ischemic brain tissue, ROS generation is accelerated by cytosolic pro-oxidant enzymes and by mitochondria, inactivation of detoxification systems, consumption of antioxidants, and failure to adequately replenish antioxidants [2]. These overproduced ROS cause macromolecular damage and activation of various pathways.
2.3. Detection and quantification of ROS
To detect and quantify various ROS in the ischemic brain, an indirect measurement method is required because of the short half-life of most ROS. One approach is to detect oxidative modification of biological targets of ROS such as lipid peroxidation, protein oxidation, or DNA oxidation. Another approach is to use reporter molecules, which are oxidized by ROS, resulting in the production of chromogenic, fluorescent, or luminescent molecules. Hydroethidine (HEt), one such reporter molecule, has been used to detect O2- in cells and tissues [13,14]. “Ethidium fluorescence”, which is the red fluorescence arising from oxidation of HEt, has been attributed to O2- trapping in cells [13,14]. However, a recent study revealed that ethidium could be generated by other ROS [15]. To specifically detect O2-, 2-hydroxyethidium (2-HE), the two-electron oxidation product of HEt [16], is a more suitable diagnostic marker than HEt [15].
Although the fluorescence spectra from 2-HE and ethidium overlap and fluorescence from 2-HE cannot be separated under a fluorescent microscope, red fluorescence caused by HEt oxidation is still a powerful tool for detecting ROS, mainly O2-. Upregulation of this red fluorescence suggests that O2- affects signaling and injury after cerebral ischemia [17-20].
A disadvantage of HEt is that reliable quantification cannot be provided with a fluorescent microscope. For specific and quantitative detection of O2-, a high-performance liquid chromatography/fluorescence assay [15], in addition to a fluorescent microscope study, may be required.
2.4. Transgenic and knockout studies of SOD
Although development of methodologies to detect and quantify ROS have enabled researchers to investigate their roles after cerebral ischemia, their causative roles in ischemic brain injury remain unclear. Advances in transgene and gene knockout (KO) technology have allowed us to investigate the contributions of ROS to molecular mechanisms of ischemic brain injury. Table 2 shows studies using cerebral ischemia models with transgenic (Tg) animals that carry human SOD genes or KO animals that are homozygously or heterozygously deficient in SOD genes.
Table 2.
Transgenic and knockout studies of superoxide dismutases using in vivo cerebral ischemia models
| Study | Animal | Model | Main findings | References |
|---|---|---|---|---|
| SOD1 +/− | Mouse | pFCI | Decreased cortical infarct (−35%) | [23] |
| SOD1 +/− | Mouse | pFCI | No protection | [84] |
| SOD1 +/− | Mouse | tFCI | Decreased infarct | [85] |
| SOD1 +/− | Mouse | tFCI | Sustained hsp70 mRNA expression | [86] |
| SOD1 +/− | Mouse | tFCI | Sustained c-fos mRNA expression | [87] |
| SOD1 +/− | Mouse | tGCI | Induction of hsp 70 | [88] |
| SOD1 +/− | Mouse | tFCI | Decreased injury (−50%) | [24] |
| SOD1 +/− | Rat | tGCI | Decreased injury (−50%) | [25] |
| SOD1 +/− | Mouse | tGCI | Decreased injury (−50%) | [26] |
| SOD1 +/− | Mouse | tFCI | Decreased DNA fragmentation | [89] |
| SOD1 +/− | Mouse | tFCI | Decreased cytochrome c release | [90] |
| SOD1 +/− | Mouse | tFCI | Decreased NF-κB expression | [32] |
| SOD1 +/− | Mouse | tFCI | Decreased activation of activator protein-1 | [91] |
| SOD1 +/− | Mouse | pFCI | No difference in infarct volume | [92] |
| SOD1 +/− | Rat | tGCI | Decreased active caspase-3, -9 | [22] |
| SOD1 +/− | Mouse | tFCI | Decreased ERK activation | [29] |
| SOD1 +/− | Mouse | tFCI | Decreased Bad activation | [33] |
| SOD1 +/− | Mouse | tFCI | Increased pAkt expression; decreased DNA fragmentation | [27] |
| SOD1 +/− | Mouse | tFCI | Decreased PARP activation | [93] |
| SOD1 +/− | Mouse | tFCI | Decreased lesion size and edema; decreased MMP-2, -9 expression | [36] |
| SOD1 +/− | Rat | tGCI | Decreased injury, PERK phosphorylation and GRP78 release | [94] |
| SOD1 +/− | Mouse | tFCI | Decreased injury, PERK phosphorylation and GRP78 release | [95] |
| SOD1 +/− | Mouse | tFCI | Decreased binding of XIAP/DNP, Smac/DNP and caspase-9/DNP | [96] |
| SOD1 +/− | Mouse | tFCI | Decreased Omi/HtrA2 activation | [96] |
| SOD1 +/− | Mouse | tFCI | Increased ILK expression and ILK/Akt complex | [97] |
| SOD1 +/− | Rat | tGCI | Inhibited ATF-4 induction and CHOP expression; decreased endoplasmic reticulum damage |
[98] |
| SOD1 +/− | Mouse | tFCI | Inhibited ATF-4 induction and CHOP expression | [98] |
| SOD1 +/− | Mouse | tFCI | Increased proteasome activity and MDM2 activation; decreased nuclear p53 | [31] |
| SOD1 +/− | Rat | tGCI | Inhibited APE/Ref-1 decrease; decreased injury | [99] |
| SOD1 +/− | Mouse | tFCI | Decreased MCP-1 and MIP-1α expression | [100] |
| SOD1 +/− | Mouse | tFCI | Decreased level of O2-; decreased NF-κB activation and phosphorylation | [20] |
| SOD1 +/− | Mouse | tFCI | Increased pPRAS, pPRAS/pAkt binding and pPRAS/14-3-3 protein binding | [101] |
| SOD1 +/− | Rat | tGCI | Increased pAkt and pGSK-3β expression | [28] |
| SOD1 +/− | Rat | tGCI | Decreased p53 translocation to mitochondria | [58] |
| SOD1 +/− | Mouse | tFCI | Decreased level of O2-; inhibited persistent upregulation of NF-κB | [19] |
| SOD1 +/− | Rat | tFCI with hyperglycemia |
Decreased MMP activity and Evans blue leakage | [102] |
| SOD1 +/− | Rat | tFCI | Decreased activity of p38, phospho-p38, Evans blue leakage, edema and infarct | [30] |
| SOD1 +/− | Rat | tGCI | Decreased PUMA activation and injury; decreased level of O2- | [18] |
| SOD1 −/− | Mouse | tFCI | Increased infarct (+40%) | [34] |
| SOD1 −/− | Mouse | tFCI | Increased lesion size and edema | [35] |
| SOD1 −/− | Mouse | tGCI | Increased cell death | [37] |
| SOD1 −/− | Mouse | pFCI | No difference in infarct volume | [92] |
| SOD1 −/− | Mouse | tFCI | Increased edema | [36] |
| SOD2 +/− | Mouse | tFCI | Decreased injury (−50%) | [38] |
| SOD2 +/− | Mouse | tFCI | Decreased vascular endothelial cell death | [39] |
| SOD2 −/+ | Mouse | pFCI | Increased infarct (+66%) | [40] |
| SOD2 −/+ | Mouse | pFCI | Increased active caspase-9 | [41] |
| SOD2 −/+ | Mouse | tFCI | Increased cytochrome c release | [42] |
| SOD2 −/+ | Mouse | pFCI | Increased O2- production | [17] |
| SOD2 −/+ | Mouse | tFCI | Increased MMP-9 expression | [103] |
| SOD2 −/+ | Mouse | tFCI | Increased MMP activity, edema, inflammation and hemorrhagic transformation | [39] |
| SOD3 +/− | Mouse | tFCI | Decreased infarct (−28%) | [43] |
| SOD3 +/− | Mouse | tGCI | Decreased injury (−48%) | [45] |
| SOD3 −/− | Mouse | tFCI | Increased infarct (+81%) | [46] |
+/−, heterozygous transgenic animals carrying human SOD genes; −/+ heterozygous knockout mutant of SOD genes; −/− homozygous knockout mutant of SOD genes.
