

SUMMARY
Age-related neurodegenerative diseases are associated with mild impairment of oxidative metabolism and accumulation of abnormal proteins. Within the cell, the mitochondria appears to be a dominant site for initiation and propagation of disease processes. Shifts in metabolism in response to mild metabolic perturbations may decrease the threshold for irreversible injury in response to ordinarily sub lethal metabolic insults. Mild impairment of metabolism accrue from and lead to increased reactive oxygen species (ROS). Increased ROS change cell signaling via post transcriptional and transcriptional changes. The cause and consequences of mild impairment of mitochondrial metabolism is one focus of this review. Many experiments in tissues from humans support the notion that oxidative modification of the α-ketoglutarate dehydrogenase complex (KGDHC) compromises neuronal energy metabolism and enhance ROS production in Alzheimer’s Disease (AD). These data suggest that cognitive decline in AD derives from the selective tricarboxylic acid (TCA) cycle abnormalities. By contrast in Huntington’s Disease (HD), a movement disorder with cognitive features distinct form AD, complex II + III abnormalities may dominate. These distinct mitochondrial abnormalities culminate in oxidative stress, energy dysfunction, and aberrant homeostasis of cytosolic calcium. Cytosolic calcium, elevations even only transiently, leads to hyperactivity of a number of enzymes. One calcium activated enzyme with demonstrated pathophysiological import in HD and AD is transglutaminase (TGase). TGase is a cross linking enzymes that can modulate transcrption, inactivate metabolic enzymes, and cause aggregation of critical proteins. Recent data indicate that TGase can silence expression of genes involved in compensating for metabolic stress. Altogether, our results suggest that increasing KGDHC via inhibition of TGase or via a host of other strategies to be described would be effective therapeutic approaches in age associated neurodegenerative diseases.
Keywords: Mitochondria, oxidative stress, transglutaminase, α-ketoglutarate dehydrogenase, calcium, reactive oxygen species
DIMINISHED METABOLISM, OXIDATIVE STRESS AND INCREASED TRANS-GLUTAMINASE (TGASE) ARE COMMON TO NEURODEGENERTATIVE DISEASES
Huntington’s disease (HD), Parkinson’s disease (PD) and Alzheimer’s disease (AD) are age-related diseases of diverse etiologies and pathologies, but abnormal mitochondrial function is common to all
All cases of Huntington Disease (HD) are caused by an expansion in the polyglutamine region of the protein huntingtin (htt). HD is one of several adult-onset neurodegenerative diseases that are caused by genes with expanded CAG triplet repeats within their coding regions and extended polyglutamine (Q) domains within the expressed proteins. How these mutations produce the clinical and pathological changes of the diseases is unknown. Mitochondrial abnormalities occur in autopsy brain samples [1] and animal models using the mitochondrial poisons malonate and 3-nitroproprionic acid (3NP) mimic human HD [2]. In PD, identification of the rare specific gene mutations that cause PD has reinforced the relevance of oxidative stress and mitochondrial dysfunction in the disease. The proteins that are associated with familial PD—PTEN-induced putative kinase 1 (PINK1), DJ-1, α-synuclein, leucine-rich repeat kinase 2, and, possibly, parkin—are either mitochondrial proteins or are associated with mitochondria, and all interface with the pathways of oxidative stress and free radical damage [3–5]. Animal models using the mitochondrial poison 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mimic human PD [6]. In AD, the rare gene mutations in amyloid precursor protein (APP) or presenilin proteins lead to increased production of amyloid β peptide (Aβ), which depresses mitochondrial function, thereby altering calcium regulation, inducing oxidative stress and generally impairing metabolism. Pathological analysis and studies of peripheral tissues demonstrate these are common features of non-genetic forms of AD as well. The brains from patients with all age-related neurodegenerative diseases show extensive damage from oxidative stress in their brains at autopsy.
Reduced metabolism can be readily related to the clinical characteristics of neurodegenerative diseases
The relation of mitochondrial changes to clinical symptomsis particularly well documented in AD. Diminished glucose metabolism always accompanies AD and the reduction occurs at early stages of the disease, even pre-symptomatically. The decline in glucose metabolism with AD was shown in some of the earliest imaging studies of the human brain and has been recognized for many decades. The reduction in glucose metabolism is feasibly linked by robustly documented mechanisms to the decline in mental function, and diminished memory is particularly vulnerable. The sensitivity of memory to altered glucose metabolism may be because of the close link of metabolism and neurotransmitters that are known to involved in memory, particularly acetylcholine [7]. In patients with APOE4 genotype that will be at greater risk to get AD later in their life, the decline precedes the clinical characteristics by decades [8]. Furthermore, mildly cognitively impaired patients that have a reduction in brain glucose metabolism all go on to develop AD [9–11]. Reductions in cerebral metabolism sufficient to impair cognition in normal individuals occur in AD. The degree of clinical disability in AD correlates well with the magnitude of the reduction in brain metabolism.
The question is no longer whether changes occur in mitochondria in neurodegenerative disease. Currently, the interesting questions are what is the cause and whether these mild changes in metabolism have any consequence on mitochondria or their ability to adapt to metabolic insults and if reversal of them can be beneficial to cells, animals or patients.
Damage from free radicals is common in brains from patients with age-related neurodegenerative disorders
Reactive Oxygen Species (ROS) are normally signaling molecules. For example, they regulate tricarboxylic acid (TCA) cycle enzymes such as KGDHC, and many transcription factors. However, if the level is too high they will damage multiple proteins. The sources of free radicals in neurons and other cells has been reviewed recently [12–13]. Changes in antioxidant enzymes or in key enzymes of glucose catabolism, especially those in the mitochondria, can lead to excess radical production. Evidence for free radical damage is extensive in brain. For example, immunostains for reactive aldehydes or peroxynititrite reveal that the damage caused by oxidative stress in AD brain is more extensive than are plaques and tangles (the defining feature of AD) [14]. Evidence of free radicals (i.e., isoprostanes) can even be observed in AD and PD urine, and these can distinguish AD and PD [15]. Increased levels of F2 isoprostane levels occur in plasma and urine of AD, but not PD patients [16]. Although the evidence for abnormal ROS in AD, PD and HD is overwhelming, the source of these ROS is not established.
In animal models of AD plaque formation, the changes in free radicals precede plaque formation. Levels of plasma, urinary and brain homogenate 8,12-iso-iPF(2alpha)-VI iso-prostanes are higher in Tg2576 than in control animals as early as 8 months of age. In contrast, a surge of Aβ 1–40 and 1–42 levels as well as Aβ deposits in Tg2576 mouse brains does not occur until 12 months of age. This suggests that brain oxidative damage contributes to AD pathogenesis before Aβ accumulation in the AD brain [15].
Increased oxidative stress also occurs in PD [6], HD[17] and PSP [18]. The questions in these diseases are similar to those for AD. The evidence for oxidative damage is overwhelming but the source of the ROS and their pathological importance is unknown. Amongst the possibilities are mitochondria, NADPH-oxidase, iNOS xanthine oxidase, monoamine oxidase and the dehydrogenase complexes such as KGDHC. The strategies for understanding the therapeutic implications are similar in these diseases although the pathologies vary. Although the underlying basis of these changes in oxidation may be primary, determining what they are has proven elusive so the strategy has been to target downstream consequences.
REDUCTIONS IN KEY MITOCHONDRIAL ENZYMES MAY UNDERLIED THE REDUCTION IN GLUCOSE METABOLISM IN NEURODEGENERATIVE DISEASES
The simplest explanation for a reduction in metabolism is decline in the activity of metabolic enzymes. The reductions in the α-ketoglutarate dehydrogenase complex (KGDHC) activity in AD brain have been particularly well documented, and the reductions appear to be a common feature of neurodegeneration of various kinds. The initial studies [19] that showed this decrease in AD have been widely replicated [20–22], and there are no contravening reports. The severity of change and their correlation with the clinical dementia rating score vary with APOE4 genotype [23], but positive correlations occur whether or not patients carry any APOE4 allele. Reductions in KGDHC occur in PD [24], HD [2, 18] and progressive supranuclear palsy [18]. The reduction in KGDHC is not limited to the areas of pathology and neuron death. The precise chemical nature of the changes in KGDHC that lead to diminished activity in brains from AD patients are unknown. Results suggest that the decline is a post-translational modification. Although the activity is diminished in all forms of AD, the immunoreactivity is selectively altered in familial AD. In patients bearing the APP670/671 mutation the immunoreactivity of E1k and E2k are diminished whereas E3 is normal [25]. The reduced activity with normal subunit protein immunoreactivity is also found in PD [24] and progressive supranuclear palsy [18]. KGDHC activity was also reduced in putamen from patients with HD [2].
The reductions in KGDHC activities do not reflect general mitochondrial changes since some TCA cycle enzymes decline while others increase. Brains were from patients with autopsy-confirmed AD who had also had recent clinical dementia ratings (CDR) before death. Significant (p < 0.01) decreases occur in the activities of the enzymes in the first part of the TCA cycle that are combined dehydrogenases and decarboxylases: the pyruvate dehydrogenase complex (−41%), isocitrate dehydrogenase (−27%), and KGDHC (−57%). On the other hand, activities of the dehydrogenases in the second half of the TCA cycle increase: succinate dehydrogenase (complex II) (+44%) and malate dehydrogenase (+54%) (p < 0.01). Activities of the other four TCA cycle enzymes are unchanged. All of the changes in TCA cycle activities correlate with the clinical state (p < 0.01), suggesting a coordinated mitochondrial alteration. The highest correlation is with pyruvate dehydrogenase complex (r = 0.77) [26]. The results are consistent with a decline in these enzymes underlying the reduction in glucose metabolism, which is coupled to the clinical state of the patient. If we understood why the reductions occur, treatments might be devised to reactivate them and hopefully thereby to restore brain metabolism. The relation of the changes in the TCA cycle enzymes to TGase and oxidative stress is discussed below.
Aβ is present in mitochondria and directly effects KGDHC and PDHC. Intracellular Aβ is present in mitochondria from brains of transgenic mice with targeted neuronal over-expression of mutant human APP and AD patients [27]. Aβ progressively accumulates in mitochondria and is associated with diminished enzymatic activity of respiratory chain complexes (III and IV) and a reduction in the rate of oxygen consumption. Mitochondria-associated Aβ was detected as early as 4 months, before extensive extracellular Aβ deposits [28]. Within the mitochondria Aβ will inhibit PDHC and KGDHC [29].
OXIDATIVE STRESS CAN LEAD TO THE AD-LIKE CHANGES IN THE TCA CYCLE
The increased oxidative stress in brains from patients that died with neurodegenerative disease and the reduction in KGDHC are likely to be related. KGDHC is very sensitive to oxidants. Inhibition occurs in cells, mitochondria or isolated enzymes by H2O2, nitric oxide, per-oxynitrite, chloroamine [30], hypochlorite [30], hydroxynonenal, hyperoxia, and acrolein [31]. Overproduction of monoamine oxidase produces increased ROS in cells and inactivates KGDHC [32]. Similarly, if oxidants are overproduced by knocking out SOD, ROS increases and KGDHC is inactivated [33].
Peroxynitrite inhibits purified KGDHC in a dose- and time-dependent manner. Loss of enzymatic activity is associated with reduced immunoreactivity of E1k and E2k subunits, but not E3, and increased immunoreactivity of each component to a nitrotyrosine antibody, suggesting nitration of a tyrosine residues. Nitrosylation was further confirmed by a combination of trypsin digestion and nano-LC-MS/MS analysis. Tyrosine residue 556 is nitrated in the E1k subunit, while no nitration is found in E2k and E3. Addition of GSH after peroxynitrite treatment of KG-DHC completely restores the KGDHC activity and the immunoreactivity of E1k and E2k subunits. The immunoreactivity of nitrotyrosine is diminished to near control level upon addition of GSH. Such decreased immunoreactivity suggests that nitrotyrosine of KGDHC is further modified by possibly converting -NO2 to -NH2 in tyrosine residues to form aminotyrosine, or denitrating -NO2 in tyrosine residues. Taken together, the data directly demonstrate the nitration of tyrosine residue of KGDHC. The findings indicate that the similar modifications of KGDHC may also occur in cultured cells, and regulation of KGDHC activity by these processes may be of significance in AD pathology as well as in normal regulation of KGDHC activity [34].
In addition, KGDHC can be inactivated by other means. KGDHC is very sensitive to other toxins such as cisplastin [35], MPTP [36], and 3-NP. In addition, KGDHC can be inactivated by transglutaminase [37].
TRANSGLUTAMINASE (TGase) IS ELEVATED IN MULTIPLE NEURODEGENER-ATIVE DISEASES
TGase catalyzes a covalent bond between peptide-bound glutamine residues and either lysine-bound peptide residues or mono- or polyamines (Figure 1). Multiple lines of evidence suggest that TGase is involved in neurodegenerative diseases including frontotemporal dementia, AD, progressive supranuclear palsy (PSP), HD, and PD [38]. Transcription of TGase is increased by head trauma and stroke and glutamate toxicity [39] and by hypoxia (Wykoff). In age-related neurodegenerative diseases TGase enzyme activity is increased in selectively vulnerable brain regions, TGase proteins are associated with inclusion bodies characteristic of the diseases, and prominent proteins in the inclusion bodies are modified by TGase enzymes including tau, alpha-synuclein, and huntingtin protein. Evidence for N(epsilon)-(gamma-L-glutamyl)-L-lysine (GGEL) linkages is apparent in brain and CSF samples from AD and HD patients [40–42]. Four gamma-glutamylpoly amines are present in normal human CSF. All four gamma-glutamylamines are elevated in HD CSF. These findings support the suggestion that gamma-glutamyl-polyamines are reflective of TGase activity in human brain, that polyamination is an important post-translational modification of brain proteins, and that TGase-catalyzed modification of proteins is increased in HD brain [42]. Thus, a wide range of studies suggest that activation and/or increased transcripton of TGase could have wide ranging effects and serve as a therapeutic target.
