

Abstract
Spondylo-megaepiphyseal-metaphyseal dysplasia (SMMD) is a rare skeletal dysplasia with only a few cases reported in the literature. Affected individuals have a disproportionate short stature with a short and stiff neck and trunk. The limbs appear relatively long and may show flexion contractures of the distal joints. The most remarkable radiographic features are the delayed and impaired ossification of the vertebral bodies as well as the presence of large epiphyseal ossification centers and wide growth plates in the long tubular bones. Numerous pseudoepiphyses of the short tubular bones in hands and feet are another remarkable feature of the disorder. Genome wide homozygosity mapping followed by a candidate gene approach resulted in the elucidation of the genetic cause in three new consanguineous families with SMMD. Each proband was homozygous for a different inactivating mutation in NKX3-2, a homeobox-containing gene located on chromosome 4p15.33. Striking similarities were found when comparing the vertebral ossification defects in SMMD patients with those observed in the Nkx3-2 null mice. Distinguishing features were the asplenia found in the mutant mice and the radiographic abnormalities in the limbs only observed in SMMD patients. The absence of the latter anomalies in the murine model may be due to the perinatal death of the affected animals. This study illustrates that NKX3-2 plays an important role in endochondral ossification of both the axial and appendicular skeleton in humans. In addition, it defines SMMD as yet another skeletal dysplasia with autosomal-recessive inheritance and a distinct phenotype.
Main Text
The formation of the axial skeleton in vertebrate organisms starts with the generation of paired somites along both sides of the neural tube and the notochord, followed by the compartimentation of these somites into dermatomyotome and sclerotome.1,2 Sclerotomal cells subsequently differentiate into chondroblasts to form vertebral templates. These cartilaginous models further grow and undergo endochondral ossification, resulting in mature vertebral bodies.3 Defects at each step of this developmental process may result in different types of vertebral anomalies. Defects at the initial step of somitogenesis will result in abnormal formation (patterning) of the vertebral bodies, whereas defects later on will result in abnormal growth and ossification of otherwise well-segmented vertebrae.
The study of human skeletal disorders that affect the vertebral column can give us more insights into the genes that regulate axial skeletogenesis. For example, the identification of DLL3 (MIM 602768) mutations in patients with spondylocostal dysostosis type 1 (SCDO1 [MIM 277300]) has highlighted the crucial role of notch signaling in human somitogenesis.4 Individuals affected with SCDO1 have multiple vertebral segmentation defects throughout the entire spine. The important role of type 2 collagen, the major fibrillar constituent of cartilage matrix, in endochondral ossification is exemplified by the lack of vertebral ossification in fetuses with severe achondrogenesis type 2 (ACG2 [MIM 200610]). The latter condition is caused by COL2A1 (MIM 120140) missense mutations that substitute a glycine residue within the triple helical domain of the type 2 procollagen chain.5
This study reports the mapping of the causal gene for a rare bone dysplasia affecting vertebral ossification and delineated for the first time in 1985 by Silverman and Reiley.6 These authors reported eight patients with a condition resembling severe cleidocranial dysplasia (CCD [MIM 119600]) and suggested the term spondylo-megaepiphyseal-metaphyseal dysplasia (SMMD) to distinguish this new entity from CCD. Recurrence in siblings and consanguinity in the parents suggested an autosomal-recessive mode of inheritance. It took almost 20 years before another patient was reported.7 With only nine cases being described up to now, SMMD remains a very rare condition, not listed in OMIM nor incorporated in the 2006 version of the Nosology and Classification of Genetic Skeletal Disorders.8 Nevertheless, the disorder has a distinct clinical and radiographic phenotype. Affected individuals have a short stature with a short trunk and relatively long limbs. The head is often large with a broad face and widely spaced eyes. Mobility in the neck and back is usually limited. With the exception of one boy, all reported patients have a normal intelligence. The most striking features on radiographs are the little or no ossification of the vertebral bodies and the presence of large epiphyseal ossification centers that are separated by wide cartilage zones from the adjacent and irregular metaphyses. The defective ossification in the axial skeleton mostly involves the vertebral bodies with preservation of the pedicles and neural arches that are often unfused posteriorly. Pelvic abnormalities include delayed ossification of the pubic bones, widened triradiate cartilages, and coxa vara with large capital femoral epiphyses. The ribs are usually short and appear to originate remotely from the vertebral column. Multiple pseudoepiphyses in hands and feet are often observed and may suggest a disturbance in the determination of proximal-distal polarity in tubular bones.