Abbreviations used are: pFCI, permanent focal cerebral ischemia; tFCI, transient focal cerebral ischemia; tGCI, transient global cerebral ischemia; APE, apurinic/apyrimidinic endonuclease; ATF-4, activating transcription factor-4; CHOP, C/EBP homologous protein; DNP, 2,4-dinitrophenylhydrazone; GRP78, glucose-regulated protein 78; ILK, integrin-linked kinase; MCP-1, monocyte chemoattractant protein 1; MIP-1α, macrophage inflammatory protein-1α; MMP, matrix metalloproteinase; NF-κB, nuclear factor-κB; pAkt, phosphorylated Akt; PERK, phosphorylation of RNA-dependent protein kinase-like endoplasmic reticulum eukaryotic initiation factor 2α kinase; PARP, poly(ADP-ribose) polymerase; pGSK-3β, phosphorylated glycogen synthase kinase-3β; pPRAS, phosphorylated proline-rich Akt substrate; PUMA, p53-upregulated modulator of apoptosis; Ref-1, redox factor-1; Smac, second mitochondria-derived activator of caspases; XIAP, X chromosome-linked inhibitor of apoptosis protein.
SOD1 is neuroprotective. In heterozygous SOD1 Tg animals that carry the human SOD1 gene, SOD1 activity increased (a three-fold increase in SOD1 Tg mice [21] and an approximate four-fold increase in SOD1 Tg rats [22]) compared with wild-type (Wt) animals. In SOD1 Tg animals, a 35-50% decrease in infarct volume is usually observed after focal cerebral ischemia (FCI) [23,24]. After transient global cerebral ischemia (tGCI), delayed neuronal cell death decreases to about 50% in SOD1 Tg animals [25,26]. Regulation of various pathways contributes to neuroprotection, including activation of the phosphoinositide 3-kinase (PI3-K) pathway [27,28], and inhibition of the mitogen-activated protein kinase (MAPK) -related pathway [29,30], and the p53 signaling [18,31], nuclear factor-κB [19,20,32], and mitochondria-dependent apoptotic [22,29,33] pathways. Moreover, infarct volume and edema levels decrease after FCI in homozygous SOD1 KO mice [34-36], while cell death increases after tGCI [37].
SOD2 also has important neuroprotective roles. Heterozygous SOD2 Tg mice carrying the human SOD2 gene showed decreased injury [38] and reduced vascular endothelial cell death [39] after FCI. Moreover, infarct volume [40], brain edema [39], O2- production [17], matrix metalloproteinase-9 activity [39], caspase-9 activation [41], and cytochrome c release [42] increase after FCI in SOD2 KO mice compared with Wt mice. Furthermore, hemorrhagic transformation after transient FCI (tFCI) significantly increases in SOD2 KO mice [39].
Although only a few studies have used SOD3 Tg or KO mice in cerebral ischemia models, they have shown that SOD3 has neuroprotective roles. Infarct volume after FCI decreases (−28%) in SOD3 Tg mice [43] that express a five-fold higher level of SOD3 in the brain compared with Wt mice [44]. Neuronal death after tGCI also decreases (−48%) in SOD3 Tg mice [45]. In contrast, infarct after FCI in homozygous SOD3 KO mice increases (+81%) [46]. In summary, studies using various SOD Tg and KO animals imply that ROS have important roles in activating various pathways and determining the outcome after cerebral ischemia.
2.5. Mitochondrial NOS
Three canonical isoforms of NOS are well known in mammals: neuronal NOS (nNOS), inducible NOS, and endothelial NOS. Recent findings reveal that mitochondria contain their own isoform of NOS, mitochondrial NOS (mtNOS), at their inner membrane [47,48]. Since NOS isoforms are encoded not by mitochondrial DNA, but by nuclear DNA, mtNOS is thought to be synthesized in the cytosol and translocated to mitochondria [49], although the mechanism of this translocation remains unknown. mtNOS stays active because of mitochondrial Ca2+ content, in contrast to other nitric oxide sources. mtNOS continuously controls mitochondrial respiration [47,48] and is considered a key molecule of reperfusion injury [50].
The enzymatic activity of mtNOS was higher in hypoxic animals than in normoxic controls [51]. mtNOS is also considered a marker of brain aging. In aged mice, mtNOS activity was linearly correlated with neurological performance and survival [52]. Since mtNOS controls mitochondrial respiration and nitric oxide generation, it may correlate with apoptosis after stroke. Further studies may reveal the roles of mtNOS after stroke and may provide novel therapeutic strategies.
3. Ischemic neuronal apoptotic pathways (Fig. 1)
Fig. 1.

Mitochondria-dependent pathways of apoptosis in cerebral ischemia and reperfusion. After cerebral ischemia, various pathways, such as the death receptor pathway, p53 pathway, c-Jun N-terminal kinase (JNK) pathway, PI3-K pathway, and the MAPK pathway are activated. Most signaling pathways induce apoptosis with the help of pro-apoptotic proteins, such as cytochrome c, Endo G, AIF and Smac, which are stored in mitochondria.
3.1. The intrinsic pathway
After mitochondria trigger various signaling pathways by overproduction of ROS, some, but not all, apoptotic signals return to mitochondria with the help of BH3-only proteins. Then, Bcl-2 family proteins (such as cytochrome c, AIF, endonuclease G [Endo G], and second mitochondria-derived activator of caspase [Smac]) interact with each other, resulting in the release of pro-apoptotic proteins stored in the mitochondrial intermembrane space, followed by neuronal apoptosis. This pathway is called the ‘intrinsic pathway’.
3.2. Bcl-2 family protein interactions
The Bcl-2 protein family, which is a principal regulator of mitochondrial membrane integrity and function, is classified into three subgroups according to structural homology: the anti-apoptotic proteins such as Bcl-2, Bcl-XL, and Bcl-w, the pro-apoptotic proteins such as Bax and Bak, and the BH3-only proteins including Bad, Bid, Bim, Noxa, and p53-upregulated modulator of apoptosis (PUMA). Since neurons lack full-length Bak, Bax is the only pro-apoptotic protein in neurons. In response to apoptotic stimuli, specific BH3-only proteins are activated and transduce apoptotic signals to mitochondria. Studies have shown that after cerebral ischemia, BH3-only proteins were upregulated, meaning cerebral ischemia activates various apoptotic pathways.
Currently, two main ideas can explain Bcl-2 protein family interaction: the ‘direct model’ and the ‘hierarchy model’ (Fig. 2). In the direct model, anti-apoptotic proteins trap pro-apoptotic proteins. BH3-only proteins disrupt this interaction, resulting in liberation of pro-apoptotic proteins and subsequent apoptosis (Fig. 2A).
Fig. 2.