Figure 1.
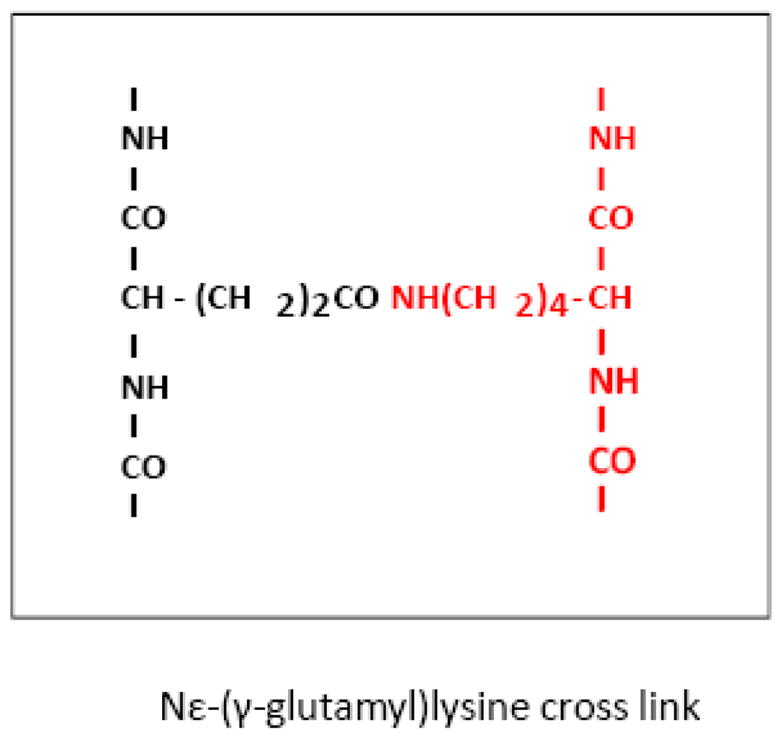
Transglutaminase (TGase) is elevated in Alzheimer’s Disease and expanded CAG-disorders
INCREASED TGase CAN ALTER METABOLISM BY TRANSLATIONAL AND POST-TRANSLATIONAL CHANGES
As described in the introduction, TGase is increased in multiple neurodegenerative diseases and in animal and chemical treatments that model these diseases. In contrast to its inactivating effect on TCA cycle enzymes, oxidative stress can upregulate TGase. For example, oxidative stress appears important in glutamate-evoked TGase upregulation in astrocyte cultures [43]. Glutamate causes a dose-dependent increase in ROS production. Pre-incubation with glutathione ethyl ester or cysteamine recovers oxidative status and reduces glutamate-increased tissue TGase. Thus, TGase up-regulation may be part of a biochemical response to oxidative stress [43]. An increase of TGase 2 expression may contribute to activation of microglia. After activation of BV-2 microglial cell lines by lipopolysaccharide, the expression of TGase 2 and iNOS increase in parallel. Furthermore, TGase inhibition reduces secretion of NO by up to 80%, in a dose-dependent manner. This observation suggests that TGase 2 controls iNOS transcription or iNOS message stability [44].
Results in cells and with isolated enzyme support a role for TGase in neurodegenerative diseases and as a possible cause of the decline in metabolism in neurodegenerative disorders. By its ability to cross link, TGase can inhibit enzymes such as glyceraldehyde-3-phosphate dehydrogenase, KGDHC [45] or aconitase [46], key enzymes of glycolysis and the TCA cycle. The cross links with aconitase have been observed in HD brains at autopsy [46], and most of the mitochondrial aconitase in HD caudate is present as high-Mr aggregates. Aconitase activity is markedly decreased in HD caudate (a region severely damaged by the disease) [47–48] and HD transgenic mice [49–50]. These findings suggest that an increase of TGase activity in HD caudate may contribute to mitochondrial dysfunction by incorporating aconitase into inactive polymers [46]. Although KGDHC is also a substrate for TGase [37], that linkage has not been shown in brains from patients that died with neurodegenerative diseases. TGase action on transcription may be due to cross linking of transcription factors to diminish gene cascades that may normally enhance mitochondrial biogenesis or function [51]. For the inactivation of mitochondrial enzymes to occur under physiological or pathological conditions, TGase must be present in the mitochondria and appropriate substrates must be present. Studies reveal that TGase is present in mitochondria from both brain and liver [52]. However, the levels are very low.
Studies that diminish TGase activity in vivo support the suggestion that TGase is pathologically important. Studies in TGase knockout mice [53] as well with the TGase inhibitor cystamine [54–55] suggest TGase activation is a critical part of the HD disease process. Understanding the mechanism for that neuroprotection is important for designing better therapeutic approaches.
TGase is a multifunctional enzyme that may have beneficial effects. TGase is up-regulated in neurons exposed to oxygen and glucose deprivation and increased TGase expression protects neurons against oxygen and glucose deprivation induced cell death independent of its trans-amidating activity. TGase protects by interacting with HIF1[56]. TGase attenuates the HIF1 hypoxic response pathway, and up-regulates the HIF-dependent proapototic gene Bnip3. These results indicate that TGase may play an important role in protecting against the delayed neuronal cell death in ischemia and stroke [56]
TGase can also affect mitochondrial function by acting at the level of transcription. Accumulating data suggests that the elevated TGase in neurodegenerative conditions may impair transcription and the ability to adapt. This implies that blocking TGase should promote transcription and be protective. Convincing evidence for this scenario has emerged in studies of HD. Transcriptional repression may be responsible for the inability of mutant huntingtin (mhtt) expressing cells to cope with the metabolic stress. Multiple groups have shown that mhtt is able to sequester needed transcription factors and other proteins necessary for transcription initiation. TGase, like mhtt, has been shown to alter specific transcription factors (e.g., Sp1) [57–58]. The transcription factor(s) found to be modified by TGase play a role in the expression of nuclear-encoded mitochondrial proteins such as cytochrome c and PGC-1 (PPARγ-coactivator 1). Numerous metabolic proteins are repressed in HD models and HD patients [59–61]. These proteins (including cytochrome c (cyt c) and PGC-1α) are under the control of nuclear transcription factors that contain glutamine (Q)-rich activation domains. Since the large number of Q residues in these activation domains provides a possible target for modification by TGase, experiments tested the ability of TGase to repress the transcription of metabolic genes (cyt c and PGC-1a) that are down-regulated in HD models and patients. Transient transfection of a promoter/reporter of cyt c reveals a basal decrease in the cyt c promoter activity between htt and mhtt expressing cells exposed to serum starvation/restimulation. Co-transfection of TGase greatly reduces cyt c promoter activity. Pharmacological inhibition, as well as genetic deletion of TGase, induces cyt c promoter activity and increases mRNA levels of cyt c and PGC-1α. Citrate synthase activity, the first step of the TCA cycle, also increases after TG inhibition [51] Whether the action of TGse on transcripton factors is a physiological regulatory mechanism that goes wrong if mutant huntingtin is present remains to be determined.
If the decreased ability of mutant huntingtin cells to respond to metabolic stress is due transcriptional repression and diminished mitochondrial activity, then increased expression of nuclear-encoded mitochondrial proteins (such as cytochrome c) should enhance mitochondrial biogenesis and increase survival of mutant huntingtin (mhtt)-encoding neurons. If the inability of mhtt-expressing cells to respond to energetic stress is due the over-activity of the enzyme TGase that has been reported in HD patients and models alike, then inhibiting TGase should increase mitogenesis and be neuroprotective. Immortalized murine striatal neurons from mice with a huntingtin gene with a normal number of repeats as well as a separate strain with a pathological level of repeats were used to test whether TGase inhibitors can be neuroprotective. Inhibition of TGase increases the transcription of key mitochondrial proteins in cells expressing mutant huntingtin. Thus, over-active TGase within mhtt-expressing cells may silence the expression of nuclear-encoded mitochondrial proteins through the modification of transcription factors, and loss of this compensatory mechanism causes these cells to be more susceptible to the metabolic stress that is seen in HD [62]. The cells expressing mutant huntingtin are more sensitive to the mitochondrial toxin, 3-NP, which induces HD like changes in animal models. 3-NP acts by inhibiting complex II in the mitochondrial respiratory chain. To test whether TGase inhibition can prevent neuronal death due to mitochondrial dysfunction, the role of the TGase inhibitor ZDON and its inactive control in preventing death were tested. ZDON, but not its analog, enters the cell and inhibits TGase [51]. ZDON is neuroprotective, but the inactive analog is not in both striatal cell lines with mutant huntingtin and in myoblasts from HD patients. The neuroprotective efficacy of ZDON correlates with its ability to inhibit TGase.
The mechanism(s) by which TGase promotes harmful effects in HD remain(s) under study. TGase can act directly at gene promoters such as PGC-1α (a coactivator) and cytochrome c to repress the ability of transcription factors to recruit the basal transcription factor machinery. TGase is bound to a complex of proteins that interact directly with the promoters of these target genes. Inhibition of TGase activity using pharmacological inhibitors or RNAi knockdown leads to increased promoter activity, mRNA levels and protein levels of the transcription factor PGC1α and the protein cytochrome c. Finally, pharmacological inhibition of TGase activity leads to robust protection of HD cell lines as well as primary neurons and HD myoblasts against the mitochondrial toxicant, 3-NP. Together these studies suggest that transglutaminase inhibition overcomes mitochondrial dysfunction in HD by facilitating the expression of genes involved in mitochondrial biogenesis and function [63].
THE α-KETOGLUTARATE DEHYDROGENASE COMPLEX(KGDHC) IS A CALCIUM SENSITIVE CENTRAL ENZYME OF METABOLISM THAT PRODUCES AND IS CONTROLLED BY ROS
A compelling body of evidence indicates that KGDHC is one of the important targets of mitochondrial injury. An understanding of the multiple roles of KGDHC [64] is required to identify the upstream (i.e., causes) and downstream (i.e., consequences) effects which may provide additional therapeutic targets. KGDHC catalyses the reaction
The substrates and the products of this reaction are central to several key metabolic and signaling pathways pathways (see Figure 2).
Figure 2.

Role of KGDHC in Brain Metabolism
The α-ketoglutarate decarboxylase subunit (E1) catalyses the rate–limiting stage of the overall KGDHC reaction [65]. In brain mitochondria, the activity of KGDHC is the lowest of the TCA cycle enzymes and may be rate limiting for overall TCA cycle activity in vivo [66–67]. Consequently, the KGDHC reaction is essential for sustaining mitochondrial oxidative energy metabolism and neuronal viability.
Mammalian KGDHC is an integral mitochondrial enzyme tightly bound to the inner mitochondrial membrane on the matrix side [68]. It binds specifically to Complex I of the mitochondrial respiratory chain [69] and may form a part of the tricarboxylic acid cycle (TCA) enzyme super-complex [70]. Structurally, KGDHC is composed of multiple copies of three enzymes: α-ketoglutarate dehydrogenase (E1k subunit, EC 1.2.4.2), dihydrolipoamide succinyl-transferase (E2k subunit, EC 2.3.1.12), and lipoamide dehydrogenase (E3 subunit, EC 1.6.4.3). The E3 subunit of KGDHC is identical to that of pyruvate dehydrogenase; it is also known as di-hydrolipoamide dehydrogenase (Dld). The other subunits are unique to KGDHC. All the subunits are encoded by nuclear genes. For enzymatic activity, E1k requires the cofactor thiamine pyrophosphate, which is tightly bound; E2k requires covalently bound dihydrolipoic acid; and E3 requires covalently bound flavin [71–73].
The composition of KGDHC and its content exhibit tissue specificity. Neurons are particularly enriched in KGDHC compared to glia [74]. Immunogold electron microscopy with an antibody against the E1k component of the KGDHC demonstrates that KGDHC is heterogeneously distributed in mitochondria within individual astrocytes originating either from cerebellum or cerebral cortex [75]. Brain E1k appears to differ from heart. In the heart complex, only the known E1k is found. However, in brain the band corresponding to the brain E1k component includes a novel E1k protein. The functional competence of the novel brain isoenzyme and different regulation of E1k and E1kl by 2-oxoglutarate are apparent from kinetic studies [76]. Brain KGDHC also differs from the heart KGDHC in the ratio to the other components, lower apparent molecular mass and stability of isolated complex.
In addition to its traditional role to produce NADH in TCA cycle, KGDHC performs another uniquely important function by providing succinyl-CoA for heme synthesis which is initiated in mitochondria [77–78]. Heme is obligatory in the assembly of ETC and several antioxidant enzymes protecting the brain from oxidative stress.
Alternative functions are known for each of the KGDHC subunits. Conditions that destabilize the E3 homodimer enabled the mouse, pig, or human enzyme to function as a pro-tease [79]. A truncated gene product (MIRTD) of E2k contributes to the biogenesis of the mitochondrial respiratory complexes [80]. E3 has diaphorase activity, being able to catalyze the oxidation of NADH to NAD by using different electron acceptors such as O2, labile ferric iron [81], nitric oxide [82], and ubiquinone [83–84]. In this capacity, E3 would primarily have a prooxidant role, achieved by reducing O2 to a superoxide radical or ferric to ferrous iron. However, the ability to scavenge nitric oxide and to reduce ubiquinone to ubiquinol suggests that the diaphorase activity of E3 may also have an antioxidant role [82–84]
The regulation of KGDHC activity is rather complex and apparently makes it an integrating hub for cellular Ca2+, metabolite, and ROS signaling systems. KGDHC is activated by low concentrations of Ca2+ and matrix ADP, and is inhibited by high NADH and its own product, succinyl–CoA [85–89]. In addition to this classical regulation, new data emerge that KGHDC activity is both controlled and controls the intracellular ROS levels. Several studies have shown that KGDHC can produce free radicals at E2k and H2O2 at E3. These become physiologically significant when the NAD+/NADH ratio in the mitochondrial matrix is low (i.e., under reducing conditions) [90–91]. This production appears to be pathologically important since inhibitors of KGDHC can protect against in vitro associated with over-production of ROS, such as ischemia [92–93] and glutamate toxicity [94] while also blocking production of free radicals.
