

Abstract
Telomerase function is critical for telomere maintenance. Mutations in telomerase components lead to telomere shortening and progressive bone marrow failure in the premature aging syndrome dyskeratosis congenita. Short telomeres are also acquired with aging, yet the role that they play in mediating age-related disease is not fully known. We generated wild-type mice that have short telomeres. In these mice, we identified hematopoietic and immune defects that resembled those present in dyskeratosis congenita patients. When mice with short telomeres were interbred, telomere length was only incrementally restored, and even several generations later, wild-type mice with short telomeres still displayed degenerative defects. Our findings implicate telomere length as a unique heritable trait that, when short, is sufficient to mediate the degenerative defects of aging, even when telomerase is wild-type.
Introduction
Telomeres are DNA-protein structures that protect chromosome ends. With cell replication, telomeres shorten successively and ultimately lead to apoptosis or permanent cell-cycle arrest. Telomeres have thus been long appreciated as a determinant of replicative senescence in cells.1 With aging, telomeres also shorten in humans, yet their role in mediating age-related disease is not fully known.
In the presence of mutant telomerase components, short telomeres cause a premature aging syndrome. In telomere-mediated syndromes, short telomeres clinically manifest as aplastic anemia in the bone marrow and progressive fibrosis in the lung and liver.2 Disease-associated mutations in telomerase components were initially identified in the context of dyskeratosis congenita (DKCX [MIM 305000]), a disorder characterized by early mortality due to bone marrow failure.3,4 Loss-of-function mutations in the essential components of telomerase, hTR, the telomerase RNA (MIM 602322), and hTERT, the catalytic reverse transcriptase (MIM 187270), lead to telomerase haploinsufficiency and autosomal-dominant inheritance of dyskeratosis congenita (DKCA [MIM 127550]).5,6 In families, the organ failure displays anticipation, an earlier and more severe onset with each generation, which is associated with progressive telomere shortening.5,7 These observations have implicated telomere length as an important modifier of disease penetrance in families that carry mutant telomerase genes. However, whether short telomeres alone, in the absence of telomerase mutations, can mediate disease with aging is not known.
Telomerase function is critical for organ homeostasis. Hematopoietic stem cells and lymphocytes are enriched for telomerase activity, suggesting that their self-renewal potential may depend on the presence of telomerase.8,9 This observation would imply that telomerase may protect against degenerative defects in these compartments by preventing telomere shortening. In approaching these questions, the study of telomerase function in mammalian models has relied on laboratory mouse strains that possess long, heterogeneous telomere lengths that do not mimic human telomere dynamics.10–13 In most laboratory strains, the average telomere length is ∼50–70 kb, compared with the average human telomere length of ∼10 kb.14 Therefore, on these strains, end organ dysfunction is present only when telomerase is null and after several generations of breeding when telomeres are short. Late-generation mTR−/− mice have organ dysfunction that manifests as a stem cell failure disorder and prominently affects tissues of high turnover: the hematopoietic system, the gastrointestinal tract, and male germ cells.10–13,15 Distinct from other laboratory strains, CAST/EiJ mice have telomere length and distribution that mimic those of humans (average telomere length ∼15 kb).16 We have previously shown that, similar to dyskeratosis congenita patients, CAST/EiJ mTR+/− mice are haploinsufficient for telomerase and develop end organ defects when telomeres are short.15,17 Wild-type littermates of late-generation heterozygous mice also inherit short telomeres.15 However, whether these short telomeres can cause clinically relevant phenotypes that resemble those of aging is not known.
Here, we show that mice that are otherwise wild-type at the telomerase locus but have short telomeres develop degenerative defects in both hematopoietic and immune systems. These defects mimic the hematopoietic and immunosenescence phenotypes present in dyskeratosis congenita patients. Our findings suggest that the short-telomere genotype (telotype)18 is a unique heritable trait, sufficient to mediate degenerative disease even when telomerase is wild-type.
Material and Methods
Mice were housed on the Johns Hopkins University School of Medicine campus, and all procedures were approved by its Institutional Animal Care and Use Committee. Blood counts and differentials were performed in a clinical lab with the use of standard antibodies: anti-Annexin V, B220, CD3, CD4, CD8, CD48, CD150, and c-kit (Becton Dickinson). Flow cytometry was performed on a FACS Calibur with the use of standard antibodies (Becton Dickinson). Quantitative fluorescence in situ hybridization (qFISH) and 5-fluorouracil studies were performed as described previously.19 IgM quantitation was performed via standard ELISA (Bethyl Laboratories). Mice were immunized with TNP-Ficoll (10 μg, Biosearch Technologies) and Imject Alum (Pierce) similarly to the methods described in Morra et al.19 We quantitated antigen-specific IgM 3 days prior to immunization and on day 7 after using ELISA (TNP-OVA, Biosearch Technologies) as described previously.19 T cell isolation was performed with EasySep (StemCell Technologies). T cells were stimulated with CD3 5 μg/mL (platebound) and CD28 10 ng/mL (eBioscience). Cells were plated at 1.5 × 106 per mL. MTT assay was performed in accordance with the manufacturer's instructions (Roche). EdU detection was performed 16 hr after incubation (Invitrogen). Tissues were prepared and fixed as described previously,15 and all pathologic analyses were performed blinded to genotype. Statistical analyses were performed with Prism software for Windows. Means were compared with Student's t test, and all p values shown are two-sided.
Results
Short Telomeres Are Inherited in wt∗ Mice
To examine the consequences of short telomeres when telomerase is wild-type, we bred CAST/EiJ mTR+/− mice successively.15 We bred mTR+/− mice to each other and assigned the generation number. For example, first-heterozygous-generation mTR+/− mice were termed HG1, second heterozygous generation as HG2, etc. To distinguish wild-type progeny from these crosses, we termed them wt∗ and indicated the generation number. For example, wild-type mice born of mTR+/− HG7 were named wt8∗1 (Figure 1A). As previously shown,15 wt∗ mice inherit the short telomeres and thus have a shorter telotype than wild-types (Figure 1B). With these short telomeres, wt∗ mice provide an opportunity to understand the contribution of short telomeres to human aging.