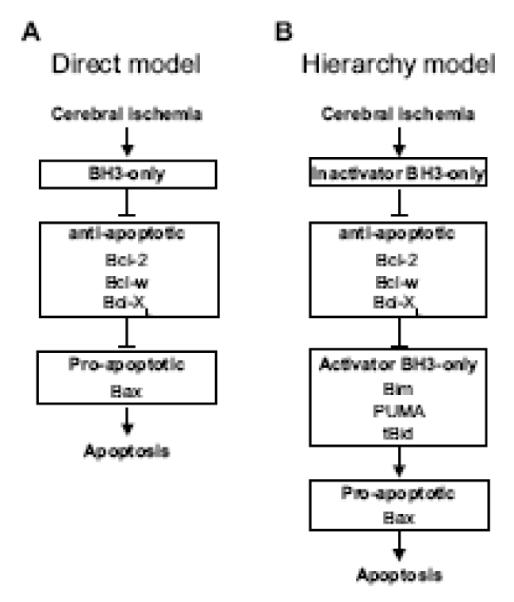
Two models of Bcl-2 protein family interaction. (A) The direct model for Bax activation. After apoptotic stimuli, specific BH3-only proteins are activated and inhibit anti-apoptotic Bcl-2 family proteins. Liberated Bax oligomerizes and triggers the release of pro-apoptotic proteins stored in the mitochondrial intermembrane space. (B) The hierarchy model for Bax activation. After apoptotic stimuli, specific inactivator BH3-only proteins are activated and inhibit anti-apoptotic Bcl-2 family proteins. Then, liberated activator BH3-only proteins interact with Bax, resulting in the release of pro-apoptotic proteins stored in the mitochondrial intermembrane space.
Recently, Kim et al. [53] advocated the ‘hierarchy model’. In this model, BH3-only proteins are subdivided into two groups: ‘activator’ and ‘inactivator’. Bim, PUMA, and truncated Bid (tBid) belong to the activator group and other BH3-only proteins belong to the inactivator group. Activator BH3-only proteins are trapped by anti-apoptotic proteins, whereas pro-apoptotic proteins are not. Inactivator BH3-only proteins disrupt this interaction, resulting in liberation of activator BH3-only proteins. Liberated activator BH3-only proteins interact with pro-apoptotic proteins, followed by apoptosis (Fig. 2B).
The Bcl-2 family plays various roles in cerebral ischemia. BH3-only proteins, including Bad [33,54,55], Bim [56,57], Noxa [57,58], PUMA [18,59], and tBid [18,60] contribute to cell death after cerebral ischemia, mainly through interactions with other Bcl-2 family members. Bax increases after tGCI [61] or FCI [62], and translocates from the cytosol to mitochondria, mediated by c-Jun N-terminal kinase with BimL [56]. Bim [56], tBid [63], and PUMA [18] have been reported to interact with Bax after cerebral ischemia, which may support the hierarchy model. After interacting with other Bcl-2 family proteins, Bax is oligomerized and activated, which triggers release of apoptotic proteins stored in the mitochondrial intermembrane space, leading to neuronal apoptosis [8,56].
3.3. Bcl-2 family downstream interactions
Proteins in the mitochondrial intermembrane space, including cytochrome c [64,65], Smac [66], AIF [67], and Endo G [68], are released after cerebral ischemia, at which time they cause transduction of apoptotic signals. Release of these proteins leads to ‘the point of no return’. Cytochrome c interacts with apoptosis activating factor-1, deoxyadenosine triphosphate, and procaspase-9, and forms the apoptosome, which activates procaspase-9 [69-71]. Caspase-9 activates procaspase-3, then caspase-3 cleaves inhibitor of caspase-activated DNase, which is an inhibitor and a chaperone of caspase-activated DNase. Liberated caspase-activated DNase damages DNA and induces apoptosis. Caspase-3 can also activate other effector caspases, which activate crucial substrates, including poly(ADP-ribose) polymerase (PARP), after cerebral ischemia [72,73]. Although PARP is involved in both apoptotic and non-apoptotic cell death, 89- and 21-kDa fragments are cleaved by caspases and are related to apoptosis after cerebral ischemia [73,74].
Smac also contributes to activation of caspases. Smac released from mitochondria binds to and neutralizes the effect of the X chromosome-linked inhibitor-of-apoptosis protein, which prevents procaspase activation and inhibits activities of activated caspases [66,74] after cerebral ischemia.
Recent reports show the importance of the caspase-independent pathways. AIF translocates from mitochondria to the nucleus and induces apoptosis after tFCI [67]. In mutant mice that express low-level AIF, infarct volume decreased (−43%) after tFCI [67]. PARP helped nuclear translocation of AIF [75]. Endo G is also known to translocate to the nucleus, causing DNA fragmentation after tFCI [68].
3.4. Upstream of the intrinsic pathway
ROS activate a number of pathways, including PI3-K, MAPK, and p53 pathways. These pathways modulate the intrinsic pathway.
3.4.1. Kinase pathway
Akt is a key molecule for neuronal death and survival after cerebral ischemia [27]. Akt is a serine/threonine kinase and a major downstream target of PI3-K. Akt phosphorylates and inactivates Bad after cerebral ischemia [55]. Since phosphorylated Bad is unable to inhibit the pro-survival Bcl-2 family proteins, Bad phosphorylation results in inactivation of the apoptotic pathway. Akt also phosphorylates procaspase-9 and caspase-9 on serine-196; procaspase-9 phosphorylation inhibits activation of procaspase-9, and caspase-9 phosphorylation inhibits protease activity [76]. Akt modulates p53 degradation through MDM2 phosphorylation [31].
Other kinases also have regulative roles in the intrinsic pathway. Phosphorylated extracellular signal-regulated kinase, which also phosphorylates and inactivates Bad, is upregulated after tFCI [29]. Protein kinase A phosphorylates and inactivates Bad after cerebral ischemia [33].
3.4.2. p53 signaling pathway
Since a number of Bcl-2 family proteins such as Bax, Bid, Noxa, PUMA, and p53AIP1 are the products of p53, p53 plays important roles in the intrinsic pathway. These Bcl-2 family proteins increase and regulate cell death after cerebral ischemia, as described in section 3.2. Recent findings suggest that p53 can activate the intrinsic pathway in a transcription-independent manner, as well as in a transcription-dependent manner [58]. p53 translocates to mitochondria and interacts with anti-apoptotic Bcl-XL, which precedes cytochrome c release after tGCI [58]. A p53 inhibitor, pifithrin-α, decreased the translocation of p53, and resulted in neuroprotection in the hippocampal CA1 subregion after tGCI [58]. In summary, p53 acts as a BH3-only protein in this transcription-independent manner in addition to transcription of apoptosis-related proteins such as Bcl-2 family proteins.
3.4.3. PIDD signaling pathway
p53 and caspase-2 are involved with stress-induced apoptosis. However, the key molecules connecting them have not been determined. Tinel and Tschopp [77] reported that p53-induced protein with a death domain (PIDD), which is a target of p53, formed a high-molecular weight protein complex with RAIDD and procaspase-2. This molecular complex is referred to the ‘PIDDosome’, in which caspase-2 is activated, similar to caspase-9 activation in the apoptosome [77]. After tGCI, the PIDDosome increased in the hippocampal CA1 subregion, followed by caspase-2 activation and Bid cleavage, which preceded neuronal death [78].
Recently, in vitro studies have presented new findings regarding this PIDD pathway. One finding is that caspase-2 can directly interact with mitochondria and activate the mitochondria-dependent apoptotic pathway [79,80]. Interestingly, this interaction occurs independently of its proteolytic activity. Another finding is that a cleaved fragment of PIDD (PIDD-C) forms protein complexes that differ from the PIDDosome. The protein complex containing PIDD-C and nuclear factor-κB has an anti-apoptotic role in response to genotoxic stress [81]. These interactions after cerebral ischemia are unknown and require further study.