We ascertained three previously unreported families with SMMD. The first family comprised two affected siblings, both children of Turkish consanguineous parents. The proband (patient IV.3) was a 20-year-old man who presented at birth with a short trunk and in whom initially the diagnosis of hypochondrogenesis was made because of defective ossification of the vertebral column. At the age of 13 years, he received a laminectomy and a ventriculoperitoneal shunting because of progressive pyramidal signs in the lower limbs caused by cervical spine stenosis and hydrocephalus. His adult height was 130 cm (span 160 cm) and head circumference 53.4 cm (−2.5 SD). Physical examination revealed a short neck, pectus carinatum, severe kyphoscoliosis, and relatively long limbs. Some of his fingers were disproportionally long and showed flexion contractures. Both thumbs were proximally implanted and subluxated (Figures 1A and 1B). He needed a wheelchair for transport over long distances and required breathing support at night. The diagnosis in his sister was made at 18 weeks' gestation on the basis of underossification of the spine on ultrasound. The pregnancy was terminated at 19 weeks.
Figure 1.
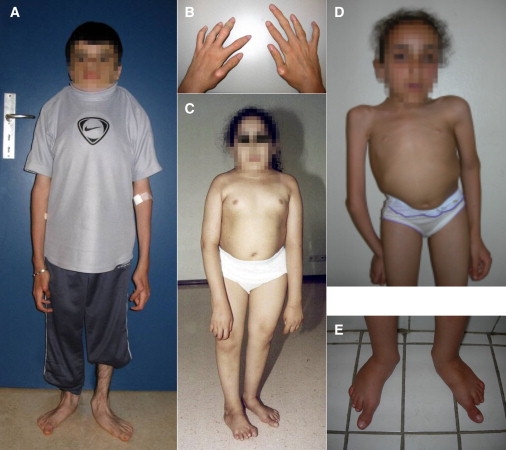
Clinical Features of the Individuals Affected with SMMD
(A and B) Proband IV.3 is wearing a neck brace because of cervical instability. His limbs are relatively long. Note the flat feet with long halluces. Both thumbs are proximally implanted and subluxated at the metacarpophalangeal joint. Complete extension of all fingers is not possible. Note the very long middle finger of the right hand.
(C) Proband IV.8 also has a short trunk and relatively long halluces.
(D and E) The proband of the third family has a kyphoscoliosis with shortening of the trunk. Her feet are remarkable with very long halluces in comparison to the other toes.
The second family comprised two affected siblings born to first-cousin parents of Turkish origin. The proband (patient IV.8) was an 18-year-old girl who was referred because of gait difficulties and back pain. She was unable to walk long distances without assistance. She was operated in childhood for bilateral hip dislocation and right clubfoot. She was short statured (height 129 cm) with a short neck and trunk. She had thoracic kyphoscoliosis and experienced limited mobility in the left shoulder. The small finger joints and elbows were hyperextensible (Figure 1C). Her affected brother died at the age of 3 months.
In the third consanguineous family of Moroccan origin, two female siblings were affected. The oldest girl was born with a low birth weight. She had a delayed motor development but was of normal intelligence. Her height at the age of 11 years was 105 cm (−6.3 SD). Clinical evaluation revealed a short trunk with short neck, scoliosis, and very long halluces (Figures 1D and 1E). The younger sister presented at birth with contractures of fingers and right wrist. Her length at 18 months was 62 cm (−7.5 SD). Radiographs in all affected individuals were characteristic for SMMD (Figure 2).
Figure 2.