Excessive entry of calcium accompanies many cell injuries and mitochondria buffer rapid increases in calcium in neurons and most other cells. The role of calcium in activation of KG-DHC has been reviewed reviewed in [95]. Mitochondria isolated from adult rat cerebral cortex and cerebellum generate extremely reactive hydroxyl (.OH) radicals, plus ascorbyl and other carbon-centered radicals when exposed to 2.5 microM Ca2+, 14 mM Na+, plus elevated ADP under normoxic conditions, circumstances that prevail in the cytoplasm of neurons during excitotoxin-induced neurodegeneration [96]. In heart, the calcium that enters the mitochondria following stimulation activates both PDHC and KGDHC [85–86, 89]. KGDHC is more sensitive than PDHC to calcium [89].
The sensitivity of the KGDHC to oxidants may also be regulatory. For example, H2O2 is known to diminish NADH production by inactivating the KGDHC [32, 90], whereas its E2k component can sense the intramitochondrial redox state which affects the E2k dihydrolipoate/lipoate ratio [97]. It has also been shown that thyil radicals can inhibit E1k and that KGDHC and thyil radicals formed at E2k lipoate residues can interact with thioredoxin [98–99]. Although arguable in light of yet insufficient evidence, the ramification of this is that KG-DHC may serve as a major regulatory element adjusting the metabolic output of TCA cycle according to the redox state of intramitochondrial thiols, pyridine nucleotides, and ROS production.
CONSEQUENCES OF MILD IMPAIRMENT OF KGDHC
Energy failure may be a late event following inhibition of KGDHC. α-Keto-β-methyl-valerate (KMV), a structural analogue of α-ketoglutarate has reasonable specificity but low potency [66]. KMV inhibits acetylcholine synthesis before impairing energy metabolism as measured by ATP levels [66]. KMV promotes translocation of mitochondrial cytochrome c to the cytosol, and leads to activation of caspase-3 before the mitochondrial membrane potential or ROS production is altered. Thus, the results suggest a decrease in KGDHC activity induces gene cell death cascades before energetic failure. This suggests that treatments that affect these gene cascades may be effective in neurodegenerative diseases that are accompanied by mild impairment of oxidative metabolism.
Succinylphosphonte (SP) and its esters are potent and specific KGDHC inhibitors. SP and its phosphonoethyl (PESP) and carboxyethyl (CESP) esters at a concentration of 0.01 mM completely inhibit isolated KGDHC. In cells, 0.01 mM SP, PESP, or CESP inhibit KGDHC by 70%. The diethyl- and tri-ethyl esters were also inhibitory in the cell system, but only after preincubation, suggesting the release of their charged groups by cellular esterases. Thus, SP and its monoethyl esters target cellular KGDHC directly, while the di- and triethyl esters are activated in intact cells. When tested on other enzymes that bind α-ketoglutarate or related α-keto acids, SP has minimal effects and its two esters (CESP and TESP) are ineffective even at concentrations one order of magnitude higher than those that inhibit cellular KGDHC activity. The high specificity in targeting KGDHC, penetration into cells, and minimal transformation by cellular enzymes indicates the usefulness of these compounds for studies of reduced KGDHC activity on neuronal and brain function [100].
Mild impairment of KGDHC by SP-esters alters the metabolic profile of intact cerebellar granule cells during incubation with [1–13C]glucose and [U-13C]glutamate. The results suggest increased α-ketoglutarate formation, increased transamination of α-ketoglutarate with valine, leucine, and GABA, and a new equilibrium position of the aspartate aminotransferase reaction. Overall, the findings also suggest that some carbon derived from alpha-ketoglutarate may bypass the block in the TCA cycle at KGDHC by means of the GABA shunt and/or conversion of valine to succinate. The results are consistent with the suggestion that impairing KGDHC increases the GABA shunt in order to maintain normal metabolism [101]. The shift in metabolism may make the cells less able to adapt to further insults.
Similar effects on metabolism were observed in cells by changing KGDHC activities by genetic manipulation of E2k. As described above, the KGDHC complex consists of three enzymes: E1k, E2k and E3. Any one of the three can be manipulated genetically to examine the consequences of a reduction in KGDHC activity. The results are complicated, but made more revealing, by the observation that manipulating each subunit does not necessarily have a parallel effect on the activity of KGDHC. Human embryonic kidney (HEK) cells were stably transfected with an E2k sense or antisense expression vector. Sense control (E2k-mRNA-100) was compared with two clones in which the mRNA was reduced to 67% of control (E2k-mRNA-67) or to 30% of control (E2k-mRNA-30). The levels of the E2k protein in clones paralleled the reduction in mRNA, and E3 proteins are not altered. Unexpectedly, the clone with the greatest reduction in E2k protein (E2k-mRNA-30) has a 40% increase in E1k protein. The activity of the complex was only 52% of normal in E2k-mRNA-67 clone, but was near normal (90%) in E2k-mRNA-30 clone [102].
Subsequent experiments tested whether a reduction in the KGDHC activity would alter cellular metabolism by comparing metabolism of [U-13C]glucose in cell line (E2k100) to one in which the KGDHC activity was about 70% of control (E2k67). After incubation of the cells with [U-13C]glucose, the E2k67 cells showed a greater increase in 13C labeling of alanine and lactate compared to the E2k100 cells, which suggests an increase in glycolysis. Increased GABA shunt in the E2k67 cells was indicated by increased 13C labeling of GABA compared to the control cells. GABA concentration as determined by HPLC also increased in the E2k67 cells compared to the control cells. The data are consistent with enhanced glycolysis and GABA shunt in response to a mild reduction in KGDHC [103]. The findings indicate that a mild change in KG-DHC activity can lead to large changes in metabolism. The changes may allow maintenance of normal energy metabolism but still make the cells less able to adapt to perturbations such as occur with oxidants.
The levels of E2k protein modify the response to oxidative stress. Subsequent experiments tested whether the response to an oxidant challenge more closely paralleled the E2k protein levels or KGDHC activity. Growth rates, increased DCF-detectable reactive oxygen species, and cell death in response to added H2O2 are proportional to E2k proteins, but not to complex activity. These results were not predicted because subunits unique to KGDHC have never been manipulated in mammalian cells. These results suggest that in addition to its essential role in metabolism, the E2k component of KGDHC may have other novel roles [102]. For example, the results are consistent with the suggestion that a truncated gene product (MIRTD) of E2k contributes to the biogenesis of the mitochondrial respiratory complexes [80].
Alterations in E2k protein levels alter gene expression. Although KGDHC activities are diminished in AD brains, the activities of MDH are increased. To test the possibility that this is secondary to the decline in KGDHC, MDH activity and message levels were determined in cells with varying E2k protein levels. Activity of MDH is increased with KGDHC deficiency but only in the E2k23 lines. Of the three lines tested, these have the greatest decrease in E2k but nearly normal KGDHC activities. In contrast to the increase in MDH activities, the message levels of both cytosolic and mitochondrial MDH decline in the E2k23 lines. H2O2 reduces KGDHC activities in cells, but increases MDH activity and the message levels for both the mitochondrial and cytosolic MDH – but only in the lines most deficient in E2k. This raises the possibility of a coordinate regulation of gene expression of the TCA cycle [104]. The interaction of KGDHC with α-ketoglutarate oxidases such as the HIF proly hydroxylases that have the same substrate (ketoglutarate ) and same product (succinate) is not clear. Thus, an increase in α-ketoglutarate due to impaired KGDHC may increase HIF prolyl hydroxylases.
Treatment of purified KGDHC with oxidized glutathione (GSSG) leads to glutathionylation of all three KGDHC subunits [105]. H2O2 converts reduced glutathione (GSH) to oxidized glutathione (GSSG), which reacts with protein thiols [104]. Cellular glutathione level has been manipulated in HEK cells by two means to determine whether the level of E2k alters the effects of glutathione on KGDHC. Both buthionine sulfoximine (BSO), which inhibits glutathione synthesis without altering redox state, and H2O2 diminished glutathione to a similar level after 24 h. However, H2O2, but not BSO, reduced KGDHC activities, and the reduction was greater in the E2k-23 line with the lowest E2k protein. These findings suggest that H2O2 moved glutathione to the protein. These findings suggest that the E2k may mediate diverse responses of KGDHC to oxidants [104]. When cardiac mitochondria are challenged with H2O2, NADH production and oxidative phosphorylation decline. Upon consumption of H2O2, mitochondrial function is restored. These alterations are due, in large part, to reversible glutathionylation and inhibition of KGDHC [106]. In addition, the differential response of KGDHC activities to BSO and H2O2 together with the in vitro interaction of KGDHC with GSSG suggests that glutathionylation is one possible mechanism underlying oxidative stress-induced inhibition of the TCA cycle [104].
The ability of KGDHC to produce H2O2 suggests that inhibiting KGDHC could be neuro-protective, while the diminished production of NADH would suggest that impairing KGDHC is damaging. As discussed above, inhibiting KGDHC under normal conditions appears damaging. However, if the cell is experiencing life threatening conditions, inhibition of KGDHC appears to protect. The first study demonstrated protection by impairing KGDHC in microglia. The KG-DHC inhibitor KMV (see above) dose-dependently reduced ROS production and LDH release from a hypoxic microglial cell line [92]. KMV also reduced ROS production and enhanced the cell viability following H2O2 but failed to reduce the SIN-1 and sodium nitroprusside (SNP) toxicity. KMV also reduced caspase-3 and -9 activation under stress. These results suggest that KMV protects BV-2 cells from stress and acts by reducing ROS production through inhibition of ROS production by KDGHC.
Inhibition of KGDHC can also be protective in neurons. KGDHC inhibitors were added to glutamate poisoned neurons. Their subsequent inhibition of KGDHC decreases the mitochondrial potential [107]. The production of O2·-was detected by reaction with hydroethidine. The distribution of the resulting fluorescence of mt-DNA-ethidium coincides with that of the mitochondrial marker Mitotracker, pointing to the mitochondrial site of the hydroethidine-detected ROS in response to glutamate. The SP inhibitors described above inhibited glutamate-induced ROS production by about 20–44%. The decreases in neuronal ROS by specific inhibitors of KG-DHC suggest that KGDHC is a source of ROS in cultured neurons responding to glutamate. However, at higher concentration the KGDHC inhibitor increased ROS production compared with glutamate alone, presumably due to secondary effects arising upon the strong KGDHC inhibition. This suggests that different mechanisms are invoked at different concentrations. They support the suggestion that inhibition of KGDHC ROS production can protect against in vitro excitotoxicity [107].
IMPAIRMENT OF KGDHC IN VIVO INDUCES OXIDATIVE STRESS AND COMPROMISES THE ABILITY OF THE BRAIN TO RESPOND TO OTHER INSULTS
Three different models test mechanistic interactions of KGHC in neurodegeneration. They are presented from the most complex to the one that only affects one enzyme. (1) thiamine deficiency (TD; i.e., vitamin B1 deficiency) which primarily affects KGDHC and transketolase (a critical enzyme of the pentose shunt), although thiamine is also a cofactor for PDHC,; (2) E3+/− mice which have reduced activities of KGHC and PDHC and (3) E2k+/− mice, which have a primary deficit in only KGDHC. A comparison of the three models helps distinguish critical variables that are necessary to cause particular deficits.
Thiamine deficiency
Thiamine deficiency (TD) is a model of neurodegeneration induced by mild impairment of oxidative metabolism. Thiamine is required for PDHC, KGDHC and transketolase. KGDHC is particularly sensitive to TD. Further, KGDHC is more sensitive to TD in aged mice than young mice [108]. TD produces a time-dependent, selective neuronal death in specific thalamic brain regions. Loss of neurons and elevation in markers of neurode-generation are preceded or accompanied by markers of oxidative stress that mimic the changes in brains from AD brain including: (1) changes in microglia including increased redox active iron, (2) induction of nitric oxide synthase and heme oxygenase-1, a marker of oxidative stress, and (3) altered processing of amyloid precursor protein that exacerbates plaque formation in an AD-like manner. Endothelial cells also show changes in early stages of TD including induction of in-tracellular adhesion molecule-1 and of endothelial NOS. Thus, the metabolic deficiency including the decline in KGDHC is sufficient to induce oxidative stress and an inflammatory response [109]. Interestingly, TGase message levels are dramatically increased (as much as 11 fold) in areas of brain damage in TD [110]. Whether the induction of TGase is secondary to the oxidative stress induced by TD has not been tested. Nor has it been tested whether impairing TGase would be protective. Mild impairment of oxidative metabolism can induce a variety of physiological changes: oxidative stress, increases in TGase message, an inflammatory response, altered APP processing and neuronal death [109].