3.4.4. Crosstalk between the intrinsic pathway and the extrinsic pathway
The extrinsic pathway is the death-receptor-mediated pathway that receives extracellular signals and transduces them to intracellular signals. Recent studies have shown that the death receptor pathway has various physiological functions as well as apoptotic roles.
The Fas pathway (Fas is a death receptor) is involved in apoptosis after cerebral ischemia. mRNA and protein levels of both Fas and the Fas ligand are upregulated after cerebral ischemia [82,83]. Mutant mice that have a loss-of-function mutation for Fas show reduced infarct volume after FCI [82]. Fas, Fas-associated death domain, and procaspase-8 form a protein complex that is referred to as the death-inducing signaling complex (DISC). DISC activates procaspase-8, similar to procaspase-9 activation by the apoptosome. Caspase-8 activation is followed by activation of caspases −3 and −10 after cerebral ischemia [83].
There is crosstalk between the intrinsic pathway and the extrinsic pathway. The key molecule involved in this crosstalk is Bid, which is also a key molecule for the p53-caspase-2 pathway as described above. Bid is truncated by caspase-8, translocates to mitochondria, and interacts with other Bcl-2 family proteins, which causes cytochrome c release followed by apoptotic cell death [60].
4. Conclusions
Numerous reports show the involvement of ROS in cell death after cerebral ischemia. ROS contribute not only to injury of macromolecules, but also to transduction of apoptotic signals. Although it is well known that various factors, including necrosis, are involved in the mechanisms of cell death after cerebral ischemia, mitochondria contribute to cell death by activating signaling pathways through ROS production and by regulating intrinsic apoptosis pathways. Future studies of these cell death mechanisms after ischemia may provide unique information regarding molecular targets for therapeutic strategies in clinical stroke.
Acknowledgements
This work was supported by National Institutes of Health grants P50 NS014543, RO1 NS025372, RO1 NS036147, and RO1 NS038653. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. We thank Liza Reola and Bernard Calagui for technical assistance and Cheryl Christensen for editorial assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem. J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J. Cereb. Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. doi:10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- [3].Chan PH. Oxygen radicals in focal cerebral ischemia. Brain Pathol. 1994;4:59–65. doi: 10.1111/j.1750-3639.1994.tb00811.x. [DOI] [PubMed] [Google Scholar]
- [4].Allen RG, Tresini M. Oxidative stress and gene regulation. Free Radic. Biol. Med. 2000;28:463–499. doi: 10.1016/s0891-5849(99)00242-7. [DOI] [PubMed] [Google Scholar]
- [5].Thannickal VJ, Day RM, Klinz SG, Bastien MC, Larios JM, Fanburg BL. Ras-dependent and -independent regulation of reactive oxygen species by mitogenic growth factors and TGF-β1. FASEB J. 2000;14:1741–1748. doi: 10.1096/fj.99-0878com. [DOI] [PubMed] [Google Scholar]
- [6].Patel RP, McAndrew J, Sellak H, White CR, Jo H, Freeman BA, Darley-Usmar VM. Biological aspects of reactive nitrogen species. Biochim. Biophys. Acta. 1999;1411:385–400. doi: 10.1016/s0005-2728(99)00028-6. doi:10.1016/S0005-2728(99)00028-6. [DOI] [PubMed] [Google Scholar]
- [7].Bredt DS. Endogenous nitric oxide synthesis: biological functions and pathophysiology. Free Radic. Res. 1999;31:577–596. doi: 10.1080/10715769900301161. [DOI] [PubMed] [Google Scholar]
- [8].Fiskum G, Murphy AN, Beal MF. Mitochondria in neurodegeneration: acute ischemia and chronic neurodegenerative diseases. J. Cereb. Blood Flow Metab. 1999;19:351–369. doi: 10.1097/00004647-199904000-00001. doi:10.1097/00004647-199904000-00001. [DOI] [PubMed] [Google Scholar]
- [9].Murphy AN, Fiskum G, Beal MF. Mitochondria in neurodegeneration: bioenergetic function in cell life and death. J. Cereb. Blood Flow Metab. 1999;19:231–245. doi: 10.1097/00004647-199903000-00001. doi:10.1097/00004647-199903000-00001. [DOI] [PubMed] [Google Scholar]
- [10].Packer JE, Slater TF, Willson RL. Direct observation of a free radical interaction between vitamin E and vitamin C. Nature. 1979;278:737–738. doi: 10.1038/278737a0. doi:10.1038/278737a0. [DOI] [PubMed] [Google Scholar]
- [11].Siesjö BK. Cell damage in the brain: a speculative synthesis. J. Cereb. Blood Flow Metab. 1981;1:155–185. doi: 10.1038/jcbfm.1981.18. [DOI] [PubMed] [Google Scholar]
- [12].Yoshida S, Abe K, Busto R, Watson BD, Kogure K, Ginsberg MD. Influence of transient ischemia on lipid-soluble antioxidants, free fatty acids and energy metabolites in rat brain. Brain Res. 1982;245:307–316. doi: 10.1016/0006-8993(82)90813-7. [DOI] [PubMed] [Google Scholar]
- [13].Bindokas VP, Jordán J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J. Neurosci. 1996;16:1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Budd SL, Castilho RF, Nicholls DG. Mitochondrial membrane potential and hydroethidine-monitored superoxide generation in cultured cerebellar granule cells. FEBS Lett. 1997;415:21–24. doi: 10.1016/s0014-5793(97)01088-0. [DOI] [PubMed] [Google Scholar]
- [15].Zhao H, Joseph J, Fales HM, Sokoloski EA, Levine RL, Vasquez-Vivar J, Kalyanaraman B. Detection and characterization of the product of hydroethidine and intracellular superoxide by HPLC and limitations of fluorescence. Proc. Natl. Acad. Sci. U. S. A. 2005;102:5727–5732. doi: 10.1073/pnas.0501719102. doi:10.1073/pnas.0501719102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhao H, Kalivendi S, Zhang H, Joseph J, Nithipatikom K, Vásquez-Vivar J, Kalyanaraman B. Superoxide reacts with hydroethidine but forms a fluorescent product that is distinctly different from ethidium: potential implications in intracellular fluorescence detection of superoxide. Free Radic. Biol. Med. 2003;34:1359–1368. doi: 10.1016/s0891-5849(03)00142-4. doi:10.1016/S0891-5849(03)00142-4. [DOI] [PubMed] [Google Scholar]
- [17].Kim GW, Kondo T, Noshita N, Chan PH. Manganese superoxide dismutase deficiency exacerbates cerebral infarction after focal cerebral ischemia/reperfusion in mice. Implications for the production and role of superoxide radicals. Stroke. 2002;33:809–815. doi: 10.1161/hs0302.103745. doi:10.1161/hs0302.103745. [DOI] [PubMed] [Google Scholar]