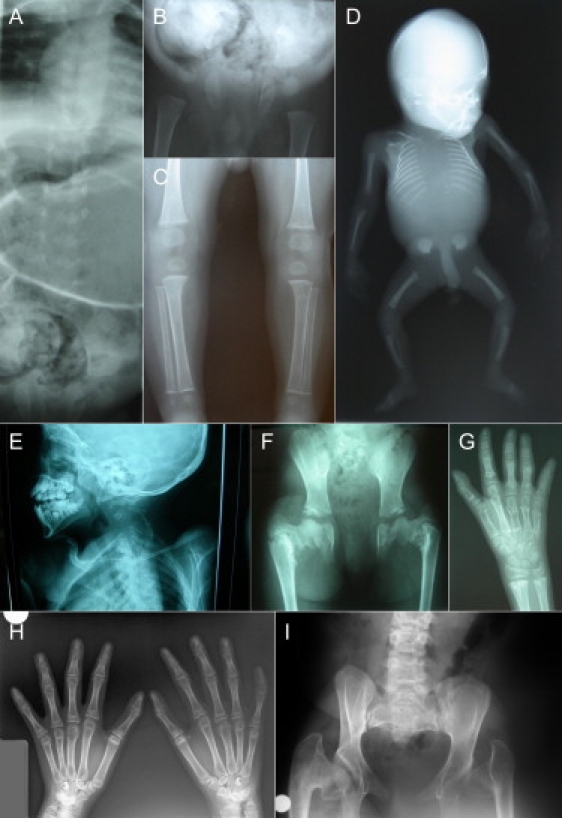
Radiographic Abnormalities in the Individuals Affected with SMMD
(A–C) Radiographs taken in proband IV.3 at the age of 10 months.
(A) The vertebral bodies are incompletely ossified and show sagittal clefts. The pedicles are well ossified in the lower thoracic and upper lumbar region. The ribs originate remotely from the vertebral column.
(B) The pelvis is abnormal with short iliac wings, widened triradiate cartilages, and absent ossification of the pubic bones. Remarkable are the round femoral epiphyses separated by a broad cartilage zone (growth plate) from their metaphyses.
(C) The knee and ankle epiphyses are also rounded and ballooned and located remotely from the metaphyses. The tubular bones are poorly modeled.
(D) Babygram of the affected sib of proband IV.3 taken at 19 weeks' gestational age showing complete absence of vertebral ossification.
(E–G) Radiographs taken in the third proband at the age of 11 years.
(E) Defective ossification of the cervical spine that mostly involves the vertebral bodies with preservation of the pedicles and neural arches.
(F) The iliac wings are tall and narrow. The pubic bones are not yet ossified. Severe coxa vara is present with metaphyseal irregularities at the acetabular roofs and femoral necks that show ossification defects.
(G) Multiple pseudoepiphyses are present, mostly visible in the metacarpals and proximal phalanges. All epiphyses are large for age. Metaphyseal irregularities are also present and in some instances resemble enchondromata, especially those at the distal radius, distal part of second and third metacarpals, and proximal part of the first to fourth proximal phalanges. The second metacarpal is very tall.
(H and I) Radiographs taken in proband IV.8 at the age of 18 years.
(H) Only few hand abnormalities are visible at this age. Pseudoepiphyses are still present in the thumb. Not all growth plates are closed. Note the tall first metacarpal and proximal phalanx and still enlarged distal epiphyses of radius and ulna.
(I) The pelvis shows hypoplastic iliac wings, severe coxa vara with dysplastic femoral heads, and incomplete ossification of the pubic rami.
To unravel the genetic defect of SMMD, we performed a whole-genome homozygosity mapping analysis in families 1 and 2 (family 3 was not available at the start of the study) (Figure 3). Informed consent was obtained for all individuals included in this study. The study was approved by the local ethical committee and is in accordance with institutional and national standards on human research. DNA of both probands was analyzed by means of a high-density SNP screening with GeneChip Human 250K Nsp arrays. We processed all genotypic information in a Microsoft Excel spreadsheet by first sorting genetic markers by chromosomal position followed by identifying all homozygous regions, taking into account a small percentage of SNP miscalls. To eliminate regions with homozygosity by state, we used an arbitrary cutoff of 500 consecutive homozygous SNPs. After this selection, 12 regions with homozygosity by descent were retained in each patient. Assuming the same gene to be implicated in both families, a selection for regions with homozygosity in both patients was made. This resulted in two candidate intervals on chromosome 4 located between 11.2 Mb to 14.5 Mb and between 21.0 Mb to 37.2 Mb, respectively (Figure 4A). Analysis of 13 microsatellite markers across both regions in all available family members suggested linkage to the smaller interval (Figure 3). Because of the low inherent power of the investigated families, no significant LOD score (Z = 1.98 for theta = 0) could be obtained. The high resolution of the performed analysis did however provide sufficient confidence to delineate a final region of interest between SNP rs11722485 and microsatellite marker D4S1602. This region contained three known genes (RAB28 [MIM 612994], NKX3-2 [MIM 602183], and FAM44A), a pseudogene, and some hypothetical genes (Figure 4B).