Figure 1.
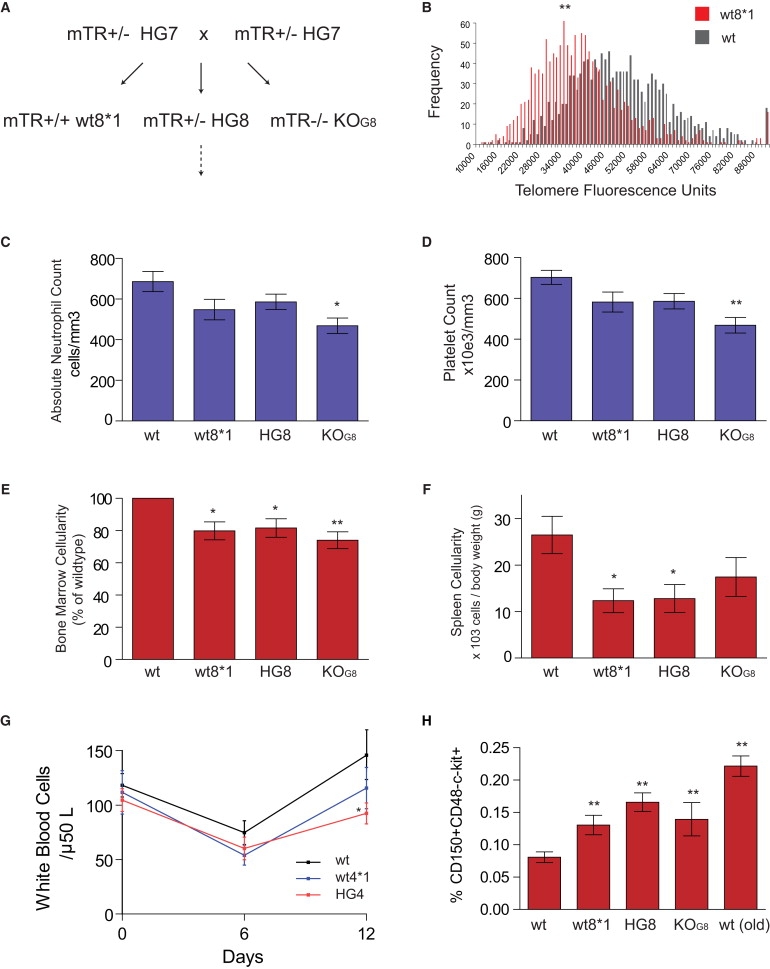
Hematopoiesis Is Ineffective in Wild-Type Mice with Short Telomeres
(A) Scheme and nomenclature of mTR+/− breeding. HG7 refers to the seventh heterozygous generation, and wt8∗1 refers to the first generation born from HG7 crosses.
(B–F) Frequency distribution of telomere lengths as measured by qFISH shows that wt∗ mice have shorter telomeres than wild-type mice (average wild-type = 50,000 telomere fluorescence units [TFU], wt∗ = 39,000 TFU; p < 0.0001; n = 3 per group). wt∗ mice have low peripheral-blood absolute neutrophil (C) and platelet counts (D). These cytopenias are associated with a hypocellular bone marrow (D) and atrophic spleen (F). (E) Peripheral white blood cell count after 5-fluorouracil injection on day 0 shows that both wt∗ and mTR+/− mice with short telomeres have lower nadir and lower day 12 white counts as compared with wild-type mice.
(G) Percentage of bone marrow cells identified by SLAM markers (CD150 and CD48) that are c-kit+. Young mice with short telomeres (2 mo), including wt∗ mice, have increased frequency of SLAM-c-kit+ cells as compared with age-matched wild-type mice. This increase is similar to the expansion of this compartment in older wild-type mice (12 mo). Mice were examined at 2 mo of age, and each experiment included at least five mice per genotype. p values < 0.05 are indicated by ∗, < 0.01 by ∗∗. Error bars represent standard error of the mean.
wt∗ Mice Have Hematopoietic Defects
Haploinsufficiency for telomerase components leads to aplastic anemia, and mTR+/− mice with short telomeres have low blood counts.15 Hematopoietic function also declines with age and manifests as progressive cytopenias, a decrease in hematopoietic organ cellularity, and a decreasing tolerance to cumulative doses of chemotherapy.20–22 To determine whether short telomeres are sufficient to mediate these defects, we examined blood counts. wt∗ mice developed low absolute neutrophil and platelet counts (Figures 1C and 1D). These cytopenias were associated with a decrease in bone marrow and spleen cellularity, consistent with a defect in bone marrow production (Figures 1E and 1F). When we challenged wt∗ mice with 5-fluorouracil, they had a more profound nadir and lower leukocyte counts at the time of expected recovery, though these defects were more profound in mTR+/− littermates (Figure 1G). The delay in recovery of white cell count after chemotherapy is consistent with a defect in bone marrow progenitor pools. Thus, even when telomerase is wild-type, the short telotype leads to a degenerative bone marrow failure syndrome.