- [18].Niizuma K, Endo H, Nito C, Myer DJ, Chan PH. Potential role of PUMA in delayed death of hippocampal CA1 neurons after transient global cerebral ischemia. Stroke. 2009;40:618–625. doi: 10.1161/STROKEAHA.108.524447. doi:10.1161/STROKEAHA.108.524447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Song YS, Lee Y-S, Narasimhan P, Chan PH. Reduced oxidative stress promotes NF-κB-mediated neuroprotective gene expression after transient focal cerebral ischemia: lymphocytotrophic cytokines and antiapoptotic factors. J. Cereb. Blood Flow Metab. 2007;27:764–775. doi: 10.1038/sj.jcbfm.9600379. doi:10.1038/sj.jcbfm.9600379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Song YS, Lee Y-S, Chan PH. Oxidative stress transiently decreases the IKK complex (IKKα, β, and γ), an upstream component of NF-κB signaling, after transient focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 2005;25:1301–1311. doi: 10.1038/sj.jcbfm.9600123. doi:10.1038/sj.jcbfm.9600123. [DOI] [PubMed] [Google Scholar]
- [21].Chan PH, Epstein CJ, Kinouchi H, Kamii H, Imaizumi S, Yang G, Chen SF, Gafni J, Carlson E. SOD-1 transgenic mice as a model for studies of neuroprotection in stroke and brain trauma. Ann. N. Y. Acad. Sci. 1994;738:93–103. doi: 10.1111/j.1749-6632.1994.tb21794.x. [DOI] [PubMed] [Google Scholar]
- [22].Sugawara T, Noshita N, Lewén A, Gasche Y, Ferrand-Drake M, Fujimura M, Morita-Fujimura Y, Chan PH. Overexpression of copper/zinc superoxide dismutase in transgenic rats protects vulnerable neurons against ischemic damage by blocking the mitochondrial pathway of caspase activation. J. Neurosci. 2002;22:209–217. doi: 10.1523/JNEUROSCI.22-01-00209.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kinouchi H, Epstein CJ, Mizui T, Carlson E, Chen SF, Chan PH. Attenuation of focal cerebral ischemic injury in transgenic mice overexpressing CuZn superoxide dismutase. Proc. Natl. Acad. Sci. U. S. A. 1991;88:11158–11162. doi: 10.1073/pnas.88.24.11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kamii H, Mikawa S, Murakami K, Kinouchi H, Yoshimoto T, Reola L, Carlson E, Epstein CJ, Chan PH. Effects of nitric oxide synthase inhibition on brain infarction in SOD-1-transgenic mice following transient focal cerebral ischemia. J. Cereb. Blood Flow Metab. 1996;16:1153–1157. doi: 10.1097/00004647-199611000-00009. doi:10.1097/00004647-199611000-00009. [DOI] [PubMed] [Google Scholar]
- [25].Chan PH, Kawase M, Murakami K, Chen SF, Li Y, Calagui B, Reola L, Carlson E, Epstein CJ. Overexpression of SOD1 in transgenic rats protects vulnerable neurons against ischemic damage after global cerebral ischemia and reperfusion. J. Neurosci. 1998;18:8292–8299. doi: 10.1523/JNEUROSCI.18-20-08292.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Murakami K, Kondo T, Epstein CJ, Chan PH. Overexpression of CuZn-superoxide dismutase reduces hippocampal injury after global ischemia in transgenic mice. Stroke. 1997;28:1797–1804. doi: 10.1161/01.str.28.9.1797. [Erratum: Stroke 28:2573, 1997] [DOI] [PubMed] [Google Scholar]
- [27].Noshita N, Sugawara T, Lewén A, Hayashi T, Chan PH. Copper-zinc superoxide dismutase affects Akt activation after transient focal cerebral ischemia in mice. Stroke. 2003;34:1513–1518. doi: 10.1161/01.STR.0000072986.46924.F4. doi:10.1161/01.STR.0000072986.46924.F4. [DOI] [PubMed] [Google Scholar]
- [28].Endo H, Nito C, Kamada H, Nishi T, Chan PH. Activation of the Akt/GSK3β signaling pathway mediates survival of vulnerable hippocampal neurons after transient global cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 2006;26:1479–1489. doi: 10.1038/sj.jcbfm.9600303. doi:10.1038/sj.jcbfm.9600303. [DOI] [PubMed] [Google Scholar]
- [29].Noshita N, Sugawara T, Hayashi T, Lewén A, Omar G, Chan PH. Copper/zinc superoxide dismutase attenuates neuronal cell death by preventing extracellular signal-regulated kinase activation after transient focal cerebral ischemia in mice. J. Neurosci. 2002;22:7923–7930. doi: 10.1523/JNEUROSCI.22-18-07923.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nito C, Kamada H, Endo H, Niizuma K, Myer DJ, Chan PH. Role of the p38 mitogen-activated protein kinase/cytosolic phospholipase A2 signaling pathway in blood—brain barrier disruption after focal cerebral ischemia and reperfusion. J. Cereb. Blood Flow Metab. 2008;28:1686–1696. doi: 10.1038/jcbfm.2008.60. doi:10.1038/jcbfm.2008.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Saito A, Hayashi T, Okuno S, Nishi T, Chan PH. Modulation of p53 degradation via MDM2-mediated ubiquitylation and the ubiquitin-proteasome system during reperfusion after stroke: role of oxidative stress. J. Cereb. Blood Flow Metab. 2005;25:267–280. doi: 10.1038/sj.jcbfm.9600028. doi:10.1038/sj.jcbfm.9600028. [DOI] [PubMed] [Google Scholar]
- [32].Huang C-Y, Fujimura M, Noshita N, Chang Y-Y, Chan PH. SOD1 down-regulates NF-κB and c-myc expression in mice after transient focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2001;21:163–173. doi: 10.1097/00004647-200102000-00008. doi:10.1097/00004647-200102000-00008. [DOI] [PubMed] [Google Scholar]
- [33].Saito A, Hayashi T, Okuno S, Ferrand-Drake M, Chan PH. Overexpression of copper/zinc superoxide dismutase in transgenic mice protects against neuronal cell death after transient focal ischemia by blocking activation of the Bad cell death signaling pathway. J. Neurosci. 2003;23:1710–1718. doi: 10.1523/JNEUROSCI.23-05-01710.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kondo T, Reaume AG, Huang T-T, Carlson E, Murakami K, Chen SF, Hoffman EK, Scott RW, Epstein CJ, Chan PH. Reduction of CuZn-superoxide dismutase activity exacerbates neuronal cell injury and edema formation after transient focal cerebral ischemia. J. Neurosci. 1997;17:4180–4189. doi: 10.1523/JNEUROSCI.17-11-04180.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kondo T, Reaume AG, Huang T-T, Murakami K, Carlson E, Chen S, Scott RW, Epstein CJ, Chan PH. Edema formation exacerbates neurological and histological outcomes after focal cerebral ischemia in CuZn-superoxide dismutase gene knockout mutant mice. Acta Neurochir. Suppl. 1997;70:62–64. doi: 10.1007/978-3-7091-6837-0_19. [DOI] [PubMed] [Google Scholar]
- [36].Kim GW, Gasche Y, Grzeschik S, Copin J-C, Maier CM, Chan PH. Neurodegeneration in striatum induced by the mitochondrial toxin 3-nitropropionic acid: role of matrix metalloproteinase-9 in early blood—brain barrier disruption? J. Neurosci. 2003;23:8733–8742. doi: 10.1523/JNEUROSCI.23-25-08733.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kawase M, Murakami K, Fujimura M, Morita-Fujimura Y, Gasche Y, Kondo T, Scott RW, Chan PH. Exacerbation of delayed cell injury after transient global ischemia in mutant mice with CuZn superoxide dismutase deficiency. Stroke. 1999;30:1962–1968. doi: 10.1161/01.str.30.9.1962. [DOI] [PubMed] [Google Scholar]