E3 deficiency
The E3 component of KGDHC is a critical subunit common to KGDHC and PDHC (as well as to the branched chain dehydrogenase complex, which however has minimal enzymic activity compared to the other two complexes). E3 is responsible for ROS production by both complexes. The E3+/− mice do not show any generalized neuron loss. They have increased oxidative stress as measured with malondialdehyde. However, mice that are deficient in E3 are more vulnerable to brain damage induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), malonate and 3-nitropropionic acid (3-NP), toxins that have been proposed as models of PD and HD, respectively. Administration of MPTP causes a greater depletion of tyrosine hydroxylase-positive neurons in the substantia nigra of E3+/− mice than in wild-type littermate controls. Striatal lesion volumes produced by malonate and 3-NP are increased in E3+/− mice [2]. Isolated brain mitochondria treated with 3-NP show that both succinate-supported respiration and membrane potential are suppressed to a greater extent in E3+/− mice. These findings provide further evidence that mitochondrial defects may contribute to the pathogenesis of neurodegenerative diseases [2].
Since E3 can be a source of free radicals in KGDHC and PDHC, a reduction in E3 may be expected to decrease ROS. Indeed, ROS mitochondria isolated from these mice produce less ROS. However, in living mice a reduction in the E3 component of KGDHC elevates free radical production as shown by an increase in malondiadehyde in their brains [2]. Further, the production of these ROS is exaggerated by toxins that are used as models of PD and HD. These data provide direct evidence that imbalance ROS metabolism occurs in animal models with only mild reductions in glucose metabolism. Thus, both TD and E3deficiency enhance the production of oxidative stress. Further, the production of these ROS is exaggerated by toxins that are used as models of PD and HD. Thus, under the metabolic conditions that have been studied in vivo, reductions in PDHC and KGDHC can initiate oxidative stress.
Reduction of E2k in mice
To determine the consequences of diminished brain KG-DHC activity, the dlst gene encoding the E2k subunit of KGDHC was disrupted. This protein is unique to KGDHC. E2k+/− heterozygous mice had lower message and protein levels for E2k, leading to reduced brain KGDHC activity by about 50%. Behavioral assessment of adult DLST+/− mice revealed abnormalities in a discriminative fear-conditioning paradigm and in high-speed rotorod performance. Spatial memory in a water maze task was unaltered. Neurons cultured from DLST+/− mice have increased resting calcium levels. Furthermore, oxidative stress as assessed by malondialdehyde is increased in DLST+/− mice. Thus, a deficiency in KG-DHC is by itself sufficient to induce oxidative stress in the brain [111].
MILD IMPAIRMENT OF METABOLISM PROMOTES PATHOLOGIES RELATED TO AGE-RELATED NEURODEGENERATIVE DISEASES
Abnormal mitochondrial function alters APP processing
Altered processing of amyloid precursor protein (APP) is prominently associated with changes in AD, but it is likely altered in other neurodegenerative diseases as well. Increased APP expression and intracellular accumulation of its toxic fragments including the carboxy terminal fragment of APP have been associated with acute neuronal death. APP C-terminal and β-amyloid domains occur in rat substantia nigra pars reticulata neurons targeted for delayed degeneration following a neurotoxic striatal lesion [112]. These findings suggest that intranuclear APP C-terminal fragments (CTF) play a role in genomic events contributing to delayed neuron degeneration [112]. These CTF also appear to be involved in neuron death following TD. During TD, CTF of APP appears in the nuclei of neurons that will die 5–6 days before neuron death [113]. Thus, very mild interruptions of metabolism and of KGDHC can lead to altered APP processing leading to accumulation of CTF of APP in the nuclei of neurons that will die.
Mild impairment of energy metabolism can also lead to the formation of Alzheimer-like plaques. In Tg19959 transgenic mice over expressing a double mutant form of APP, TD exacerbates amyloid plaque pathology, and enlarges the area occupied by plaques by 50% in cortex and by 200% in both hippocampus and thalamus. TD increases Aβ(1–42) levels by about three fold, beta-CTF (C99) levels by 33% and β-secretase (BACE1) protein levels by 43%. Thus, TD induces plaque formation throughout the brain even though neuron loss is restricted to a small region [114].
Alterations of metabolic/oxidative activity modify APP processing in a variety of different models. Genetic ablation of inducible nitric oxide synthase protects against plaque formation in mice [115]. Insulin, 2-deoxyglucose, 3-nitropropionic acid, and kainic acid induce acute energy inhibition, and simultaneously induce the BACE enzyme which cleaves APP [116]. Cerebral Aβ1–40 levels in plaque bearing mice increase to approximately 200% of control at seven days after treatment, demonstrating that energy inhibition can be amyloidogenic [116]. Energy deprivation induces phosphorylation of the translation initiation factor eIF2α to eIF2α-P, which increases the translation of BACE1 [117]. These results strongly suggest that eIF2α phosphorylation increases BACE1 levels and causes Aβ overproduction, which could be an early, initiating molecular mechanism in sporadic AD [117]. Thus, these studies demonstrate that impairing energy metabolism can promote plaque formation.
Abnormal mitochondrial function can also lead to tangle formation
A reduction in mitochondrial function can also lead to tangle formation. Tangles are hyperphosphorylated tau proteins, and a hallmark of typical pathology in AD brains. They also occur in brains from patients with Wernicke Korsakoff syndrome (i.e., human thiamine deficiency)[118]. Insulin deficiency and diabetes causes a mild hyperphosphorylation of tau at early stages, and at later stages massive hyperphosphorylation of tau occurs. However, neither mild nor massive tau phosphorylation induced tau aggregation in mice or rats, i.e., true tangles [119]. Rotenone, an inhibitor of electron transport, causes loss of neurons in the substantia nigra and in the striatum. Spherical deposits of alpha-synuclein, which accumulates in Parkinson disease, are observed in a few cells, but cells with abnormal cytoplasmic accumulations of tau immunoreactivity are significantly more numerous in the striatum after rotenone [120] or annonacin, a natural complex I inhibitor [121], treatment. Many tau+ cell bodies also stain positively for thioflavin S (which stains Alzheimer plaques and tangles), for nitrotyrosine and for ubiquitin [122]. The data suggest that chronic respiratory chain dysfunction might trigger a form of neurodegeneration in which accumulation of hyperphosphorylated tau protein predominates over deposits of α-synuclein [120]. SOD2 deficiency which increases ROS also increases tau phosphorylation [123].
Abnormal energy metabolism and oxidative stress also alter cellular calcium
Glucose metabolism and cellular calcium are closely linked. Mitochondrial calcium uptake is diminished in mitochondria from AD fibroblasts[124]. The changes in calcium regulation in mitochondria from E2k deficient mice are consistent with altered mitochondrial buffering. Endoplasmic reticulum (ER) calcium is a major calcium store within the cell, ER calcium is increased in cells bearing AD causing presenilin-1 mutations [125]. ER calcium is also increased in non-genetic forms of AD. ER calcium is also sensitive to oxidants. A screen of a variety of times and dosages of revealed that specific oxidants can alter calcium in an AD-like manner. These oxidant-induced changes can be blocked by specific antioxidants. The antioxidants that blocked the effects the effects on ER calcium also ameliorated the deficits in ER calcium in cells from AD patients [126].
KGDHC changes with aging
The most important factor for developing age-related neurodegenerative diseases is aging. One possibility for a role of KGDHC in age related neuro-degeneration is that a factor with aging making the brain vulnerable with age. In mice, the baseline activity of brain KGDHC does not change with age, but with increased age it does become more sensitive to other metabolic insults such as thiamine deficiency [108]. KGDHC activity also diminishes with age in heart, and the number of hydroxynoneal (HNE) adducts is increased [127]. Hearts from old rats exhibit significantly higher HNE-modified mitochondrial proteins when compared with those from young rats. The protein most markedly modified by HNE with age is the E2k component of KGDHC [127]. Replicative life span in Saccharomyces cerevisiae is increased by glucose limitation and by augmented NAD+. The increased life span due to glucose limitation is related to diminished mitochondrial ROS production. The data indicate that E3 is an important source of glucose-mediated preventable mitochondrial ROS production. The results substantiate the concept that E3 is an important and novel source of ROS limiting life span [128].
Neurogenesis is impaired by reductions in KGDHC
Although the precise role of neurogenesis in the adult brain is unknown, one interpretation is that it permits the brain to respond to insults. Thus, reduction in neurogenesis would make the brain less able to respond [129]. Mitochondrial dysfunction can influence the number of neural progenitor cells in the hippocampus of adult mice [111]. Neurogenesis is impaired in three models of impaired TCA cycle enzymes including E2k+/−, E3+/− [111] and thiamine deficiency [129]. Adult brain neurogenesis was assessed using immunohistochemistry for the immature neuron markers, doublecortin (Dcx) and polysialic acid-neural cell adhesion molecule, as well as a marker for proliferation, proliferating cell nuclear antigen (PCNA). Both E3 and E2k-deficient mice showed reduced Dcx-positive neuroblasts in the subgranular zone (SGZ) of the hippocampal dentate gyrus compared with wild-type mice. PCNA staining reveals decreased proliferation in the SGZ of E2k-deficient mice. An analogous pattern of change was observed in thiamine-deficient mice [129].
Mitochondrial dynamics
Extensive evidence implicates mitochondrial dynamics in neurodegenerative diseases. Whether these changes lead or follow the changes in mitochondrial function described in this review is unknown. These dynamic processes include transport of mitochondria down axons and dendrites as well as mitochondrial fission and fusion. Axonal transport is energy dependent and the movement of mitochondria can be selectively impaired by glutamate, zinc, rotenone or uncouplers [130]. Thus, any interruption of energy metabolism (i.e., a loss of KGDHC activity) may diminish transport. The resulting diminished mitochondrial function in the nerve ending would likely diminish synaptic function. Similarly, a balance of fusion and fission is necessary for normal mitochondrial and neuronal function. Changes in fission/fusion alter mitochondrial function and vice versa. Disruption of the mitochondrial fission and fusion equilibrium lead to neurodegeneration [131]. Genetic mutations in fusion proteins cause neuropathy. When genetic defects lead to lack of fusion, the enzymes encoded by the mitochondrial (mt)DNA are most affected, whereas the expression of nuclear DNA–encoded enzymes increase, apparently to compensate for the mitochondrial dysfunction. The results also show that functional mitochondria due to loss of fusion are lost in the neuronal terminals more than in the cell body [131–132]. Dominant optic atrophy is caused by mutations in the OPA1 gene, which encodes a dynamin-related GTPase required for mitochondrial fusion [133]. In Drosophila, iIncreased mitochondrial fission occurs in PD models, such as mutations in PINK1, and proteins that are mutant in genetic forms of PD promote fission, and their inhibition leads to excessive fusion [134]. In mammalian cells, PINK1 mutants or knockdown promotes excessive fission [135–137]. Parkin and PINK2 function in a pathway in which defective mitochondria are targeted for autophagy [138]. Mutant huntingtin alters mitochondrial morphology and dynamics. Both rotenone and β-amyloid initiate mitochondrial fission. Mitochondrial fission is an early event in ischemic stroke and diabetic neuropathies [139]. Recent studies show that APP, through Aβ production, causes an imbalance of mitochondrial fission/fusion that results in mitochondrial fragmentation and abnormal distribution, which contributes to mitochondrial and neuronal dysfunction [140]. In AD brains, mitochondria are distributed away from axons in pyramidal neurons, and this is replicated in cultured neurons using oligomeric Aβ[141–142]. As described in other sections of this review, mild impairment of oxidative metabolism can lead to overproduction of Aβ. Thus, mild impairment of oxidative metabolism can lead to increased Aβ which can imbalance fusion/fission. Thus, precedent exists that suggests alteration in fusion/fission could lead to alterations in mitochondrial proteins and that alterations in energy metabolism can cause changes in fusion/fission. Together they would form a cascade of events to promote neurodegeneration
THERAPEUTIC APPROACHES
The goal of these studies is to develop new therapeutic strategies. The data reviewed above suggest several innovative approaches.
1. Bypass the metabolic block
Since the degree of cognitive deterioration in AD correlates to the reduction in cerebral metabolic rate, this is a reasonable therapeutic target. An attempt to treat this abnormality in AD patients is based on feeding the patients glucose and malate to bypass the diminished activities of PDHC and KGDHC. In addition, the antioxidant resveratrol is included. Encouraging results of a preliminary double-blind clinical trial do not allow confident conclusions but do support the need for more intensive examination of this approach to AD treatment [143].
2. Reactivate KGDHC
KGDHC is diminished in brains of patients that died with neurode-generative diseases. The lack of change in protein levels suggest that this is related to a post-translational modification. Thus, reversal of the modification should be beneficial. Gluta-thionylation of KGDHC due to H2O2 reverses when H2O2 is diminished [106]. Nitration of isolated KGDHC can also be reversed by glutathione [105]. The inactivation of KGDHC in cells by 4-hydroxy-2-nonenal can be protected against by DL-lipoamide, DL-lipoic acid, reduced glutathione and cysteine [144]. Prolonged treatment with trolox (a vitamin E like compound) increases KGDHC by three fold [126]. In vivo treatment with particular lipoic acid derivatives can increase heart KGDHC two to three fold [145]. Although reversing the inactivation of one enzyme may seem like a too focused approach, one should remember that even relatively small increases in the activity of enzymes involved in a number of inborn errors of metabolism have been associated with clinically significant benefits. Furthermore, it is likely that compounds that reactivate KGDHC may also reactivate other processes that have been inactivated by similar mechanisms. For example, the same conditions that inactivate KGDHC alter endoplasmic reticulum calcium in a manner that is reminiscent of AD [126].