Hematopoietic Progenitor Defects in Wild-Type Mice with Short Telomeres
Although stem cell function declines with age, in certain mouse strains the frequency of hematopoietic stem cells increases with age.23 To examine whether wt∗ mice have an increase in stem cell number, we assayed the frequency of SLAM cells.24,25 Consistent with their progenitor phenotype, SLAM cells in CAST/EiJ mice express the c-kit antigen and are enriched by depleting committed lineages (Figure S1).24 We also found that the frequency of SLAM cells increased in aging CAST/EiJ mice, similar to what has been described for other strains (Figure 1H).23 To examine whether young wt∗ mice develop an age-related phenotype in the hematopoietic stem cell compartment, we quantitated the frequency of CD150+CD48−c-kit+ cells and found that they had a nearly 2-fold increase in comparison to age-matched wild-types (Figure 1H). Therefore, although hematopoiesis is impaired in wt∗ mice, the pool of hematopoietic stem cells increases, suggesting that these cells may have defects in proliferative expansion.
wt∗ Mice Develop Lymphocyte Defects that Mirror Immunosenescence
Lymphocyte function declines with age in humans, and cells rely on self-renewal and proliferative potential to maintain antigen specificity throughout adult life. To test whether short telomeres can mediate immunosenescence, we examined lymphocyte compartments. wt∗ mice had a profound peripheral B cell lymphopenia that led to a significant decrease in IgM production (Figures 2A and 2B). When challenged with immunization, wt∗ mice failed to mount a robust immune response after antigen exposure (Figure 2C). mTR+/− mice with short telomeres also developed similar defects, consistent with observations in dyskeratosis congenita patients who have defects in antibody production and humoral immunity (Figures 2A–2C).26 T cell function also declines with age, and CD4 lymphopenia in the presence of an intact CD8 count is characteristic of immunosenescence.27 We therefore examined T cell numbers in wt∗ mice and found that short telomeres led to a general T cell lymphopenia (Figure 2D). Specifically, wt∗ mice developed an asymmetric lymphopenia with a more severe deficit in CD4 cells in comparison to CD8 cells (Figure 2E). mTR+/− mice with short telomeres developed similar patterns of preferential CD4 lymphopenia. This is noteworthy given the predilection of dyskeratosis congenita patients to an opportunistic infection spectrum similar to that of patients with the acquired immunodeficiency syndrome (e.g., Pneumocystis carinii).28,29 Therefore, short telomeres in wt∗ mice are sufficient to cause features of the combined immunodeficiency associated with aging. These findings are particularly significant given the association of short telomeres with an increased mortality due to infection in elderly individuals,30 and they suggest that short telomeres can directly mediate an immunodeficient state.
Figure 2.
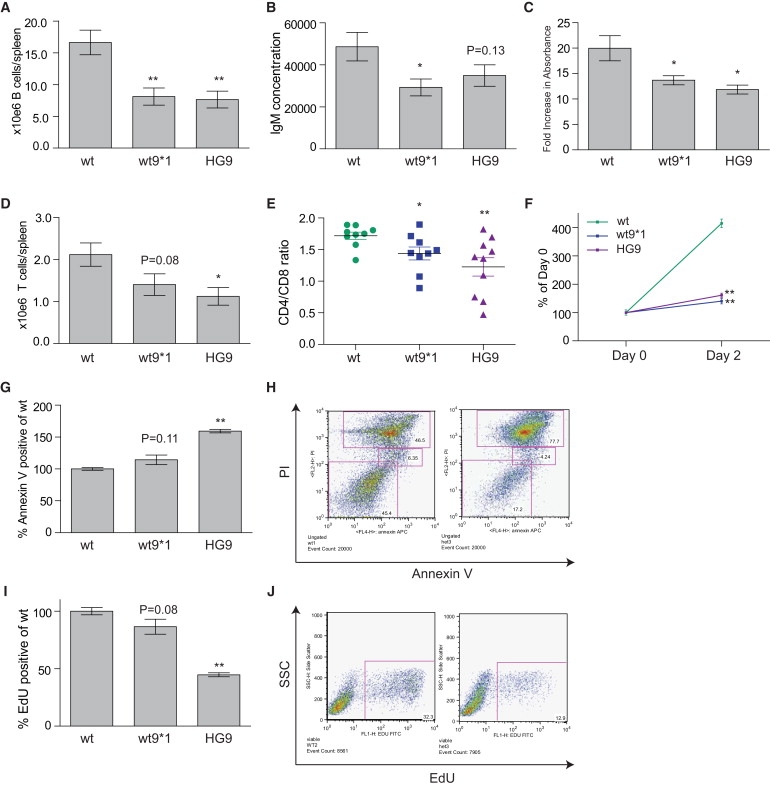
wt∗ Mice Have Lymphocyte Defects that Mirror Immunosenescence
(A and B) Absolute B cell lymphopenia in the spleen (A) is associated with low serum IgM levels (B).
(C) Fold increase over baseline in antigen-specific IgM 1 wk after immunization. Baseline IgM levels were measured in each mouse 3 days prior to injection.
(D and E) Absolute T cell lymphopenia in splenocytes is skewed and favors loss of CD4 cells (D), as shown by the decrease in CD4/CD8 ratio (E).
(F–J) T cells from wt∗ and mTR+/− mice with short telomeres fail to expand after CD3-CD28 stimulation, as quantitated by the MTT assay (F). This failure to expand is associated with an increase in the proportion of Annexin-V-positive apoptotic cells (G), as well as a decrease in the proportion of live cells in S-phase (I). Representative flow cytometry plots for the quantitation shown in (G) and (I) are show in (H) and (J), respectively. Mice were examined at 3–6 mo of age.
wt∗ Mice Have Proliferative Defects in T Lymphocytes
To determine whether short telomeres can cause qualitative T cell defects when telomerase is wild-type, we isolated and stimulated T cells. T cell activation and proliferation are known to be associated with a burst in telomerase activity that sustains clonal expansion after antigen stimulation.11,31,32 We therefore reasoned that wild-type telomerase may sustain the proliferative capacity of T cells even when telomeres are short. When we stimulated wt∗ T cells in vitro, they failed to expand in comparison to cells from true wild-types, and in fact responded similarly to those of mTR+/− mice (Figure 2F). To examine the underlying mechanism, we assayed for apoptosis, and we found that although there were no baseline differences (not shown), wt∗ T cells had an increase in the apoptotic fraction after stimulation in comparison to wild-type cells (Figures 2G and 2H). Because short telomeres also impair cell-cycle progression,33 we examined cell-cycle profiles. We found that wt∗ T cells had fewer cells in S-phase in comparison to wild-type cells (Figures 2I and 2J). This was associated with an accumulation at G2-M, a checkpoint that is well characterized in response to telomere shortening.33–35 These defects were all more prominent in mTR+/− mice. Thus, when telomeres are short, T cell proliferation cannot be sustained and short telomeres cause qualitative defects by inducing both apoptosis and impaired cell-cycle progression, even when telomerase is wild-type.