- [38].Keller JN, Kindy MS, Holtsberg FW, Clair DK, Yen H-C, Germeyer A, Steiner SM, Bruce-Keller AJ, Hutchins JB, Mattson MP. Mitochondrial manganese superoxide dismutase prevents neural apoptosis and reduces ischemic brain injury: suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction. J. Neurosci. 1998;18:687–697. doi: 10.1523/JNEUROSCI.18-02-00687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Maier CM, Hsieh L, Crandall T, Narasimhan P, Chan PH. Evaluating therapeutic targets for reperfusion-related brain hemorrhage. Ann. Neurol. 2006;59:929–938. doi: 10.1002/ana.20850. doi:10.1002/ana.20850. [DOI] [PubMed] [Google Scholar]
- [40].Murakami K, Kondo T, Kawase M, Li Y, Sato S, Chen SF, Chan PH. Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J. Neurosci. 1998;18:205–213. doi: 10.1523/JNEUROSCI.18-01-00205.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Fujimura M, Morita-Fujimura Y, Kawase M, Copin J-C, Calagui B, Epstein CJ, Chan PH. Manganese superoxide dismutase mediates the early release of mitochondrial cytochrome c and subsequent DNA fragmentation after permanent focal cerebral ischemia in mice. J. Neurosci. 1999;19:3414–3422. doi: 10.1523/JNEUROSCI.19-09-03414.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Noshita N, Sugawara T, Fujimura M, Morita-Fujimura Y, Chan PH. Manganese superoxide dismutase affects cytochrome c release and caspase-9 activation after transient focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 2001;21:557–567. doi: 10.1097/00004647-200105000-00010. doi:10.1097/00004647-200105000-00010. [DOI] [PubMed] [Google Scholar]
- [43].Sheng H, Bart RD, Oury TD, Pearlstein RD, Crapo JD, Warner DS. Mice overexpressing extracellular superoxide dismutase have increased resistance to focal cerebral ischemia. Neuroscience. 1999;88:185–191. doi: 10.1016/s0306-4522(98)00208-5. [DOI] [PubMed] [Google Scholar]
- [44].Oury TD, Ho Y-S, Piantadosi CA, Crapo JD. Extracellular superoxide dismutase, nitric oxide, and central nervous system O2 toxicity. Proc. Natl. Acad. Sci. U. S. A. 1992;89:9715–9719. doi: 10.1073/pnas.89.20.9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Sheng H, Kudo M, Mackensen GB, Pearlstein RD, Crapo JD, Warner DS. Mice overexpressing extracellular superoxide dismutase have increased resistance to global cerebral ischemia. Exp. Neurol. 2000;163:392–398. doi: 10.1006/exnr.2000.7363. doi:10.1006/exnr.2000.7363. [DOI] [PubMed] [Google Scholar]
- [46].Sheng H, Brady TC, Pearlstein RD, Crapo JD, Warner DS. Extracellular superoxide dismutase deficiency worsens outcome from focal cerebral ischemia in the mouse. Neurosci. Lett. 1999;267:13–16. doi: 10.1016/s0304-3940(99)00316-x. [DOI] [PubMed] [Google Scholar]
- [47].Giulivi C, Poderoso JJ, Boveris A. Production of nitric oxide by mitochondria. J. Biol. Chem. 1998;273:11038–11043. doi: 10.1074/jbc.273.18.11038. doi:10.1074/jbc.273.18.11038. [DOI] [PubMed] [Google Scholar]
- [48].Tatoyan A, Giulivi C. Purification and characterization of a nitric-oxide synthase from rat liver mitochondria. J. Biol. Chem. 1998;273:11044–11048. doi: 10.1074/jbc.273.18.11044. doi:10.1074/jbc.273.18.11044. [DOI] [PubMed] [Google Scholar]
- [49].Finocchietto PV, Franco MC, Holod S, Gonzalez AS, Converso DP, Arciuch V.G. Antico, Serra MP, Poderoso JJ, Carreras MC. Mitochondrial nitric oxide synthase: a master piece of metabolic adaptation, cell growth, transformation, and death. Exp. Biol. Med. 2009;234:1020–1028. doi: 10.3181/0902-MR-81. doi:10.3181/0902-MR-81. [DOI] [PubMed] [Google Scholar]
- [50].Ignarro LJ. Heart mtNOS, a key mediator of oxidative injury in ischemia/reperfusion. J. Mol. Cell. Cardiol. 2007;43:409–410. doi: 10.1016/j.yjmcc.2007.07.053. [Editorial] doi:10.1016/j.yjmcc.2007.07.053. [DOI] [PubMed] [Google Scholar]
- [51].Lacza Z, Puskar M, Figueroa JP, Zhang J, Rajapakse N, Busija DW. Mitochondrial nitric oxide synthase is constitutively active and is functionally upregulated in hypoxia. Free Radic. Biol. Med. 2001;31:1609–1615. doi: 10.1016/s0891-5849(01)00754-7. [DOI] [PubMed] [Google Scholar]
- [52].Boveris A, Navarro A. Brain mitochondrial dysfunction in aging. IUBMB Life. 2008;60:308–314. doi: 10.1002/iub.46. [DOI] [PubMed] [Google Scholar]
- [53].Kim H, Rafiuddin-Shah M, Tu H-C, Jeffers JR, Zambetti GP, Hsieh JJ-D, Cheng EH-Y. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat. Cell Biol. 2006;8:1348–1358. doi: 10.1038/ncb1499. doi:10.1038/ncb1499. [DOI] [PubMed] [Google Scholar]
- [54].Jiang P, Du W, Heese K, Wu M. The Bad guy cooperates with good cop p53: Bad is transcriptionally up-regulated by p53 and forms a Bad/p53 complex at the mitochondria to induce apoptosis. Mol. Cell. Biol. 2006;26:9071–9082. doi: 10.1128/MCB.01025-06. doi:10.1128/MCB.01025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kamada H, Nito C, Endo H, Chan PH. Bad as a converging signaling molecule between survival PI3-K/Akt and death JNK in neurons after transient focal cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 2007;27:521–533. doi: 10.1038/sj.jcbfm.9600367. doi:10.1038/sj.jcbfm.9600367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Okuno S, Saito A, Hayashi T, Chan PH. The c-Jun N-terminal protein kinase signaling pathway mediates Bax activation and subsequent neuronal apoptosis through interaction with Bim after transient focal cerebral ischemia. J. Neurosci. 2004;24:7879–7887. doi: 10.1523/JNEUROSCI.1745-04.2004. doi:10.1523/JNEUROSCI.1745-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Inta I, Paxian S, Maegele I, Zhang W, Pizzi M, Spano P, Sarnico I, Muhammad S, Herrmann O, Inta D, Baumann B, Liou H-C, Schmid RM, Schwaninger M. Bim and Noxa are candidates to mediate the deleterious effect of the NF-κB subunit RelA in cerebral ischemia. J. Neurosci. 2006;26:12896–12903. doi: 10.1523/JNEUROSCI.3670-06.2006. doi:10.1523/jneurosci.3670-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Endo H, Kamada H, Nito C, Nishi T, Chan PH. Mitochondrial translocation of p53 mediates release of cytochrome c and hippocampal CA1 neuronal death after transient global cerebral ischemia in rats. J. Neurosci. 