3. Inhibit TGase activity
Numerous papers try to block the actions of TGase in order to treat multiple disorders including skin and gastrointestinal disorders. The inhibitors of TGase are generally directed to blocking the cross linking. The inhibitors would act to block the crosslinking of enzymes such KGDHC and aconitase, and also to alleviate the inhibition of transcription. The compound used the most with the CNS is cystamine. Treatment in R6/2 transgenic HD mice, using the transglutaminase inhibitor cystamine, significantly extends survival, improves body weight and motor performance, and delays the neuropathological sequelae [54–55]. Free N[Sigma]-([gamma]-L-glutamyl)-L-lysine (GGEL), a specific biochemical marker of TGase activity, is markedly elevated in the neocortex and caudate nucleus in HD patients. TGase activity and GGEL levels are significantly altered in HD mice. Both TGase and GGEL immunoreactivities colocalized to huntingtin aggregates. Cystamine treatment normalized transglutaminase and GGEL levels in R6/2 mice. These findings are consistent with the hypothesis that trans-glutaminase activity may play a role in the pathogenesis of HD, and they identify cystamine as a potential therapeutic strategy for treating HD patients [54–55]. However, the specificity of cystamine is questionable, so the studies need to be replicated with more specific TGase inhibitors that enter the brain.
Rather than being a direct inhibitor of TGase in brain, cystamine may provide an alternative substrate for TGase. Cystamine [[beta]-mercaptoethanolamine (MEA) disulfide] is reduced within cells to MEA which is largely responsible for the in vivo effects of its disulfide precursor. The amine group of MEA acts as a TGase substrate resulting in the formation of N[beta]-([gamma]-l-glutamyl)-MEA bonds. The formation of such bonds would compete with the generation of other TG-catalyzed reactions that may contribute to neurodegeneration such as polyamination, protein cross-linking, deamination and the covalent attachment of ceramide to proteins. Structure-function studies also indicated that the mercapto group of MEA significantly influences the substrate behavior of this compound [146]. Whether the more specific inhibitor of TGase, ZDON, that overcomes transcriptional inhibition associated with mutant huntingtin can be beneficial in mice or humans is unknown [51]
4. Prevent upregulation of TGase
The ability of glutathione to prevent glutamate-evoked TGase upregulation was tested in astrocyte cultures. A 24 h exposure to glutamate caused a dose-dependent depletion of glutathione intracellular content and increased the ROS production. Pre-incubation with glutathione ethyl ester or cysteamine recovered oxidative status and was effective in significantly reducing glutamate-increased TGase. These data suggest that TGase upregulation may be part of a biochemical response to oxidative stress induced by a prolonged exposure of astrocyte cultures to glutamate [43]. Thus, the available data suggest that both the KGDHC deficit and the induction of transglutaminase increase in neurodegenerative diseases can be “treated” with glutathione. One way to increase glutathione is by administration of tri-terpenoids, or other compounds, which activate NrF2/ARE pathway which activates the enzymes involved in glutathione synthesis [147]
5. Act on redox sensitive transcription factors
Chronic exposure to ROS in neurodegenerative diseases likely activates cascades of genes. Even if initially protective, prolonged activation may be damaging. Thus, therapeutic approaches based on modulation of these gene cascades may lead to effective therapies. Genes involved in several pathways including antioxidant defense, detoxification, inflammation, etc. are induced in response to oxidative stress in neurodegeneration. However, genes that are associated with energy metabolism, which is necessary for normal brain function, are mostly down-regulated. Redox sensitive transcription factors such as activator protein-1 (AP-1), nuclear factor κB (NF-κB), specificity protein-1 and hypoxia-inducible factor (HIF) are important in redox-dependent gene regulation. PPARγ co-activator (PGC-1α) is a co-activator of several transcription factors and is a potent stimulator of mitochondrial biogenesis and respiration. Down-regulated expression of PGC-1α has been implicated in HD and in several HD animal models [59–61]. Its role in regulation of ROS metabolism makes it a potential candidate player between ROS, mitochondria and neurodegenerative diseases[148]. Potential therapeutic strategies based on the updated understandings of redox state-dependent gene regulation may be productive [149].
Therapies normally associated with antioxidant or energy metabolism may work by more general gene mechanisms. For example, results show a protective effect of two mitochondrial an-tioxidant/nutrients, R-03B12013lipoic acid and acetyl-L-carnitine in a chronic rotenone-induced cellular model of PD. When combined, lipoic acid and acetylcarnitne work at 100 to 1000 fold lower concentrations than they did individually. The combined pretreatment increases mitochondrial biogenesis and decreases production of ROS reactive oxygen species through the up-regulation of the peroxisome proliferator-activated receptor-γ (PGC-1α) - a possible underlying mechanism [150].
Experiments with HIF1–1 provide a specific example of this approach. The transcription factor hypoxia-inducible factor-1 (HIF1–1) mediates the activation of a large cassette of genes involved in adaptation to hypoxia in surviving neurons after stroke. Pharmacological or molecular approaches that inhibit HIF prolyl 4 hydroxylases lead to profound sparing of brain tissue and enhanced recovery of function. The strategy appears to involve HIF-dependent and HIF-independent pathways and more than 70 genes and proteins activated transcriptionally and post-transcriptionally [151]. One of the genes activated by HIF is TGase [152], and TGase regulates HIF [56].
CONCLUSION
A surprisingly large number of features of age-related neurodegenerative diseases can be accounted for by changes in KGDHC, TGase, oxidative stress and calcium (Figure 4). Therapeutic approaches that ameliorate these deficits are likely to be therapeutically useful.
Figure 4.
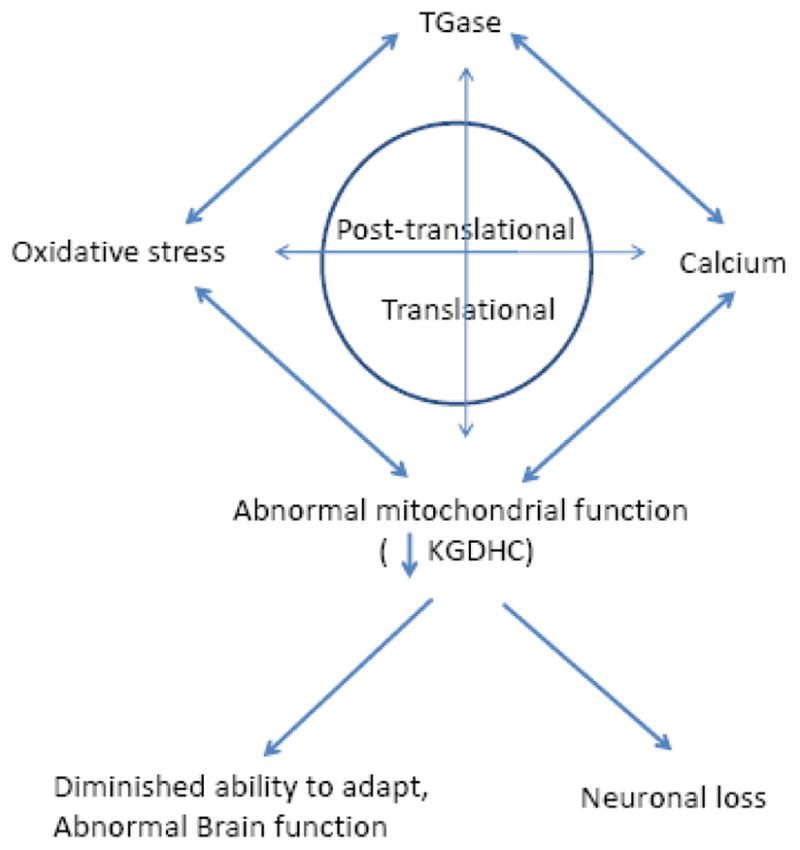
Interaction of TGase, oxidative stress, calcium lead to abnormal mitochondrial function and diminished ability to adapt.
Figure 3.

Subunit composition of KGDHC. KGDHC produces oxidants or NADH depending on redox state. Low NAD+ promotes ROS formation
Acknowledgments
Supported by National Institute on Aging PP-AG14930 and the Burke Medical Research Institute
Abbreviations
- AD
Alzheimer’s Disease
- APP
amyloid precursor
- Aβ
protein amyloid β peptide
- KGDHC
α-ketoglutarate dehydrogenase complex
- ETC
electron transport chain
- htt
huntingtin
- HD
Huntington’s Disease
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- 3NP
3-nitroproprionic acid
- PD
Parkinson’s disease
- PSP
progressive supranuclear palsy
- ROS
reactive oxygen species
- TGase
transglutaminase
- TCA
tricarboxylic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Browne SE. Mitochondria and Huntington’s disease pathogenesis: insight from genetic and chemical models. Ann N Y Acad Sci. 2008;1147:358–382. doi: 10.1196/annals.1427.018. [DOI] [PubMed] [Google Scholar]
- 2.Klivenyi P, Starkov AA, Calingasan NY, Gardian G, Browne SE, Yang L, Bubber P, Gibson GE, Patel MS, Beal MF. Mice deficient in dihydrolipoamide dehydrogenase show increased vulnerability to MPTP, malonate and 3-nitropropionic acid neurotoxicity. J Neurochem. 2004;88:1352–1360. doi: 10.1046/j.1471-4159.2003.02263.x. [DOI] [PubMed] [Google Scholar]
- 3.Schapira AHV. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. The Lancet Neurology. 2008;7:97–109. doi: 10.1016/S1474-4422(07)70327-7. [DOI] [PubMed] [Google Scholar]
- 4.Thomas B, Beal MF. Parkinson’s disease. Hum Mol Genet. 2007;16:R183–194. doi: 10.1093/hmg/ddm159. [DOI] [PubMed] [Google Scholar]
- 5.Banerjee R, Starkov AA, Beal MF, Thomas B. Mitochondrial dysfunction in the limelight of Parkinson’s disease pathogenesis. Biochim Biophys Acta. 2009;1792:651–663. doi: 10.1016/j.bbadis.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou C, Huang Y, Przedborski S. Oxidative stress in Parkinson’s disease: a mechanism of pathogenic and therapeutic significance. Ann N Y Acad Sci. 2008;1147:93–104. doi: 10.1196/annals.1427.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joseph JA, Gibson GE. Coupling of neuronal function to oxygen and glucose metabolism through changes in neurotransmitter dynamics as revealed with aging, hypoglycemia and hypoxia. In: Gibson GE, Dienel G, editors. Handbook of Neurochemistry and Molecular Biology. 3rd Edition Volume 5. Brain energetics from genes to metabolites to cells: Integration of molecular and cellular processes. Vol. 4. 2007. pp. 297–320. [Google Scholar]
- 8.Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc Natl Acad Sci U S A. 2004;101:284–289. doi: 10.1073/pnas.2635903100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Santi S, de Leon MJ, Rusinek H, Convit A, Tarshish CY, Roche A, Tsui WH, Kandil E, Boppana M, Daisley K, Wang GJ, Schlyer D, Fowler J. Hippocampal formation glucose metabolism and volume losses in MCI and AD. Neurobiol Aging. 2001;22:529–539. doi: 10.1016/s0197-4580(01)00230-5. [DOI] [PubMed] [Google Scholar]
- 10.de Leon MJ, Convit A, Wolf OT, Tarshish CY, DeSanti S, Rusinek H, Tsui W, Kandil E, Scherer AJ, Roche A, Imossi A, Thorn E, Bobinski M, Caraos C, Lesbre P, Schlyer D, Poirier J, Reisberg B, Fowler J. Prediction of cognitive decline in normal elderly subjects with 2-[(18)F]fluoro-2-deoxy-D-glucose/poitron-emission tomography (FDG/PET) Proc Natl Acad Sci U S A. 2001;98:10966–10971. doi: 10.1073/pnas.191044198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alexander GE, Chen K, Pietrini P, Rapoport SI, Reiman EM. Longitudinal PET Evaluation of Cerebral Metabolic Decline in Dementia: A Potential Outcome Measure in Alzheimer’s Disease Treatment Studies. Am J Psychiatry. 2002;159:738–745. doi: 10.1176/appi.ajp.159.5.738. [DOI] [PubMed] [Google Scholar]
- 12.Starkov AA. The role of mitochondria in ROS metabolism and signaling. Ann NY Acad Sci. 2008;1147:37–52. doi: 10.1196/annals.1427.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brookes PS. Mitochondrial production of oxidants and their role in the regulation of cellular processes. In: Dienel GEGaG., editor. Handbook of Neurochemistry and Molecular Biology. 3rd Edition Volume 5. Brain energetics from genes to metabolites to cells: Integration of molecular and cellular processes. Vol. 4. 2007. pp. 519–548. [Google Scholar]
- 14.Calingasan NY, Uchida K, Gibson GE. Protein-bound acrolein: a novel marker of oxidative stress in Alzheimer’s disease. J Neurochem. 1999;72:751–756. doi: 10.1046/j.1471-4159.1999.0720751.x. [DOI] [PubMed] [Google Scholar]
- 15.Pratico D, MYLV, Trojanowski JQ, Rokach J, Fitzgerald GA. Increased F2-isoprostanes in Alzheimer’s disease: evidence for enhanced lipid peroxidation in vivo. FASEB J. 1998;12:1777–1783. doi: 10.1096/fasebj.12.15.1777. [DOI] [PubMed] [Google Scholar]
- 16.Connolly J, Siderowf A, Clark CM, Mu D, Pratico D. F2 isoprostane levels in plasma and urine do not support increased lipid peroxidation in cognitively impaired Parkinson disease patients. Cogn Behav Neurol. 2008;21:83–86. doi: 10.1097/WNN.0b013e31817995e7. [DOI] [PubMed] [Google Scholar]
- 17.Stack EC, Matson WR, Ferrante RJ. Evidence of oxidant damage in Huntington’s disease: translational strategies using antioxidants. Ann N Y Acad Sci. 2008;1147:79–92. doi: 10.1196/annals.1427.008. [DOI] [PubMed] [Google Scholar]