Clinical Consequences of Bone Marrow Failure in Mice with Short Telomeres
To examine the clinical consequences of short telomeres, we examined survival. mTR−/− mice showed progressive worsening of a survival defect with each generation, similar to what was previously shown (Figure 3A).15 In fact, the generation number, a surrogate for telomere length, could precisely predict the median survival of mTR−/− mice (R2 = 0.95, p < 0.0001) (Figure 3B). Late generation mTR+/−mice also had an increased incidence of premature death. In these mice, similar to mTR−/− mice, the survival defect displayed genetic anticipation (Figure 3C). Consistent with a pattern of anticipation, the Mendelian ratio of mTR−/− pups from late-generation mTR+/− parents decreased successively. By the tenth generation of heterozygous breeding, only 9% of pups had the mTR−/− genotype, compared with an expected 25% (p = 0.004, chi-square test). We also examined whether wt∗ mice had a survival defect and were able to document several premature deaths in the first 6 mo of life. However, these trends have not reached statistical significance and need to be verified in ongoing long-term prospective studies. Together our data indicate that, at least when mice are null or haploinsufficient for telomerase, short telomeres can predict the onset of a fatal illness.
Figure 3.
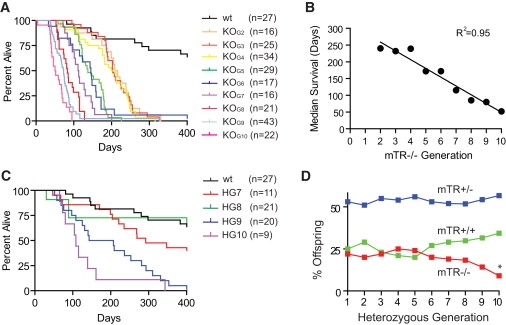
The Short Telotype Predicts Survival
(A) Kaplan-Meier survival plot of mTR−/− mice that died prematurely. Median survival for wild-type, KOG2, and KOG10 mice was 476, 212, and 51 days, respectively (p < 0.0001 for both compared to wild-type; log-rank test).
(B) Median survival of mTR−/− mice is predicted by the generation number, a surrogate for telomere length with R2 as shown (p < 0.0001, Pearson's test).
(C) Survival plot of heterozygous mice that died prematurely. Median survival was 464, 292, 175, and 108 days for HG7–HG10 mice, respectively (p = 0.6, 0.006, and 0.003 for HG8, HG9, and HG10 compared with HG7, respectively; log-rank test).
(D) Mendelian ratio of mTR−/− pups from mTR+/− parents in successive generations (total n = 3091 pups examined). By the tenth generation of breeding, 9% of pups had the mTR−/− genotype, in comparison to an expected 25% (p = 0.004; chi-square test).
Short Telomeres Do Not Promote Tumorigenesis on the CAST/EiJ Background
Short telomeres have been implicated in genomic instability and in promoting tumorigenesis.12,36 To examine whether mice with short telomeres on the CAST/EiJ background have an increased incidence of tumors, we performed careful necropsies. As previously shown,15 mTR−/− mice had microadenomas in the lower gastrointestinal tract. However, these small lesions were localized to the mucosa and did not invade the basement membrane. Microadenomas were notably absent in wt∗ and mTR+/− mice up to the tenth generation. Furthermore, we did not detect any evidence of gross or microscopic malignancy in early- or late-generation mTR−/−, mTR+/−, and wt∗ mice that we examined (50 mice per group). To determine whether short telomeres cause genomic instability, we examined metaphases prepared from primary cells from CAST/EiJ mice with short telomeres. We also did not detect spontaneous chromosome fusions in any of the three groups of mice (n = 20 mice per group, 20 metaphases per mouse). Together, these data indicate that in the presence of an intact DNA damage response, and in the CAST/EiJ strain that possesses a human telomere length and distribution, the most prominent clinical consequences of short telomeres manifest as degenerative disease.
Bone Marrow Failure as the Primary Cause of Death
Humans with short telomeres have an 8-fold increased risk of mortality due to infection, and wt∗ mice share with mTR+/− and mTR−/− mice all of the hematopoietic and immune defects, albeit less severely. To examine the cause of death in these mice, we performed careful histopathology. Two features consistently distinguished mTR−/− and mTR+/− mice from wild-type mice. Mice with short telomeres had evidence of severe extramedulllary hematopoiesis (0 of 9 wild-type, 5 of 5 mTR−/−, and 4 of 5 mTR+/− mice; p = 0.01 and p = 0.02, respectively, as compared to wild-type; chi-square test) (Figure 4A). Extramedullary hematopoiesis is a compensatory response to bone marrow failure. Additionally, mTR−/− and mTR+/− mice had evidence of necrotizing typhlocolitis, an infection that is usually superimposed on areas of mucosal atrophy (Figure 4B). Typhlocolitis is a superinfection of the gastrointestinal tract that leads to microperforartion and death from bacteremia.37 Typhlocolitis has been best described in the setting of bone marrow suppression and mucosal injury after chemotherapy, as well as in aplastic anemia.37 mTR−/− and mTR+/− mice have known mucosal atrophy,15 and wt∗ mice also had mucosal blunting and villous shortening (0 of 9 wild-types, 3 of 6 wt9∗1; p = 0.02; chi-square test) (Figure 4C). Wt∗ mice also had evidence of enteritis and extramedullary hematopoiesis, though these defects were less severe than those in null mice. Our analyses suggest that the primary cause of death in mice with short telomeres is opportunistic infection due to typhlocolitis, related to impaired bone marrow function and superimposed on mucosal defects in the gastrointestinal tract.