Figure 3.
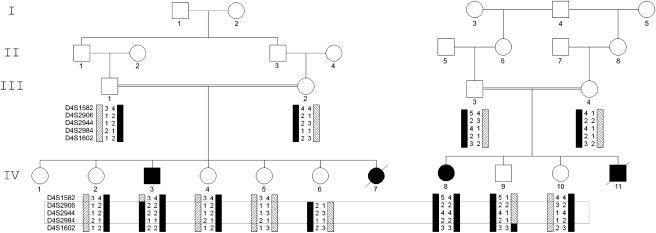
SMMD Pedigrees with Genotypes
Pedigrees of two consanguineous families, each containing one SMMD patient (filled icon) available for analysis. Black bars indicate the haplotype that segregates with SMMD. The candidate region (framed area) is defined by recombinations between markers D4S1582 and D4S2906 in patient IV.3 and between D4S2984 and D4S1602 in individual IV.9.
Figure 4.
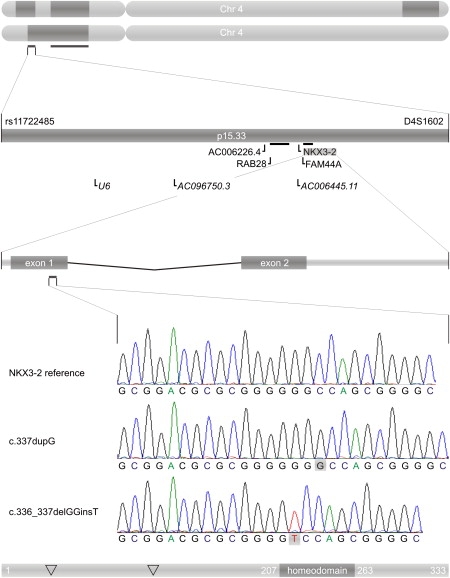
From Genome to Mutation
The top shows an ideogram of chromosome 4 for patients IV.3 (top) and IV.8 (bottom) with homozygous regions in dark gray. Horizontal bars below the chromosomes indicate overlapping homozygous regions. Shown underneath is a zoom in on the linkage interval between SNP rs11722485 and microsatellite D4S1602. Genes, pseudogenes, and hypothetical genes are indicated below the chromosomal interval. NKX3-2 is located at the minus strand of chromosome 4. Below that is a zoom in on the genetic structure of the NKX3-2 gene with two exons. Light-gray bars represent untranslated regions; dark-gray bars indicate coding sequences. Underneath that is the sequence of the NKX3-2 gene for a normal control (top), patient IV.3 (middle) and patient IV.8 (bottom). The bottom shows a NKX3-2 protein with indication of the homeobox domain and the positions (∇) at which SMMD mutations have been found: p.Ala35fs and p.Ala113fs (twice).
NKX3-2 was selected as a good candidate gene because its murine homolog was demonstrated to be essential for proper axial skeletogenesis.9,10 Sequence analysis of the NKX3-2 gene revealed two different homozygous mutations in the two SMMD probands. Both mutations (c.336_337 delGGinsT in patient IV.8 and c.337 dupG in patient IV.3) result in a frameshift with a premature stop codon in the first exon (Figure 4D). Sequence analysis of the NKX3-2 gene in the two affected sisters of family 3 revealed homozygosity for a third inactivating mutation in the first exon (c.104_110 delCGCCCGG, data not shown). These nucleotide changes were not found in a set of 210 control alleles, demonstrating that they are not common in the general population (allele frequency below 1%, 80% power11).