- 18.Albers DS, Augood SJ, Park LC, Browne SE, Martin DM, Adamson J, Hutton M, Standaert DG, Vonsattel JP, Gibson GE, Beal MF. Frontal lobe dysfunction in progressive supranuclear palsy: evidence for oxidative stress and mitochondrial impairment. J Neurochem. 2000;74:878–881. doi: 10.1046/j.1471-4159.2000.740878.x. [DOI] [PubMed] [Google Scholar]
- 19.Gibson GE, Sheu KF, Blass JP, Baker A, Carlson KC, Harding B, Perrino P. Reduced activities of thiamine-dependent enzymes in the brains and peripheral tissues of patients with Alzheimer’s disease. Archives of neurology. 1988;45:836–840. doi: 10.1001/archneur.1988.00520320022009. [DOI] [PubMed] [Google Scholar]
- 20.Butterworth RF, Besnard AM. Thiamine-dependent enzyme changes in temporal cortex of patients with Alzheimer’s disease. Metab Brain Dis. 1990;5:179–184. doi: 10.1007/BF00997071. [DOI] [PubMed] [Google Scholar]
- 21.Mastrogiacoma F, Lindsay JG, Bettendorff L, Rice J, Kish SJ. Brain protein and alpha-ketoglutarate dehydrogenase complex activity in Alzheimer’s disease. Ann Neurol. 1996;39:592–598. doi: 10.1002/ana.410390508. [DOI] [PubMed] [Google Scholar]
- 22.Terwel D, Bothmer J, Wolf E, Meng F, Jolles J. Affected enzyme activities in Alzheimer’s disease are sensitive to antemortem hypoxia. J Neurol Sci. 1998;161:47–56. doi: 10.1016/s0022-510x(98)00240-8. [DOI] [PubMed] [Google Scholar]
- 23.Gibson GE, Haroutunian V, Zhang H, Park LC, Shi Q, Lesser M, Mohs RC, Sheu RK, Blass JP. Mitochondrial damage in Alzheimer’s disease varies with apolipoprotein E genotype. Ann Neurol. 2000;48:297–303. [PubMed] [Google Scholar]
- 24.Gibson GE, Kingsbury AE, Xu H, Lindsay JG, Daniel S, Foster OJ, Lees AJ, Blass JP. Deficits in a tricarboxylic acid cycle enzyme in brains from patients with Parkinson’s disease. Neurochem Int. 2003;43:129–135. doi: 10.1016/s0197-0186(02)00225-5. [DOI] [PubMed] [Google Scholar]
- 25.Gibson GE, Zhang H, Sheu KF, Bogdanovich N, Lindsay JG, Lannfelt L, Vestling M, Cowburn RF. Alpha-ketoglutarate dehydrogenase in Alzheimer brains bearing the APP670/671 mutation. Ann Neurol. 1998;44:676–681. doi: 10.1002/ana.410440414. [DOI] [PubMed] [Google Scholar]
- 26.Bubber P, Haroutunian V, Fisch G, Blass JP, Gibson GE. Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann Neurol. 2005;57:695–703. doi: 10.1002/ana.20474. [DOI] [PubMed] [Google Scholar]
- 27.Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science. 2004;304:448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 28.Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005;19:2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- 29.Casley CS, Canevari L, Land JM, Clark JB, Sharpe MA. Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J Neurochem. 2002;80:91–100. doi: 10.1046/j.0022-3042.2001.00681.x. [DOI] [PubMed] [Google Scholar]
- 30.Jeitner TM, Xu H, Gibson GE. Inhibition of the alpha-ketoglutarate dehydrogenase complex by the myeloperoxidase products, hypochlorous acid and mono-N-chloramine. J Neurochem. 2005;92:302–310. doi: 10.1111/j.1471-4159.2004.02868.x. [DOI] [PubMed] [Google Scholar]
- 31.Pocernich CB, Butterfield DA. Acrolein inhibits NADH-linked mitochondrial enzyme activity: implications for Alzheimer’s disease. Neurotoxicity research. 2003;5:515–520. doi: 10.1007/BF03033161. [DOI] [PubMed] [Google Scholar]
- 32.Kumar MJ, Nicholls DG, Andersen JK. Oxidative alpha-ketoglutarate dehydrogenase inhibition via subtle elevations in monoamine oxidase B levels results in loss of spare respiratory capacity: implications for Parkinson’s disease. J Biol Chem. 2003;278:46432–46439. doi: 10.1074/jbc.M306378200. [DOI] [PubMed] [Google Scholar]
- 33.Hinerfeld D, Traini MD, Weinberger RP, Cochran B, Doctrow SR, Harry J, Melov S. Endogenous mitochondrial oxidative stress: neurodegeneration, proteomic analysis, specific respiratory chain defects, and efficacious antioxidant therapy in superoxide dismutase 2 null mice. J Neurochem. 2004;88:657–667. doi: 10.1046/j.1471-4159.2003.02195.x. [DOI] [PubMed] [Google Scholar]
- 34.Shi Q, Hui X, Deng H, Yu H, Je Y, Estevez AG, Gibson GE. Inactivation of the mitochondrial α-ketoglutarate dehydrogenase complex by peroxynitrite. Society for Neuoscience Abstracts. 2008 [Google Scholar]
- 35.Zhang L, Cooper AJ, Krasnikov BF, Xu H, Bubber P, Pinto JT, Gibson GE, Hanigan MH. Cisplatin-induced toxicity is associated with platinum deposition in mouse kidney mitochondria in vivo and with selective inactivation of the alpha-ketoglutarate dehydrogenase complex in LLC-PK1 cells. Biochemistry. 2006;45:8959–8971. doi: 10.1021/bi060027g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McNaught KS, Altomare C, Cellamare S, Carotti A, Thull U, Carrupt PA, Testa B, Jenner P, Marsden CD. Inhibition of alpha-ketoglutarate dehydrogenase by isoquinoline derivatives structurally related to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) Neuroreport. 1995;6:1105–1108. doi: 10.1097/00001756-199505300-00008. [DOI] [PubMed] [Google Scholar]
- 37.Cooper AJL, Sheu KFR, Burke JR, Onodera O, Strittmatter WJ, Roses AD, Blass JP. Transglutaminase-catalyzed inactivation of glyceraldehyde 3-phosphate dehydrogenase and α-ketoglutarate dehydrogenase complex by polyglutamine domains of pathological length. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:12604–12609. doi: 10.1073/pnas.94.23.12604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Junn E, Ronchetti RD, Quezado MM, Kim SY, Mouradian MM. Tissue transglutaminase-induced aggregation of α-synuclein: Implications for Lewy body formation in Parkinson’s disease and dementia with Lewy bodies. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:2047–2052. doi: 10.1073/pnas.0438021100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ientile R, Caccamo D, Marciano MC, Currò M, Mannucci C, Campisi A, Calapai G. Transglutaminase activity and transglutaminase mRNA transcripts in gerbil brain ischemia. Neuroscience Letters. 2004;363:173–177. doi: 10.1016/j.neulet.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 40.Jeitner TM, Bogdanov MB, Matson WR, Daikhin Y, Yudkoff M, Folk JE, Steinman L, Browne SE, Beal MF, Blass JP, Cooper AJ. N(epsilon)-(gamma-L-glutamyl)-L-lysine (GGEL) is increased in cerebrospinal fluid of patients with Huntington’s disease. J Neurochem. 2001;79:1109–1112. doi: 10.1046/j.1471-4159.2001.00673.x. [DOI] [PubMed] [Google Scholar]
- 41.Jeitner TM, Pinto JT, Krasnikov BF, Horswill M, Cooper AJ. Transglutaminases and neurodegeneration. J Neurochem. 2009;109(Suppl 1):160–166. doi: 10.1111/j.1471-4159.2009.05843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jeitner TM, Matson WR, Folk JE, Blass JP, Cooper AJ. Increased levels of gamma-glutamylamines in Huntington disease CSF. J Neurochem. 2008;106:37–44. doi: 10.1111/j.1471-4159.2008.05350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Campisi A, Caccamo D, Li Volti G, Currò M, Parisi G, Avola R, Vanella A, Ientile R. Glutamate-evoked redox state alterations are involved in tissue transglutaminase upregulation in primary astrocyte cultures. FEBS Letters. 2004;578:80–84. doi: 10.1016/j.febslet.2004.10.074. [DOI] [PubMed] [Google Scholar]
- 44.Park KC, Chung KC, Kim YS, Lee J, Joh TH, Kim SY. Transglutaminase 2 induces nitric oxide synthesis in BV-2 microglia. Biochemical and Biophysical Research Communications. 2004;323:1055–1062. doi: 10.1016/j.bbrc.2004.08.204. [DOI] [PubMed] [Google Scholar]
- 45.Cooper AJ, Sheu KR, Burke JR, Onodera O, Strittmatter WJ, Roses AD, Blass JP. Transglutaminase-catalyzed inactivation of glyceraldehyde 3-phosphate dehydrogenase and alpha-ketoglutarate dehydrogenase complex by polyglutamine domains of pathological length. Proc Natl Acad Sci U S A. 1997;94:12604–12609. doi: 10.1073/pnas.94.23.12604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim SY, Marekov L, Bubber P, Browne SE, Stavrovskaya I, Lee J, Steinert PM, Blass JP, Beal MF, Gibson GE, Cooper AJ. Mitochondrial aconitase is a transglutaminase 2 substrate: transglutamination is a probable mechanism contributing to high-molecular-weight aggregates of aconitase and loss of aconitase activity in Huntington disease brain. Neurochem Res. 2005;30:1245–1255. doi: 10.1007/s11064-005-8796-x. [DOI] [PubMed] [Google Scholar]
- 47.Schapira AH. Mitochondrial involvement in Parkinson’s disease, Huntington’s disease, hereditary spastic paraplegia and Friedreich’s ataxia. Biochim Biophys Acta. 1999;1410:159–170. doi: 10.1016/s0005-2728(98)00164-9. [DOI] [PubMed] [Google Scholar]
- 48.Tabrizi SJ, Cleeter MW, Xuereb J, Taanman JW, Cooper JM, Schapira AH. Biochemical abnormalities and excitotoxicity in Huntington’s disease brain. Ann Neurol. 1999;45:25–32. doi: 10.1002/1531-8249(199901)45:1<25::aid-art6>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 49.Tabrizi SJ, Workman J, Hart PE, Mangiarini L, Mahal A, Bates G, Cooper JM, Schapira AH. Mitochondrial dysfunction and free radical damage in the Huntington R6/2 transgenic mouse. Ann Neurol. 2000;47:80–86. doi: 10.1002/1531-8249(200001)47:1<80::aid-ana13>3.3.co;2-b. [DOI] [PubMed] [Google Scholar]
- 50.Perluigi M, Poon HF, Maragos W, Pierce WM, Klein JB, Calabrese V, Cini C, De Marco C, Butterfield DA. Proteomic analysis of protein expression and oxidative modification in r6/2 transgenic mice: a model of Huntington disease. Mol Cell Proteomics. 2005;4:1849–1861. doi: 10.1074/mcp.M500090-MCP200. [DOI] [PubMed] [Google Scholar]
- 51.Mcconoughey SJ, Niatsetkaya Z, Karpisheva K, CAJL, Krasnikov BF, BMF, RRR Inhibition of transglutaminase 2 increases the transcription of key mitochondrial proteins in cells expressing mutant huntingtin. Soc Neuroscience. 2008:642.10. [Google Scholar]
- 52.Krasnikov BF, Kim SY, McConoughey SJ, Ryu H, Xu H, Stavrovskaya I, Iismaa SE, Mearns BM, Ratan RR, Blass JP, Gibson GE, Cooper AJ. Transglutaminase activity is present in highly purified nonsynaptosomal mouse brain and liver mitochondria. Biochemistry. 2005;44:7830–7843. doi: 10.1021/bi0500877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mastroberardino PG, Iannicola C, Nardacci R, Bernassola F, De Laurenzi V, Melino G, Moreno S, Pavone F, Oliverio S, Fesus L, Piacentini M. ‘Tissue’ transglutaminase ablation reduces neuronal death and prolongs survival in a mouse model of Huntington’s disease. Cell Death Differ. 2002;9:873–880. doi: 10.1038/sj.cdd.4401093. [DOI] [PubMed] [Google Scholar]
- 54.Dedeoglu A, Kubilus JK, Jeitner TM, Matson SA, Bogdanov M, Kowall NW, Matson WR, Cooper AJ, Ratan RR, Beal MF, Hersch SM, Ferrante RJ. Therapeutic effects of cystamine in a murine model of Huntington’s disease. J Neurosci. 2002;22:8942–8950. doi: 10.1523/JNEUROSCI.22-20-08942.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karpuj MV, Becher MW, Springer JE, Chabas D, Youssef S, Pedotti R, Mitchell D, Steinman L. Prolonged survival and decreased abnormal movements in transgenic model of Huntington disease, with administration of the transglutaminase inhibitor cystamine. Nat Med. 2002;8:143–149. doi: 10.1038/nm0202-143. [DOI] [PubMed] [Google Scholar]
- 56.Filiano AJ, Bailey CDC, Tucholski J, Gundemir S, Johnson GVW. Transglutaminase 2 protects against ischemic insult, interacts with HIF1{beta}, and attenuates HIF1 signaling. FASEB J. 2008;22:2662–2675. doi: 10.1096/fj.07-097709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cha JH. Transcriptional signatures in Huntington’s disease. Prog Neurobiol. 2007;83:228–248. doi: 10.1016/j.pneurobio.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Benn CL, Sun T, Sadri-Vakili G, McFarland KN, DiRocco DP, Yohrling GJ, Clark TW, Bouzou B, Cha JH. Huntingtin modulates transcription, occupies gene promoters in vivo, and binds directly to DNA in a polyglutamine-dependent manner. J Neurosci. 2008;28:10720–10733. doi: 10.1523/JNEUROSCI.2126-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 60.Weydt P, Pineda VV, Torrence AE, Libby RT, Satterfield TF, Lazarowski ER, Gilbert ML, Morton GJ, Bammler TK, Strand AD, Cui L, Beyer RP, Easley CN, Smith AC, Krainc D, Luquet S, Sweet IR, Schwartz MW, La Spada AR. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration. Cell Metab. 2006;4:349–362. doi: 10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 61.Chaturvedi RK, Adhihetty P, Shukla S, Hennessy T, Calingasan N, Yang L, Starkov A, Kiaei M, Cannella M, Sassone J, Ciammola A, Squitieri F, Beal MF. Impaired PGC-1{alpha} function in muscle in Huntington’s disease. Hum Mol Genet. 2009 doi: 10.1093/hmg/ddp243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mcconoughey ZNSJ, Karpisheva K, Cooper AJL, Krasnikov BF, Beal MF, Ratan RR. Inhibition of transglutaminase 2 increases the transcription of key mitochondrial proteins in cells expressing mutant huntingtin. Soc Neuroscience. 2008:642.10. [Google Scholar]