Figure 4.
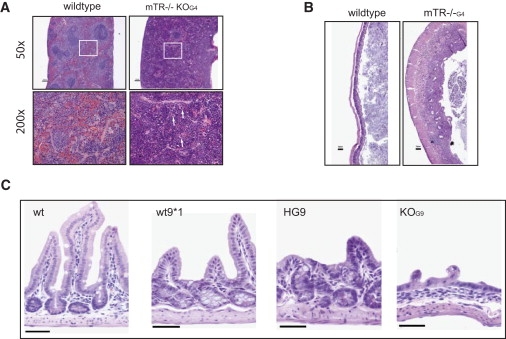
Histopathology Identifies Mucosal Defects and Bone Marrow Failure as the Cause of Death in Mice with Short Telomeres
(A) Photomicrographs of spleen histology in wild-type and mTR−/− mice showing effacement of the normal white-red pulp architecture and replacement with myelo-erythropoiesis as evidenced by the presence of megakaryocytes (arrows) in the inlet at 200X.
(B) Representative photomicrograph of typhlocolitis lesion seen in mTR−/− mice. Compared to wild-type mice (left panel), the lamina propria and submucosa is infiltrated by neutrophils in the cecum, indicating an ongoing infection (right). There was evidence of colitis in 100% of mTR−/− mice examined (8 of 8).
(C) Micrographs from mid-intestine showing gastrointestinal mucosal defects in wt∗ mice. Compared with wild-type mice, in which no abnormalities were ever seen (0 of 9), wt∗ mice had evidence of villous blunting (3 of 6). In mTR+/− and mTR−/− mice with short telomeres, more-severe defects were noted, including severe villar blunting adjacent to areas of crypt hyperplasia, as well as regions of complete villous atrophy and crypt loss, which were present in all mTR−/− mice examined, as illustrated.
Telomerase Dose Is Limiting in Development
The fact that first-generation wt∗ mice have short telomeres implies that when telomerase is haploinsufficient, telomere-length equilibrium cannot be established after one generation.15 To examine whether wild-type telomere length could be reestablished and to determine the rate of telomere elongation when telomerase is wild-type, we bred wt∗ mice to each other in two independent lines (Figure 5A and Figure S2A). Although telomerase elongated the shortest telomeres, it surprisingly did so only incrementally across each generation (Figure 5B and Figure S2B). In fact, telomere-length analysis showed that it took as many generations for telomeres to return to wild-type length as it took for them to shorten in heterozygous breeding. For example, wt5∗1 telomeres were approaching wild-type lengths after four generations of breeding (Figure 5B). Although the degenerative phenotype became less severe with successive generations of wt∗ breeding, in one line, even two generations later, there was evidence of organ dysfunction that was never seen in wild-type mice (Figures 5C and 5D and Figures S2C and S2D). These data indicate that although wild-type telomere lengths can eventually be reestablished, telomerase levels are tightly regulated in development and lead to incremental elongation across each generation.
Figure 5.
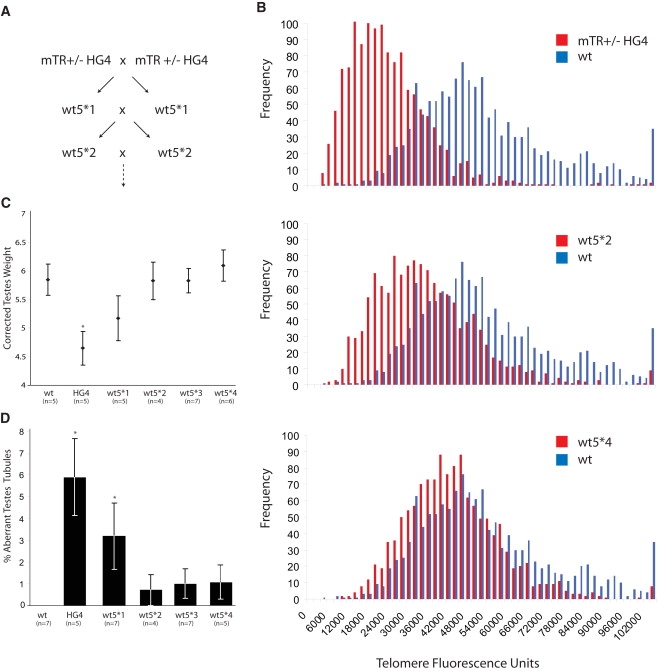
Wild-Type Telomerase Elongates Telomeres Incrementally across Generations; Example of mTR+/− HG4 Family Is Shown
(A) Breeding scheme of wt∗ mice with nomenclature.
(B–D) Frequency distribution of telomere length as examined by qFISH on metaphase splenocytes for each wt∗ generation, compared with true wild-types, are shown in each panel (n = 2 mice per group). Degenerative phenotypes resolve with successive telomere elongation, as shown by an increase in testes weight (C) and the decreasing frequency of aberrant tubules in (D). The number of examined mice is shown below each column, and testes weight was corrected for body weight. Mice were examined at 12 mo of age.