To investigate the effect of NKX3-2 mutations in these patients, we performed an expression assay with real-time quantitative PCR (qPCR). We cultured skin fibroblasts of patient IV.8 and treated half of the cultures with cycloheximide (CHX) to block nonsense -mediated decay. Comparison of NKX3-2 expression in both culture types revealed increased levels of NKX3-2 after CHX treatment (Figure 5A), demonstrating the presence of partial (∼50%) nonsense-mediated decay of the mutant transcripts. This effect of CHX treatment was not observed in the normal skin fibroblasts. In addition, higher levels of NKX3-2 transcripts were observed in the skin fibroblasts of patient IV.8 compared to controls (Figure 5B). The upregulation of mutant NKX3-2 mRNA may indicate a disturbed autoregulatory loop. The truncated NKX3-2 protein that lacks the homeodomain (encoded by exon 2) may impair the regulatory feedback of its own transcriptional activation. Performing gain- and loss-of-function experiments in presomitic mesoderm explants from chicken embryos, Zeng et al. have shown evidence for a positive regulatory loop between Nkx3-2 and Sox9 (MIM 608160), which is important for somitic chondrogenesis.12
Figure 5.
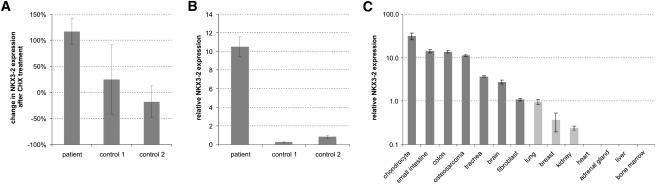
NKX3-2 Expression in Human Tissues
NKX3-2 expression analysis was performed with the qPCR assay with RTprimerDB ID #8115, a combination of expressed ALU repeats (Alu Sq and Alu Sx) (Witt et al., unpublished data) and traditional reference genes (HPRT1 and YWHAZ) for normalization (geNorm30 M stability measure < 0.5) and the qbasePLUS software for data analysis. Error bars represent the propagated standard error on repeated measures
(A) Relative change in NKX3-2 expression after cycloheximide (CHX) treatment. No effect of CHX treatment can be seen in normal skin fibroblasts (+25% and −18%), whereas in fibroblasts of patient IV.8, CHX treatment leads to a doubling in NKX3-2 expression. These results indicate a partial (∼54%) nonsense mediated decay.
(B) Despite the partial NMD, patient fibroblasts show a 10- to 40-fold higher NKX3-2 expression than similar cultures from normal controls.
(C) Cross-tissue comparison of NKX3-2 expression. The highest levels of NKX3-2 expression were found in cartilage, bone (osteosarcoma) and gut (small intestine and colon), whereas moderate expression was observed in trachea and brain. Normal skin fibroblasts only showed low levels of the NKX3-2 gene. NKX3-2 expression was also close to the detection limit for lung, breast, and kidney (one out of four PCR replicates failed to amplify) and was undetectable in adrenal gland, heart, bone marrow, and liver. Details on the samples used for NKX3-2 expression profiling are listed in Table S1 available online.
The NKX3-2 (BAPX1) gene is located on chromosome band 4p15.33 and codes for a homeobox-containing protein of 333 amino acids (Figure 4E).10,13 It belongs to the NK-2 family of homeobox-containing genes, which is the second-most-abundant super class of homeobox genes after the HOX class. This gene family has previously been implicated in other human disorders such as congenital anomalies of the heart (NKX2-6 [MIM 611770] and NKX2-5 [MIM 600584]) (MIM 217095,14187500,15 and 10890016), cancer (NKX2-1 [MIM 600635 and 21198017]), developmental anomalies of the eye (NKX5-3 [MIM 142992 and 61210918]) and in forms of choreoathetosis and hypothyroidism (NKX2-1 and NKX2-5) [MIM 610978,19 118700,20 and 22525021].