- 63.Mcconoughey S, Chavez J, Cooper A, Krasnikov B, Beal M, Ratan R. Society for Neuoscience Abstracts. 2007. Release of transcriptional repression created by transglutaminase is protective in mutant huntingtin expressing; p. 764.761. [Google Scholar]
- 64.Gibson GE, Blass JP, Beal MF, Bunik V. The alpha-ketoglutarate-dehydrogenase complex: a mediator between mitochondria and oxidative stress in neurodegeneration. Mol Neurobiol. 2005;31:43–63. doi: 10.1385/MN:31:1-3:043. [DOI] [PubMed] [Google Scholar]
- 65.Waskiewicz DE, Hammes GG. Elementary steps in the reaction mechanism of the alpha-ketoglutarate dehydrogenase multienzyme complex from Escherichia coli: kinetics of succinylation and desuccinylation. Biochemistry. 1984;23:3136–3143. doi: 10.1021/bi00309a005. [DOI] [PubMed] [Google Scholar]
- 66.Gibson GE, Blass JP. Inhibition of acetylcholine synthesis and of carbohydrate utilization by maple-syrup-urine disease metabolites. J Neurochem. 1976;26:1073–1078. doi: 10.1111/j.1471-4159.1976.tb06988.x. [DOI] [PubMed] [Google Scholar]
- 67.Sheu KF, Blass JP. The alpha-ketoglutarate dehydrogenase complex. Annals of the New York Academy of Sciences. 1999;893:61–78. doi: 10.1111/j.1749-6632.1999.tb07818.x. [DOI] [PubMed] [Google Scholar]
- 68.Maas E, Bisswanger H. Localization of the alpha-oxoacid dehydrogenase multienzyme complexes within the mitochondrion. FEBS Lett. 1990;277:189–190. doi: 10.1016/0014-5793(90)80840-f. [DOI] [PubMed] [Google Scholar]
- 69.Sumegi B, Srere PA. Complex I binds several mitochondrial NAD-coupled dehydrogenases. Journal of Biological Chemistry. 1984;259:15040–15045. [PubMed] [Google Scholar]
- 70.Lyubarev AE, Kurganov BI. Supramolecular organization of tricarboxylic acid cycle enzymes. Biosystems. 1989;22:91–102. doi: 10.1016/0303-2647(89)90038-5. [DOI] [PubMed] [Google Scholar]
- 71.Koike K, Hamada M, Tanaka N, Otsuka KI, Ogasahara K, Koike M. Properties and subunit composition of the pig heart 2-oxoglutarate dehydrogenase. Journal of Biological Chemistry. 1974;249:3836–3842. [PubMed] [Google Scholar]
- 72.Reed LJ, Hackert ML. Structure-function relationships in dihydrolipoamide acyltransferases. Journal of Biological Chemistry. 1990;265:8971–8974. [PubMed] [Google Scholar]
- 73.Patel MS, Roche TE. Molecular biology and biochemistry of pyruvate dehydrogenase complexes. FASEB Journal. 1990;4:3224–3233. doi: 10.1096/fasebj.4.14.2227213. [DOI] [PubMed] [Google Scholar]
- 74.Calingasan NY, Baker H, Sheu KF, Gibson GE. Distribution of the alpha-ketoglutarate dehydrogenase complex in rat brain. Journal of Comparative Neurology. 1994;346:461–479. doi: 10.1002/cne.903460309. [DOI] [PubMed] [Google Scholar]
- 75.Waagepetersen HS, Hansen GH, Fenger K, Lindsay JG, Gibson G, Schousboe A. Cellular mitochondrial heterogeneity in cultured astrocytes as demonstrated by immunogold labeling of alpha-ketoglutarate dehydrogenase. Glia. 2006;53:225–231. doi: 10.1002/glia.20276. [DOI] [PubMed] [Google Scholar]
- 76.Bunik V, Kaehne T, Degtyarev D, Shcherbakova T, Reiser G. Novel isoenzyme of 2-oxoglutarate dehydrogenase is identified in brain, but not in heart. FEBS Journal. 2008;275:4990–5006. doi: 10.1111/j.1742-4658.2008.06632.x. [DOI] [PubMed] [Google Scholar]
- 77.Atamna H, Frey WH. 2nd, A role for heme in Alzheimer’s disease: heme binds amyloid beta and has altered metabolism. Proc Natl Acad Sci U S A. 2004;101:11153–11158. doi: 10.1073/pnas.0404349101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Atamna H, Liu J, Ames BN. Heme Deficiency Selectively Interrupts Assembly of Mitochondrial Complex IV in Human Fibroblasts. RELEVANCE TO AGING. J Biol Chem. 2001;276:48410–48416. doi: 10.1074/jbc.M108362200. [DOI] [PubMed] [Google Scholar]
- 79.Babady NE, Pang YP, Elpeleg O, Isaya G. Cryptic proteolytic activity of dihydrolipoamide dehydrogenase. Proc Natl Acad Sci U S A. 2007;104:6158–6163. doi: 10.1073/pnas.0610618104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kanamori T, Nishimaki K, Asoh S, Ishibashi Y, Takata I, Kuwabara T, Taira K, Yamaguchi H, Sugihara S, Yamazaki T, Ihara Y, Nakano K, Matuda S, Ohta S. Truncated product of the bifunctional DLST gene involved in biogenesis of the respiratory chain. The EMBO journal. 2003;22:2913–2923. doi: 10.1093/emboj/cdg299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Petrat F, Paluch S, Dogruoz E, Dorfler P, Kirsch M, Korth HG, Sustmann R, de Groot H. Reduction of Fe(III) ions complexed to physiological ligands by lipoyl dehydrogenase and other flavoenzymes in vitro: implications for an enzymatic reduction of Fe(III) ions of the labile iron pool. The Journal of biological chemistry. 2003;278:46403–46413. doi: 10.1074/jbc.M305291200. [DOI] [PubMed] [Google Scholar]
- 82.Igamberdiev AU, Bykova NV, Ens W, Hill RD. Dihydrolipoamide dehydrogenase from porcine heart catalyzes NADH-dependent scavenging of nitric oxide. FEBS Lett. 2004;568:146–150. doi: 10.1016/j.febslet.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 83.Olsson JM, Xia L, Eriksson LC, Bjornstedt M. Ubiquinone is reduced by lipoamide dehydrogenase and this reaction is potently stimulated by zinc. FEBS Lett. 1999;448:190–192. doi: 10.1016/s0014-5793(99)00363-4. [DOI] [PubMed] [Google Scholar]
- 84.Xia L, Bjornstedt M, Nordman T, Eriksson LC, Olsson JM. Reduction of ubiquinone by lipoamide dehydrogenase. An antioxidant regenerating pathway. European journal of biochemistry/FEBS. 2001;268:1486–1490. doi: 10.1046/j.1432-1327.2001.02013.x. [DOI] [PubMed] [Google Scholar]
- 85.Hamada M, Koike K, Nakaula Y, Hiraoka T, Koike M. A kinetic study of the alpha-keto acid dehydrogenase complexes from pig heart mitochondria. Journal of Biochemistry. 1975;77:1047–1056. doi: 10.1093/oxfordjournals.jbchem.a130805. [DOI] [PubMed] [Google Scholar]
- 86.McMinn CL, Ottaway JH. Studies on the mechanism and kinetics of the 2-oxoglutarate dehydrogenase system from pig heart. Biochemical Journal. 1977;161:569–581. doi: 10.1042/bj1610569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kiselevsky YV, Ostrovtsova SA, Strumilo SA. Kinetic characterization of the pyruvate and oxoglutarate dehydrogenase complexes from human heart. Acta Biochimica Polonica. 1990;37:135–139. [PubMed] [Google Scholar]
- 88.LaNoue KF, Schoolwerth AC, Pease AJ. Ammonia formation in isolated rat liver mitochondria. Journal of Biological Chemistry. 1983;258:1726–1734. [PubMed] [Google Scholar]
- 89.Wan B, LaNoue KF, Cheung JY, Scaduto RC., Jr Regulation of citric acid cycle by calcium. Journal of Biological Chemistry. 1989;264:13430–13439. [PubMed] [Google Scholar]
- 90.Tretter L, Adam-Vizi V. Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase. J Neurosci. 2004;24:7771–7778. doi: 10.1523/JNEUROSCI.1842-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS, Beal MF. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J Neurosci. 2004;24:7779–7788. doi: 10.1523/JNEUROSCI.1899-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huang HM, Ou HC, CHEN HL, HOU RC, JENG KCG. Protective Effect of alpha-Keto-Beta-Methyl-n-Valeric Acid on BV-2 Microglia under Hypoxia or Oxidative Stress. Annals of the New York Academy of Sciences. 2005;1042:272–278. doi: 10.1196/annals.1338.049. [DOI] [PubMed] [Google Scholar]
- 93.Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zundorf G, Kahlert S, Bunik VI, Reiser G. alpha-Ketoglutarate dehydrogenase contributes to production of reactive oxygen species in glutamate-stimulated hippocampal neurons in situ. Neuroscience. 2009;158:610–616. doi: 10.1016/j.neuroscience.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 95.Hansford RG, Zorov D. Role of mitochondrial calcium transport in the control of substrate oxidation. Mol Cell Biochem. 1998;184:359–369. [PubMed] [Google Scholar]
- 96.Dykens JA. Isolated cerebral and cerebellar mitochondria produce free radicals when exposed to elevated CA2+ and Na+: implications for neurodegeneration. Journal of Neurochemistry. 1994;63:584–591. doi: 10.1046/j.1471-4159.1994.63020584.x. [DOI] [PubMed] [Google Scholar]
- 97.Bunik VI. 2-Oxo acid dehydrogenase complexes in redox regulation. European Journal of Biochemistry. 2003;270:1036–1042. doi: 10.1046/j.1432-1033.2003.03470.x. [DOI] [PubMed] [Google Scholar]
- 98.Bunik VI. 2-Oxo acid dehydrogenase complexes in redox regulation. European journal of biochemistry/FEBS. 2003;270:1036–1042. doi: 10.1046/j.1432-1033.2003.03470.x. [DOI] [PubMed] [Google Scholar]
- 99.Gourlay LJ, Bhella D, Kelly SM, Price NC, Lindsay JG. Structure-function analysis of recombinant substrate protein 22 kDa (SP-22). A mitochondrial 2-CYS peroxiredoxin organized as a decameric toroid. The Journal of biological chemistry. 2003;278:32631–32637. doi: 10.1074/jbc.M303862200. [DOI] [PubMed] [Google Scholar]
- 100.Bunik VI, Denton TT, Xu H, Thompson CM, Cooper AJ, Gibson GE. Phosphonate analogues of alpha-ketoglutarate inhibit the activity of the alpha-ketoglutarate dehydrogenase complex isolated from brain and in cultured cells. Biochemistry. 2005;44:10552–10561. doi: 10.1021/bi0503100. [DOI] [PubMed] [Google Scholar]
- 101.Santos SS, Gibson GE, Cooper AJ, Denton TT, Thompson CM, Bunik VI, Alves PM, Sonnewald U. Inhibitors of the alpha-ketoglutarate dehydrogenase complex alter [1–13C]glucose and [U-13C]glutamate metabolism in cerebellar granule neurons. J Neurosci Res. 2006;83:450–458. doi: 10.1002/jnr.20749. [DOI] [PubMed] [Google Scholar]
- 102.Shi Q, Chen HL, Xu H, Gibson GE. Reduction in the E2k subunit of the alpha-ketoglutarate dehydrogenase complex has effects independent of complex activity. J Biol Chem. 2005;280:10888–10896. doi: 10.1074/jbc.M409064200. [DOI] [PubMed] [Google Scholar]
- 103.Shi Q, Risa Ø, Sonnewald U, Gibson GE. Mild reduction in the activity of the α-ketoglutarate dehydrogenase complex elevates GABA shunt and glycolysis. J Neurochem. 2009 doi: 10.1111/j.1471-4159.2009.05955.x. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shi Q, Xu H, Kleinman WA, Gibson GE. Novel functions of the alpha-ketoglutarate dehydrogenase complex may mediate diverse oxidant-induced changes in mitochondrial enzymes associated with Alzheimer’s disease. Biochim Biophys Acta. 2008;1782:229–238. doi: 10.1016/j.bbadis.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Applegate MA, Humphries KM, Szweda LI. Reversible inhibition of alpha-ketoglutarate dehydrogenase by hydrogen peroxide: glutathionylation and protection of lipoic acid. Biochemistry. 2008;47:473–478. doi: 10.1021/bi7017464. [DOI] [PubMed] [Google Scholar]
- 107.Zündorf G, Kahlert S, Bunik VI, Reiser G. [alpha]-Ketoglutarate dehydrogenase contributes to production of reactive oxygen species in glutamate-stimulated hippocampal neurons in situ. Neuroscience. 2009;158:610–616. doi: 10.1016/j.neuroscience.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 108.Freeman GB, Nielsen PE, Gibson GE. Effect of age on behavioral and enzymatic changes during thiamin deficiency. Neurobiol Aging. 1987;8:429–434. doi: 10.1016/0197-4580(87)90037-6. [DOI] [PubMed] [Google Scholar]