Discussion
The phenotypes that we identify in wt∗ mice provide a model for understanding the role of short telomeres in human aging. In the hematopoietic and immune systems, short telomeres are sufficient to induce the age-associated degenerative changes that resemble those seen in dyskeratosis congenita. The presence of defects in tissues of high turnover indicates that short telomeres impair stem cell and lymphocyte proliferation even when the genes for telomerase are wild-type. Although telomerase activity is enriched in these compartments, this enrichment does not render to them an indefinite functional capacity. The clinical significance of the bone marrow defects that we identify is highlighted by the fact that short telomeres provoke a life-threatening illness in mTR+/− and mTR−/− mice and may also contribute to a survival defect in wt∗ mice. Short telomeres have been associated with premature death due to infection in elderly individuals,30 and our data indicate that short telomeres alone are sufficient to mediate the immunodeficiency of aging and probably its consequent morbidities.
We did not identify an increased incidence of tumors in CAST/EiJ mice with short telomeres. Aside from noninvasive microadenomas which were present in mTR−/− mice, we did not observe any evidence of a neoplastic process in mTR null, heterozygous, or wt∗ mice. Additionally, we did not observe any evidence of genomic instability. Our data are in contrast to prior studies in the C57BL/6 background, which noted an increase in spontaneous tumors in mTR−/− mice.12,36 They indicate that, on the Castaneus genetic background, which possesses human telomere dynamics, and in the presence of an intact DNA damage response, short telomeres primarily mediate degenerative organ failure. Our observations are consistent with several studies in tumor-prone mouse models that have demonstrated that short telomeres are indeed protective and play a powerful tumor-suppressor role.38–43 Also, in dyskeratosis congenita, the primary cause of mortality in more than 90% of cases is organ failure.44 Although dyskeratosis congenita patients do have an increased incidence of cancer,45 the predominant phenotype is degenerative, and cancer-related mortality is less common. Collectively, this evidence suggests that short telomeres have their most prominent clinical consequences in degenerative phenotypes that lead to organ failure. The molecular mechanisms by which short telomeres may, rarely, be associated with an increase in tumor incidence remain to be fully elucidated.
Finally, our data indicate that telomere length is a unique heritable trait. Because telomerase levels are tightly regulated in development, parental telomere length partly determines telomere-length heterogeneity across populations.46 Supporting these observations, in some human families that carry mutant telomerase genes, wild-type progeny also have short telomeres in comparison to those of age-matched controls.47 The fact that telomerase dose is limiting across generations may have particular significance for attempts to use adult somatic cells, which may have short telomeres, in stem-cell-based applications. In this setting, short telomeres could theoretically limit the long-term viability of these cells, even when telomerase dose is restored. Given that the spectrum of telomere-mediated disease is broad and includes idiopathic pulmonary fibrosis as well as cryptogenic forms of liver cirrhosis,48–50 our data implicate the short telotype as sufficient to induce features of dyskeratosis congenita in otherwise wild-type individuals.
Supplemental Data
Supplemental Data include two figures and can be found with this article online at http://www.cell.com/AJHG.
Supplemental Data
Web Resources
The URL for data presented herein is as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nih.gov/Omim
Acknowledgments
We are grateful to Curt Civin, Steve Desiderio, and Katie Whartenby, as well as lab members, for helpful discussions. We thank Ling-Yang Hao for setting up the wt∗ crosses and Brendan Cormack for critical comments on the manuscript. This work was supported by National Institutes of Health grants K08CA118416 (M.A.) and RO1AG27406 (C.W.G.), the Kimmel Foundation (M.A.), and a Maryland Stem Cell Research Fellowship (J.K.A.). E.M.P. received support from a Medical Scientist Training Program grant (T32GM007309).
References
- 1.Harley C.B., Futcher A.B., Greider C.W. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 2.Armanios M. Syndromes of telomere shortening. Annu. Rev. Genomics Hum. Genet. 2009;10:45–61. doi: 10.1146/annurev-genom-082908-150046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heiss N.S., Knight S.W., Vulliamy T.J., Klauck S.M., Wiemann S., Mason P.J., Poustka A., Dokal I. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat. Genet. 1998;19:32–38. doi: 10.1038/ng0598-32. [DOI] [PubMed] [Google Scholar]
- 4.Mitchell J.R., Wood E., Collins K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature. 1999;402:551–555. doi: 10.1038/990141. [DOI] [PubMed] [Google Scholar]
- 5.Armanios M., Chen J.L., Chang Y.P., Brodsky R.A., Hawkins A., Griffin C.A., Eshleman J.R., Cohen A.R., Chakravarti A., Hamosh A. Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc. Natl. Acad. Sci. USA. 2005;102:15960–15964. doi: 10.1073/pnas.0508124102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vulliamy T., Marrone A., Goldman F., Dearlove A., Bessler M., Mason P.J., Dokal I. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature. 2001;413:432–435. doi: 10.1038/35096585. [DOI] [PubMed] [Google Scholar]