2006;26:7974–7983. doi: 10.1523/JNEUROSCI.0897-06.2006. doi:10.1523/JNEUROSCI.0897-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Reimertz C, Kögel D, Rami A, Chittenden T, Prehn JHM. Gene expression during ER stress—induced apoptosis in neurons: induction of the BH3-only protein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. J. Cell Biol. 2003;162:587–597. doi: 10.1083/jcb.200305149. doi:10.1083/jcb.200305149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Plesnila N, Zinkel S, Le DA, Amin-Hanjani S, Wu Y, Qiu J, Chiarugi A, Thomas SS, Kohane DS, Korsmeyer SJ, Moskowitz MA. BID mediates neuronal cell death after oxygen/glucose deprivation and focal cerebral ischemia. Proc. Natl. Acad. Sci. U. S. A. 2001;98:15318–15323. doi: 10.1073/pnas.261323298. doi:10.1073/pnas.261323298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Krajewski S, Mai JK, Krajewska M, Sikorska M, Mossakowski MJ, Reed JC. Upregulation of Bax protein levels in neurons following cerebral ischemia. J. Neurosci. 1995;15:6364–6376. doi: 10.1523/JNEUROSCI.15-10-06364.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Gillardon F, Lenz C, Waschke KF, Krajewski S, Reed JC, Zimmermann M, Kuschinsky W. Altered expression of Bcl-2, Bcl-X, Bax, and c-Fos colocalizes with DNA fragmentation and ischemic cell damage following middle cerebral artery occlusion in rats. Mol. Brain Res. 1996;40:254–260. doi: 10.1016/0169-328x(96)00059-9. [DOI] [PubMed] [Google Scholar]
- [63].Polster BM, Fiskum G. Mitochondrial mechanisms of neural cell apoptosis. J. Neurochem. 2004;90:1281–1289. doi: 10.1111/j.1471-4159.2004.02572.x. doi:10.1111/j.1471-4159.2004.02572.x. [DOI] [PubMed] [Google Scholar]
- [64].Fujimura M, Morita-Fujimura Y, Murakami K, Kawase M, Chan PH. Cytosolic redistribution of cytochrome c after transient focal cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 1998;18:1239–1247. doi: 10.1097/00004647-199811000-00010. doi:10.1097/00004647-199811000-00010. [DOI] [PubMed] [Google Scholar]
- [65].Sugawara T, Fujimura M, Morita-Fujimura Y, Kawase M, Chan PH. Mitochondrial release of cytochrome c corresponds to the selective vulnerability of hippocampal CA1 neurons in rats after transient global cerebral ischemia. J. Neurosci. 1999;19(RC39):1–6. doi: 10.1523/JNEUROSCI.19-22-j0002.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Saito A, Hayashi T, Okuno S, Ferrand-Drake M, Chan PH. Interaction between XIAP and Smac/DIABLO in the mouse brain after transient focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2003;23:1010–1019. doi: 10.1097/01.WCB.0000080702.47016.FF. doi:10.1097/01.WCB.0000080702.47016.FF. [DOI] [PubMed] [Google Scholar]
- [67].Culmsee C, Zhu C, Landshamer S, Becattini B, Wagner E, Pellechia M, Blomgren K, Plesnila N. Apoptosis-inducing factor triggered by poly(ADP-ribose) polymerase and Bid mediates neuronal cell death after oxygen—glucose deprivation and focal cerebral ischemia. J. Neurosci. 2005;25:10262–10272. doi: 10.1523/JNEUROSCI.2818-05.2005. doi:10.1523/jneurosci.2818-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Lee BI, Lee DJ, Cho KJ, Kim GW. Early nuclear translocation of endonuclease G and subsequent DNA fragmentation after transient focal cerebral ischemia in mice. Neurosci. Lett. 2005;386:23–27. doi: 10.1016/j.neulet.2005.05.058. doi:10.1016/j.neulet.2005.05.058. [DOI] [PubMed] [Google Scholar]
- [69].Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- [70].Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c—dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- [71].Yoshida H, Kong Y-Y, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–750. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- [72].Krupinski J, Lopez E, Marti E, Ferrer I. Expression of caspases and their substrates in the rat model of focal cerebral ischemia. Neurobiol. Dis. 2000;7:332–342. doi: 10.1006/nbdi.2000.0310. doi:10.1006/nbdi.2000.0310. [DOI] [PubMed] [Google Scholar]
- [73].Chaitanya GV, Babu PP. Differential PARP cleavage: an indication of heterogeneous forms of cell death and involvement of multiple proteases in the infarct of focal cerebral ischemia in rat. Cell. Mol. Neurobiol. 2009;29:563–573. doi: 10.1007/s10571-009-9348-8. doi:10.1007/s10571-009-9348-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Ferrer I, Planas AM. Signaling of cell death and cell survival following focal cerebral ischemia: life and death struggle in the penumbra. J. Neuropathol. Exp. Neurol. 2003;62:329–339. doi: 10.1093/jnen/62.4.329. [DOI] [PubMed] [Google Scholar]
- [75].Yu S-W, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, Dawson VL. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc. Natl. Acad. Sci. U. S. A. 2006;103:18314–18319. doi: 10.1073/pnas.0606528103. doi:10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. doi:10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- [77].Tinel A, Tschopp J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science. 2004;304:843–846. doi: 10.1126/science.1095432. doi:10.1126/science.1095432. [DOI] [PubMed] [Google Scholar]
- [78].Niizuma K, Endo H, Nito C, Myer DJ, Kim GS, Chan PH. The PIDDosome mediates delayed death of hippocampal CA1 neurons after transient global cerebral ischemia in rats. Proc. Natl. Acad. Sci. U. S. A. 2008;105:16369–16374. doi: 10.1073/pnas.0806222105. doi:10.1073/pnas.0806222105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Robertson JD, Gogvadze V, Kropotov A, Vakifahmetoglu H, Zhivotovsky B, Orrenius S. Processed caspase-2 can induce mitochondria-mediated apoptosis independently of its enzymatic activity. EMBO Rep. 2004;5:643–648. doi: 10.1038/sj.embor.7400153. doi:10.1038/sj.embor.7400153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Enoksson M, Robertson JD, Gogvadze V, Bu P, Kropotov A, Zhivotovsky B, Orrenius S. Caspase-2 permeabilizes the outer mitochondrial membrane and disrupts the binding of cytochrome c to anionic phospholipids. J. Biol. Chem. 2004;279:49575–49578. doi: 10.1074/jbc.C400374200. doi:10.1074/jbc.C400374200. [DOI] [PubMed] [Google Scholar]
- [81].Tinel A, Janssens S, Lippens S, Cuenin S, Logette E, Jaccard B, Quadroni M, Tschopp J. Autoproteolysis of PIDD marks the bifurcation between pro-death caspase-2 and pro-survival NF-κB pathway. EMBO J. 2007;26:197–208. doi: 10.1038/sj.emboj.7601473. doi:10.1038/sj.emboj.7601473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Rosenbaum DM, Gupta G, D’Amore J, Singh M, Weidenheim K, Zhang H, Kessler JA. Fas (CD95/APO-1) plays a role in the pathophysiology of focal cerebral ischemia. J. Neurosci. Res. 2000;61:686–692. doi: 10.1002/1097-4547(20000915)61:6<686::AID-JNR12>3.0.CO;2-7. doi:10.1002/1097-4547(20000915)61:6<686::AID-JNR12>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- [83].Jin K, Graham SH, Mao X, Nagayama T, Simon RP, Greenberg DA. Fas (CD95) may mediate delayed cell death in hippocampal CA1 sector after global cerebral ischemia. J. Cereb. Blood Flow Metab. 2001;21:1411–1421. doi: 10.1097/00004647-200112000-00005. doi:10.1097/00004647-200112000-00005. [DOI] [PubMed] [Google Scholar]