- 109.Karuppagounder SS, Gibson GE. Thiamine deficiency: a model of metabolic encephalopathy and of selective neuronal vulnerability. In: McCandless DW, editor. Metabolic Encephalopathy. Springer Press; 2008. pp. 235–260. [Google Scholar]
- 110.Vemuganti R, Kalluri H, Yi JH, Bowen KK, Hazell AS. Gene expression changes in thalamus and inferior colliculus associated with inflammation, cellular stress, metabolism and structural damage in thiamine deficiency. Eur J Neurosci. 2006;23:1172–1188. doi: 10.1111/j.1460-9568.2006.04651.x. [DOI] [PubMed] [Google Scholar]
- 111.Calingasan NY, Ho DJ, Wille EJ, Campagna MV, Ruan J, Dumont M, Yang L, Shi Q, Gibson GE, Beal MF. Influence of mitochondrial enzyme deficiency on adult neurogenesis in mouse models of neurodegenerative diseases. Neuroscience. 2008;153:986–996. doi: 10.1016/j.neuroscience.2008.02.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.DeGiorgio LA, DeGiorgio N, Milner TA, Conti B, Volpe BT. Neurotoxic APP C-terminal and beta-amyloid domains colocalize in the nuclei of substantia nigra pars reticulata neurons undergoing delayed degeneration. Brain Res. 2000;874:137–146. doi: 10.1016/s0006-8993(00)02545-2. [DOI] [PubMed] [Google Scholar]
- 113.Karuppagounder SS, Xu H, Pechman D, Chen LH, DeGiorgio LA, Gibson GE. Translocation of amyloid precursor protein C-terminal fragment(s) to the nucleus precedes neuronal death due to thiamine deficiency-induced mild impairment of oxidative metabolism. Neurochem Res. 2008;33:1365–1372. doi: 10.1007/s11064-008-9594-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Karuppagounder SS, Xu H, Shi Q, Chen LH, Pedrini S, Pechman D, Baker H, Beal MF, Gandy SE, Gibson GE. Thiamine deficiency induces oxidative stress and exacerbates the plaque pathology in Alzheimer’s mouse model. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2007.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nathan C, Calingasan N, Nezezon J, Ding A, Lucia MS, La Perle K, Fuortes M, Lin M, Ehrt S, Kwon NS, Chen J, Vodovotz Y, Kipiani K, Beal MF. Protection from Alzheimer’s-like disease in the mouse by genetic ablation of inducible nitric oxide synthase. J Exp Med. 2005;202:1163–1169. doi: 10.1084/jem.20051529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Velliquette RA, O’Connor T, Vassar R. Energy inhibition elevates beta-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: possible early events in Alzheimer’s disease pathogenesis. J Neurosci. 2005;25:10874–10883. doi: 10.1523/JNEUROSCI.2350-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.O’Connor T, Sadleir KR, Maus E, Velliquette RA, Zhao J, Cole SL, Eimer WA, Hitt B, Bembinster LA, Lammich S, Lichtenthaler SF, Hebert SS, De Strooper B, Haass C, Bennett DA, Vassar R. Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron. 2008;60:988–1009. doi: 10.1016/j.neuron.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cullen KM, Halliday GM. Neurofibrillary tangles in chronic alcoholics. Neuropathol Appl Neurobiol. 1995;21:312–318. doi: 10.1111/j.1365-2990.1995.tb01065.x. [DOI] [PubMed] [Google Scholar]
- 119.Planel E, Tatebayashi Y, Miyasaka T, Liu L, Wang L, Herman M, Yu WH, Luchsinger JA, Wadzinski B, Duff KE, Takashima A. Insulin Dysfunction Induces In Vivo Tau Hyperphosphorylation through Distinct Mechanisms. J Neurosci. 2007;27:13635–13648. doi: 10.1523/JNEUROSCI.3949-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hoglinger GU, Lannuzel A, Khondiker ME, Michel PP, Duyckaerts C, Feger J, Champy P, Prigent A, Medja F, Lombes A, Oertel WH, Ruberg M, Hirsch EC. The mitochondrial complex I inhibitor rotenone triggers a cerebral tauopathy. J Neurochem. 2005;95:930–939. doi: 10.1111/j.1471-4159.2005.03493.x. [DOI] [PubMed] [Google Scholar]
- 121.Escobar-Khondiker M, Hollerhage M, Muriel MP, Champy P, Bach A, Depienne C, Respondek G, Yamada ES, Lannuzel A, Yagi T, Hirsch EC, Oertel WH, Jacob R, Michel PP, Ruberg M, Hoglinger GU. Annonacin, a natural mitochondrial complex I inhibitor, causes tau pathology in cultured neurons. J Neurosci. 2007;27:7827–7837. doi: 10.1523/JNEUROSCI.1644-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ogawa O, Lee HG, Zhu X, Raina A, Harris PL, Castellani RJ, Perry G, Smith MA. Increased p27, an essential component of cell cycle control, in Alzheimer’s disease. Aging Cell. 2003;2:105–110. doi: 10.1046/j.1474-9728.2003.00042.x. [DOI] [PubMed] [Google Scholar]
- 123.Melov S, Adlard PA, Morten K, Johnson F, Golden TR, Hinerfeld D, Schilling B, Mavros C, Masters CL, Volitakis I, Li QX, Laughton K, Hubbard A, Cherny RA, Gibson B, Bush AI. Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS ONE. 2007;2:e536. doi: 10.1371/journal.pone.0000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kumar U, Dunlop DM, Richardson JS. Mitochondria from Alzheimer’s fibroblasts show decreased uptake of calcium and increased sensitivity to free radicals. Life Sci. 1994;54:1855–1860. doi: 10.1016/0024-3205(94)90142-2. [DOI] [PubMed] [Google Scholar]
- 125.Ito E, Oka K, Etcheberrigaray R, Nelson TJ, McPhie DL, Tofel-Grehl B, Gibson GE, Alkon DL. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91:534–538. doi: 10.1073/pnas.91.2.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gibson GE, Zhang H, Xu H, Park LCH, Jeitner TM. Oxidative stress increases internal calcium stores and reduces a key mitochondrial enzyme. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2002;1586:177–189. doi: 10.1016/s0925-4439(01)00091-6. [DOI] [PubMed] [Google Scholar]
- 127.Moreau Rg, Heath S-HD, Doneanu CE, Lindsay JG, Hagen TM. Age-Related Increase in 4-Hydroxynonenal Adduction to Rat Heart α-Ketoglutarate Dehydrogenase Does Not Cause Loss of Its Catalytic Activity. Antioxidants & Redox Signaling. 2003;5:517–527. doi: 10.1089/152308603770310167. [DOI] [PubMed] [Google Scholar]
- 128.Tahara EB, Barros MH, Oliveira GA, Netto LE, Kowaltowski AJ. Dihydrolipoyl dehydrogenase as a source of reactive oxygen species inhibited by caloric restriction and involved in Saccharomyces cerevisiae aging. Faseb J. 2006 doi: 10.1096/fj.06-6686com. [DOI] [PubMed] [Google Scholar]
- 129.Zhao N, Zhong C, Wang Y, Zhao Y, Gong N, Zhou G, Xu T, Hong Z. Impaired hippocampal neurogenesis is involved in cognitive dysfunction induced by thiamine deficiency at early pre-pathological lesion stage. Neurobiology of Disease. 2008;29:176–185. doi: 10.1016/j.nbd.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 130.Chang DTW, Honick AS, Reynolds IJ. Mitochondrial Trafficking to Synapses in Cultured Primary Cortical Neurons. J Neurosci. 2006;26:7035–7045. doi: 10.1523/JNEUROSCI.1012-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Manfredi G, Beal MF. Merging mitochondria for neuronal survival. Nat Med. 2007;13:1140–1141. doi: 10.1038/nm1007-1140. [DOI] [PubMed] [Google Scholar]
- 132.Chen H, McCaffery JM, Chan DC. Mitochondrial Fusion Protects against Neurodegeneration in the Cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 133.Zanna C, Ghelli A, Porcelli AM, Karbowski M, Youle RJ, Schimpf S, Wissinger B, Pinti M, Cossarizza A, Vidoni S, Valentino ML, Rugolo M, Carelli V. OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain. 2008;131:352–367. doi: 10.1093/brain/awm335. [DOI] [PubMed] [Google Scholar]
- 134.Yang Y, Ouyang Y, Yang L, Beal MF, McQuibban A, Vogel H, Lu B. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci U S A. 2008;105:7070–7075. doi: 10.1073/pnas.0711845105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lutz AK, Exner N, Fett ME, Schlehe JS, Kloos K, Laemmermann K, Brunner B, Kurz-Drexler A, Vogel F, Reichert AS, Bouman L, Vogt-Weisenhorn D, Wurst W, Tatzelt J, Haass C, Winklhofer KF. Loss of parkin or PINK1 function increases DRP1-dependent mitochondrial fragmentation. J Biol Chem. 2009:M109.035774. doi: 10.1074/jbc.M109.035774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dagda RK, Cherra SJ, III, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 Function Promotes Mitophagy through Effects on Oxidative Stress and Mitochondrial Fission. J Biol Chem. 2009;284:13843–13855. doi: 10.1074/jbc.M808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sandebring A, Thomas KJ, Beilina A, van der Brug M, Cleland MM, Ahmad R, Miller DW, Zambrano I, Cowburn RF, Behbahani H, Cedazo-Minguez A, Cookson MR. Mitochondrial alterations in PINK1 deficient cells are influenced by calcineurin-dependent dephosphorylation of dynamin-related protein 1. PLoS ONE. 2009;4:e5701. doi: 10.1371/journal.pone.0005701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Knott Andrew B, Bossy-Wetzel Ella. Impairing the Mitochondrial Fission and Fusion Balance: A New Mechanism of Neurodegeneration. Annals of the New York Academy of Sciences. 2008;1147:283–292. doi: 10.1196/annals.1427.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, Casadesus G, Zhu X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A. 2008;105:19318–19323. doi: 10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, Zhu X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci. 2009;29:9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wang X, Su B, Zheng L, Perry G, Smith MA, Zhu X. The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J Neurochem. 2009;109(Suppl 1):153–159. doi: 10.1111/j.1471-4159.2009.05867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Blass JP. A New Approach to Treating Alzheimer’s Disease. Annals of the New York Academy of Sciences. 2008;1147:122–128. doi: 10.1196/annals.1427.022. [DOI] [PubMed] [Google Scholar]
- 144.Korotchkina LG, Yang HS, Tirosh O, Packer L, Patel MS. Protection by thiols of the mitochondrial complexes from 4-hydroxy-2-nonenal. Free Radical Biology and Medicine. 2001;30:992–999. doi: 10.1016/s0891-5849(01)00491-9. [DOI] [PubMed] [Google Scholar]
- 145.Sudheesh NP, Ajith TA, Janaradhanan KK, Krishnan CV. Palladium α-lipoic acid complex formulation enhances activities of Krebs cycle dehydrogenases and respiratory complexes I–IV in the heart of aged rats. Food and Chemical Toxicology. 2009 doi: 10.1016/j.fct.2009.05.032. [DOI] [PubMed] [Google Scholar]
- 146.Jeitner TM, Delikatny EJ, Ahlqvist J, Capper H, Cooper AJL. Mechanism for the inhibition of transglutaminase 2 by cystamine. Biochemical Pharmacology. 2005;69:961–970. doi: 10.1016/j.bcp.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 147.Yang L, Calingasan NY, Thomas B, Chaturvedi RK, Kiaei M, Wille EJ, Liby KT, Williams C, Royce D, Risingsong R, Musiek ES, Morrow JD, Sporn M, Beal MF. Neuroprotective effects of the triterpenoid, CDDO methyl amide, a potent inducer of Nrf2-mediated transcription. PLoS ONE. 2009;4:e5757. doi: 10.1371/journal.pone.0005757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 149.Shi Q, Gibson GE. Oxidative Stress and Transcriptional Regulation in Alzheimer Disease. Alzheimer Disease & Associated Disorders. 2007;21:276–291. doi: 10.1097/WAD.0b013e31815721c3. 210.1097/WAD.1090b1013e31815721c31815723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zhang H, Jia H, Liu J, Ao N, Yan B, Shen W, Wang X, Li X, Luo C, Liu j. Combined R-α–lipoic acid and acetyl-L-carnitine exerts efficient preventative effects in a cellular model of Parkinson’s disease. Journal of Cellular and Molecular Medicine. :9999, 2008. doi: 10.1111/j.1582-4934.2008.00390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ratan RR, Siddiq A, Smirnova N, Karpisheva K, Haskew-Layton R, McConoughey S, Langley B, Estevez A, Huerta PT, Volpe B, Roy S, Sen CK, Gazaryan I, Cho S, Fink M, LaManna J. Harnessing hypoxic adaptation to prevent, treat, and repair stroke. J Mol Med. 2007;85:1331–1338. doi: 10.1007/s00109-007-0283-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lee JW, Bae SH, Jeong JW, Kim SH, Kim KW. Hypoxia-inducible factor (HIF-1)alpha: its protein stability and biological functions. Exp Mol Med. 2004;36:1–12. doi: 10.1038/emm.2004.1. [DOI] [PubMed] [Google Scholar]