- 7.Vulliamy T., Marrone A., Szydlo R., Walne A., Mason P.J., Dokal I. Disease anticipation is associated with progressive telomere shortening in families with dyskeratosis congenita due to mutations in TERC. Nat. Genet. 2004;36:447–449. doi: 10.1038/ng1346. [DOI] [PubMed] [Google Scholar]
- 8.Morrison S.J., Prowse K.R., Ho P., Weissman I.L. Telomerase activity in hematopoietic cells is associated with self-renewal potential. Immunity. 1996;5:207–216. doi: 10.1016/s1074-7613(00)80316-7. [DOI] [PubMed] [Google Scholar]
- 9.Chiu C.P., Dragowska W., Kim N.W., Vaziri H., Yui J., Thomas T.E., Harley C.B., Lansdorp P.M. Differential expression of telomerase activity in hematopoietic progenitors from adult human bone marrow. Stem Cells. 1996;14:239–248. doi: 10.1002/stem.140239. [DOI] [PubMed] [Google Scholar]
- 10.Blasco M.A., Lee H.W., Hande M.P., Samper E., Lansdorp P.M., DePinho R.A., Greider C.W. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 1997;91:25–34. doi: 10.1016/s0092-8674(01)80006-4. [DOI] [PubMed] [Google Scholar]
- 11.Lee H.W., Blasco M.A., Gottlieb G.J., Horner J.W., Greider C.W., DePinho R.A. Essential role of mouse telomerase in highly proliferative organs. Nature. 1998;392:569–574. doi: 10.1038/33345. [DOI] [PubMed] [Google Scholar]
- 12.Rudolph K.L., Chang S., Lee H.W., Blasco M., Gottlieb G.J., Greider C., DePinho R.A. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell. 1999;96:701–712. doi: 10.1016/s0092-8674(00)80580-2. [DOI] [PubMed] [Google Scholar]
- 13.Herrera E., Samper E., Martin-Caballero J., Flores J.M., Lee H.W., Blasco M.A. Disease states associated with telomerase deficiency appear earlier in mice with short telomeres. EMBO J. 1999;18:2950–2960. doi: 10.1093/emboj/18.11.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kipling D., Cooke H.J. Hypervariable ultra-long telomeres in mice. Nature. 1990;347:400–402. doi: 10.1038/347400a0. [DOI] [PubMed] [Google Scholar]
- 15.Hao L.Y., Armanios M., Strong M.A., Karim B., Feldser D.M., Huso D., Greider C.W. Short telomeres, even in the presence of telomerase, limit tissue renewal capacity. Cell. 2005;123:1121–1131. doi: 10.1016/j.cell.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 16.Hemann M.T., Greider C.W. Wild-derived inbred mouse strains have short telomeres. Nucleic Acids Res. 2000;28:4474–4478. doi: 10.1093/nar/28.22.4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hathcock K.S., Hemann M.T., Opperman K.K., Strong M.A., Greider C.W., Hodes R.J. Haploinsufficiency of mTR results in defects in telomere elongation. Proc. Natl. Acad. Sci. USA. 2002;99:3591–3596. doi: 10.1073/pnas.012549799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Makovets S., Williams T.L., Blackburn E.H. The telotype defines the telomere state in Saccharomyces cerevisiae and is inherited as a dominant non-Mendelian characteristic in cells lacking telomerase. Genetics. 2008;178:245–257. doi: 10.1534/genetics.107.083030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morra M., Barrington R.A., Abadia-Molina A.C., Okamoto S., Julien A., Gullo C., Kalsy A., Edwards M.J., Chen G., Spolski R. Defective B cell responses in the absence of SH2D1A. Proc. Natl. Acad. Sci. USA. 2005;102:4819–4823. doi: 10.1073/pnas.0408681102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Segal J.B., Moliterno A.R. Platelet counts differ by sex, ethnicity, and age in the United States. Ann. Epidemiol. 2006;16:123–130. doi: 10.1016/j.annepidem.2005.06.052. [DOI] [PubMed] [Google Scholar]
- 21.Hartsock R.J., Smith E.B., Petty C.S. Normal Variations With Aging Of The Amount Of Hematopoietic Tissue In Bone Marrow From The Anterior Iliac Crest. A Study Made From 177 Cases Of Sudden Death Examined By Necropsy. Am. J. Clin. Pathol. 1965;43:326–331. doi: 10.1093/ajcp/43.4.326. [DOI] [PubMed] [Google Scholar]
- 22.Smith T.J., Khatcheressian J., Lyman G.H., Ozer H., Armitage J.O., Balducci L., Bennett C.L., Cantor S.B., Crawford J., Cross S.J. 2006 update of recommendations for the use of white blood cell growth factors: an evidence-based clinical practice guideline. J. Clin. Oncol. 2006;24:3187–3205. doi: 10.1200/JCO.2006.06.4451. [DOI] [PubMed] [Google Scholar]
- 23.Schlessinger D., Van Zant G. Does functional depletion of stem cells drive aging? Mech. Ageing Dev. 2001;122:1537–1553. doi: 10.1016/s0047-6374(01)00299-8. [DOI] [PubMed] [Google Scholar]
- 24.Kiel M.J., Yilmaz O.H., Iwashita T., Terhorst C., Morrison S.J. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 25.Yilmaz O.H., Kiel M.J., Morrison S.J. SLAM family markers are conserved among hematopoietic stem cells from old and reconstituted mice and markedly increase their purity. Blood. 2006;107:924–930. doi: 10.1182/blood-2005-05-2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Knudson M., Kulkarni S., Ballas Z.K., Bessler M., Goldman F. Association of immune abnormalities with telomere shortening in autosomal-dominant dyskeratosis congenita. Blood. 2005;105:682–688. doi: 10.1182/blood-2004-04-1673. [DOI] [PubMed] [Google Scholar]
- 27.Weng N.P. Aging of the immune system: how much can the adaptive immune system adapt? Immunity. 2006;24:495–499. doi: 10.1016/j.immuni.2006.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rose C., Kern W.V. Another case of Pneumocystis carinii pneumonia in a patient with dyskeratosis congenita (Zinsser-Cole-Engman syndrome) Clin. Infect. Dis. 1992;15:1056–1057. doi: 10.1093/clind/15.6.1056. [DOI] [PubMed] [Google Scholar]