- [84].Chan PH, Kamii H, Yang G, Gafni J, Epstein CJ, Carlson E, Reola L. Brain infarction is not reduced in SOD-1 transgenic mice after a permanent focal cerebral ischemia. Neuroreport. 1993;5:293–296. doi: 10.1097/00001756-199312000-00028. [DOI] [PubMed] [Google Scholar]
- [85].Yang G, Chan PH, Chen J, Carlson E, Chen SF, Weinstein P, Epstein CJ, Kamii H. Human copper-zinc superoxide dismutase transgenic mice are highly resistant to reperfusion injury after focal cerebral ischemia. Stroke. 1994;25:165–170. doi: 10.1161/01.str.25.1.165. [DOI] [PubMed] [Google Scholar]
- [86].Kamii H, Kinouchi H, Sharp FR, Koistinaho J, Epstein CJ, Chan PH. Prolonged expression of hsp70 mRNA following transient focal cerebral ischemia in transgenic mice overexpressing CuZn-superoxide dismutase. J. Cereb. Blood Flow Metab. 1994;14:478–486. doi: 10.1038/jcbfm.1994.59. [DOI] [PubMed] [Google Scholar]
- [87].Kamii H, Kinouchi H, Sharp FR, Epstein CJ, Sagar SM, Chan PH. Expression of c-fos mRNA after a mild focal cerebral ischemia in SOD-1 transgenic mice. Brain Res. 1994;662:240–244. doi: 10.1016/0006-8993(94)90818-4. [DOI] [PubMed] [Google Scholar]
- [88].Kondo T, Murakami K, Honkaniemi J, Sharp FR, Epstein CJ, Chan PH. Expression of hsp70 mRNA is induced in the brain of transgenic mice overexpressing human CuZn-superoxide dismutase following transient global cerebral ischemia. Brain Res. 1996;737:321–326. doi: 10.1016/0006-8993(96)00949-3. [DOI] [PubMed] [Google Scholar]
- [89].Fujimura M, Morita-Fujimura Y, Narasimhan P, Copin J-C, Kawase M, Chan PH. Copper-zinc superoxide dismutase prevents the early decrease of apurinic/apyrimidinic endonuclease and subsequent DNA fragmentation after transient focal cerebral ischemia in mice. Stroke. 1999;30:2408–2415. doi: 10.1161/01.str.30.11.2408. [DOI] [PubMed] [Google Scholar]
- [90].Fujimura M, Morita-Fujimura Y, Noshita N, Sugawara T, Kawase M, Chan PH. The cytosolic antioxidant copper/zinc-superoxide dismutase prevents the early release of mitochondrial cytochrome c in ischemic brain after transient focal cerebral ischemia in mice. J. Neurosci. 2000;20:2817–2824. doi: 10.1523/JNEUROSCI.20-08-02817.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Huang C-Y, Fujimura M, Chang Y-Y, Chan PH. Overexpression of copper-zinc superoxide dismutase attenuates acute activation of activator protein-1 after transient focal cerebral ischemia in mice. Stroke. 2001;32:741–747. doi: 10.1161/01.str.32.3.741. [DOI] [PubMed] [Google Scholar]
- [92].Fujimura M, Morita-Fujimura Y, Copin J-C, Yoshimoto T, Chan PH. Reduction of copper,zinc-superoxide dismutase in knockout mice does not affect edema or infarction volumes and the early release of mitochondrial cytochrome c after permanent focal cerebral ischemia. Brain Res. 2001;889:208–213. doi: 10.1016/s0006-8993(00)03134-6. [DOI] [PubMed] [Google Scholar]
- [93].Narasimhan P, Fujimura M, Noshita N, Chan PH. Role of superoxide in poly(ADP-ribose) polymerase upregulation after transient cerebral ischemia. Mol. Brain Res. 2003;113:28–36. doi: 10.1016/s0169-328x(03)00062-7. doi:10.1016/S0169-328X(03)00062-7. [DOI] [PubMed] [Google Scholar]
- [94].Hayashi T, Saito A, Okuno S, Ferrand-Drake M, Dodd RL, Nishi T, Maier CM, Kinouchi H, Chan PH. Oxidative damage to the endoplasmic reticulum is implicated in ischemic neuronal cell death. J. Cereb. Blood Flow Metab. 2003;23:1117–1128. doi: 10.1097/01.WCB.0000089600.87125.AD. doi:10.1097/01.WCB.0000089600.87125.AD. [DOI] [PubMed] [Google Scholar]
- [95].Hayashi T, Saito A, Okuno S, Ferrand-Drake M, Dodd RL, Chan PH. Oxidative injury to the endoplasmic reticulum in mouse brains after transient focal ischemia. Neurobiol. Dis. 2004;15:229–239. doi: 10.1016/j.nbd.2003.10.005. doi:10.1016/j.nbd.2003.10.005. [DOI] [PubMed] [Google Scholar]
- [96].Saito A, Hayashi T, Okuno S, Nishi T, Chan PH. Modulation of the Omi/HtrA2 signaling pathway after transient focal cerebral ischemia in mouse brains that overexpress SOD1. Mol. Brain Res. 2004;127:89–95. doi: 10.1016/j.molbrainres.2004.05.012. doi:10.1016/j.molbrainres.2004.05.012. [DOI] [PubMed] [Google Scholar]
- [97].Saito A, Hayashi T, Okuno S, Nishi T, Chan PH. Oxidative stress affects the integrin-linked kinase signaling pathway after transient focal cerebral ischemia. Stroke. 2004;35:2560–2565. doi: 10.1161/01.STR.0000144653.32853.ed. doi:10.1161/01.STR.0000144653.32853.ed. [DOI] [PubMed] [Google Scholar]
- [98].Hayashi T, Saito A, Okuno S, Ferrand-Drake M, Dodd RL, Chan PH. Damage to the endoplasmic reticulum and activation of apoptotic machinery by oxidative stress in ischemic neurons. J. Cereb. Blood Flow Metab. 2005;25:41–53. doi: 10.1038/sj.jcbfm.9600005. doi:10.1038/sj.jcbfm.9600005. [DOI] [PubMed] [Google Scholar]
- [99].Narasimhan P, Sugawara T, Liu J, Hayashi T, Noshita N, Chan PH. Overexpression of human copper/zinc-superoxide dismutase in transgenic animals attenuates the reduction of apurinic/apyrimidinic endonuclease expression in neurons after in vitro ischemia and after transient global cerebral ischemia. J. Neurochem. 2005;93:351–358. doi: 10.1111/j.1471-4159.2005.03039.x. doi:10.1111/j.1471-4159.2005.03039.x. [DOI] [PubMed] [Google Scholar]
- [100].Nishi T, Maier CM, Hayashi T, Saito A, Chan PH. Superoxide dismutase 1 overexpression reduces MCP-1 and MIP-1α expression after transient focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2005;25:1312–1324. doi: 10.1038/sj.jcbfm.9600124. doi:10.1038/sj.jcbfm.9600124. [DOI] [PubMed] [Google Scholar]
- [101].Saito A, Hayashi T, Okuno S, Nishi T, Chan PH. Modulation of proline-rich Akt substrate survival signaling pathways by oxidative stress in mouse brains after transient focal cerebral ischemia. Stroke. 2006;37:513–517. doi: 10.1161/01.STR.0000198826.56611.a2. doi:10.1161/01.STR.0000198826.56611.a2. [DOI] [PubMed] [Google Scholar]
- [102].Kamada H, Yu F, Nito C, Chan PH. Influence of hyperglycemia on oxidative stress and matrix metalloproteinase-9 activation after focal cerebral ischemia/reperfusion in rats. Relation to blood-brain barrier dysfunction. Stroke. 2007;38:1044–1049. doi: 10.1161/01.STR.0000258041.75739.cb. doi:10.1161/01.STR.0000258041.75739.cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Maier CM, Hsieh L, Yu F, Bracci P, Chan PH. Matrix metalloproteinase-9 and myeloperoxidase expression. Quantitative analysis by antigen immunohistochemistry in a model of transient focal cerebral ischemia. Stroke. 2004;35:1169–1174. doi: 10.1161/01.STR.0000125861.55804.f2. doi:10.1161/01.STR.0000125861.55804.f2. [DOI] [PubMed] [Google Scholar]