- 29.Lee B.W., Yap H.K., Quah T.C., Chong A., Seah C.C. T cell immunodeficiency in dyskeratosis congenita. Arch. Dis. Child. 1992;67:524–526. doi: 10.1136/adc.67.4.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cawthon R.M., Smith K.R., O'Brien E., Sivatchenko A., Kerber R.A. Association between telomere length in blood and mortality in people aged 60 years or older. Lancet. 2003;361:393–395. doi: 10.1016/S0140-6736(03)12384-7. [DOI] [PubMed] [Google Scholar]
- 31.Weng N.P., Levine B.L., June C.H., Hodes R.J. Regulated expression of telomerase activity in human T lymphocyte development and activation. J. Exp. Med. 1996;183:2471–2479. doi: 10.1084/jem.183.6.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buchkovich K.J., Greider C.W. Telomerase regulation during entry into the cell cycle in normal human T cells. Mol. Biol. Cell. 1996;7:1443–1454. doi: 10.1091/mbc.7.9.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.d'Adda di Fagagna F., Reaper P.M., Clay-Farrace L., Fiegler H., Carr P., Von Zglinicki T., Saretzki G., Carter N.P., Jackson S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 34.Enomoto S., Glowczewski L., Berman J. MEC3, MEC1, and DDC2 are essential components of a telomere checkpoint pathway required for cell cycle arrest during senescence in Saccharomyces cerevisiae. Mol. Biol. Cell. 2002;13:2626–2638. doi: 10.1091/mbc.02-02-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.IJpma A.S., Greider C.W. Short telomeres induce a DNA damage response in Saccharomyces cerevisiae. Mol. Biol. Cell. 2003;14:987–1001. doi: 10.1091/mbc.02-04-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Artandi S.E., Chang S., Lee S.L., Alson S., Gottlieb G.J., Chin L., DePinho R.A. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature. 2000;406:641–645. doi: 10.1038/35020592. [DOI] [PubMed] [Google Scholar]
- 37.Davila M.L. Neutropenic enterocolitis. Curr. Treat. Options Gastroenterol. 2006;9:249–255. doi: 10.1007/s11938-006-0043-2. [DOI] [PubMed] [Google Scholar]
- 38.Greenberg R.A., Chin L., Femino A., Lee K.H., Gottlieb G.J., Singer R.H., Greider C.W., DePinho R.A. Short dysfunctional telomeres impair tumorigenesis in the INK4a(delta2/3) cancer-prone mouse. Cell. 1999;97:515–525. doi: 10.1016/s0092-8674(00)80761-8. [DOI] [PubMed] [Google Scholar]
- 39.Rudolph K.L., Millard M., Bosenberg M.W., DePinho R.A. Telomere dysfunction and evolution of intestinal carcinoma in mice and humans. Nat. Genet. 2001;28:155–159. doi: 10.1038/88871. [DOI] [PubMed] [Google Scholar]
- 40.Wong K.K., Maser R.S., Bachoo R.M., Menon J., Carrasco D.R., Gu Y., Alt F.W., DePinho R.A. Telomere dysfunction and Atm deficiency compromises organ homeostasis and accelerates ageing. Nature. 2003;421:643–648. doi: 10.1038/nature01385. [DOI] [PubMed] [Google Scholar]
- 41.Qi L., Strong M.A., Karim B.O., Armanios M., Huso D.L., Greider C.W. Short telomeres and ataxia-telangiectasia mutated deficiency cooperatively increase telomere dysfunction and suppress tumorigenesis. Cancer Res. 2003;63:8188–8196. [PubMed] [Google Scholar]
- 42.Feldser D.M., Greider C.W. Short telomeres limit tumor progression in vivo by inducing senescence. Cancer Cell. 2007;11:461–469. doi: 10.1016/j.ccr.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo X., Deng Y., Lin Y., Cosme-Blanco W., Chan S., He H., Yuan G., Brown E.J., Chang S. Dysfunctional telomeres activate an ATM-ATR-dependent DNA damage response to suppress tumorigenesis. EMBO J. 2007;26:4709–4719. doi: 10.1038/sj.emboj.7601893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dokal I., Vulliamy T. Dyskeratosis congenita: its link to telomerase and aplastic anaemia. Blood Rev. 2003;17:217–225. doi: 10.1016/s0268-960x(03)00020-1. [DOI] [PubMed] [Google Scholar]
- 45.Alter B.P., Giri N., Savage S.A., Rosenberg P.S. Cancer in dyskeratosis congenita. Blood. 2009;113:6549–6557. doi: 10.1182/blood-2008-12-192880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Njajou O.T., Cawthon R.M., Damcott C.M., Wu S.H., Ott S., Garant M.J., Blackburn E.H., Mitchell B.D., Shuldiner A.R., Hsueh W.C. Telomere length is paternally inherited and is associated with parental lifespan. Proc. Natl. Acad. Sci. USA. 2007;104:12135–12139. doi: 10.1073/pnas.0702703104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goldman F., Bouarich R., Kulkarni S., Freeman S., Du H.Y., Harrington L., Mason P.J., Londono-Vallejo A., Bessler M. The effect of TERC haploinsufficiency on the inheritance of telomere length. Proc. Natl. Acad. Sci. USA. 2005;102:17119–17124. doi: 10.1073/pnas.0505318102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Armanios M.Y., Chen J.J., Cogan J.D., Alder J.K., Ingersoll R.G., Markin C., Lawson W.E., Xie M., Vulto I., Phillips J.A. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2007;356:1317–1326. doi: 10.1056/NEJMoa066157. [DOI] [PubMed] [Google Scholar]
- 49.Tsakiri K.D., Cronkhite J.T., Kuan P.J., Xing C., Raghu G., Weissler J.C., Rosenblatt R.L., Shay J.W., Garcia C.K. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. USA. 2007;104:7552–7557. doi: 10.1073/pnas.0701009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alder J.K., Chen J.J., Lancaster L., Danoff S., Su S.C., Cogan J.D., Vulto I., Xie M., Qi X., Tuder R.M. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. USA. 2008;105:13051–13056. doi: 10.1073/pnas.0804280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.