In Drosophila, the homolog Bagpipe is required for the specification of the visceral mesoderm during midgut musculature formation.22 In vertebrates, its expression in essentially all cartilaginous condensations that undergo endochondral bone formation suggests an additional role in skeletogenesis. Mouse Nkx3-2 expression is detected initially at embryonic day (E) 8.5 in the splanchnic mesoderm adjacent to the prospective gut endoderm and in the sclerotomal portion of the somites. At E10.5, Nkx3-2 is detected in limb mesenchyme and the first branchial arch that becomes restricted to the precursor of Meckel's cartilage.10 In human embryos at day 52 (mouse equivalent E13.5), NKX3-2 expression is visible in the gut mesoderm, in the sclerotome, and in the cartilaginous templates of developing bones in the limbs. With RNA in situ hybridization, no expression is observed in the nervous system and neurogenic neural crest-derived tissues.10 Because of its expression in the early stage of chondrogenesis, Nkx3-2 is regarded as one of the earliest markers for the sclerotome lineage that forms the axial skeleton. We analyzed the expression of NKX3-2 across different adult cell types (Figure 5C). The highest expression was observed in chondrocytes and gut. In contrast to the previous report, we were able to demonstrate NKX3-2 expression in the nervous system (brain) with the more sensitive and quantitative qPCR assay.
Nkx3-2−/− mice are affected by a perinatally lethal skeletal dysplasia. They are asplenic and have a shorter vertebral column with a normal number of vertebrae that appear more compact. The migration of mesenchymal cells toward the notochord is normal, but the subsequent organization and differentiation of these cells (after E12.5) to form the cartilaginous templates of the prevertebrae is impaired.23,24 The vertebral bodies are not fused in the midline and may lack ossification centers, mainly in the cervical region. The neural arches are less affected and only reduced in size.9,23,24 Despite the expression of Nkx3-2 in all cartilaginous condensations of the developing limb, no defects are observed in the appendicular skeleton at any prenatal stage.
Several studies have indicated essential roles for Shh, Bmps, and the transcription factors Pax1, Pax9, Nkx3-2, Runx2, and Sox9 in the signaling cascade regulating axial chondrogenesis (MIM 600725, 167411, 167416, and 600211). Shh signaling from the notochord and floor plate induces a competence in somitic cells for subsequent BMP2 (MIM 112261) signals to activate chondrogenesis.25,26 This chondrogenic competence is established by the induction of both Sox9 and Nkx3-2 and a positive regulatory loop between these two transcription factors.12 This results in the expression of early cartilage differentiation markers such as aggrecan, collagen type II, and epiphycan. Subsequently, and by influence of Sox9 and Nkx3-2, Runx2 will promote cartilage maturation and endochondral ossification by inducing Ihh and collagen type X expression in (pre)hypertrophic chondrocytes.27 The stimulation of Nkx3-2 expression by Shh is, at least in part, mediated by the transcription factors Pax1 and Pax9.28 Forced expression of Nkx3-2 in presomitic mesoderm is also found to be sufficient to induce chondrogenesis in the presence of BMP signals.12
Studies in chick embryo limb buds have shown that Nkx3-2 is initially expressed in all cartilaginous cells in the limb bud soon after mesenchymal condensation but as chondrocyte maturation begins, expression becomes restricted to the immature proliferative chondrocytes. The Nkx3-2 expression declines in the prehypertrophic cells expressing Ihh and is at trace levels in mature hypertrophic chondrocytes expressing collagen type X. In addition, overexpression of Nkx3-2 inhibits chondrocyte maturation.29 Taken together, these experiments indicate that Nkx3-2 plays an important role in early somitic chondrogenesis and control of chondrocyte maturation during limb development.
Our findings that homozygous inactivating mutations in the NKX3-2 gene result in SMMD are largely in agreement with the phenotype observed in the Nkx3-2 knockout mice. Defective ossification of the anterior parts of the vertebral bodies is present in both the Nkx3-2 null mice and individuals with SMMD. On the other hand, there are also differences between the human SMMD and the murine Nkx3-2−/− phenotypes. Skeletal defects in the appendicular skeleton are found in patients with SMMD but have not been reported in the mutant mice. This discrepancy may be explained by the perinatal death of Nkx3-2−/− mice, before defects in the appendicular skeleton become apparent. In patients with SMMD, the radiographic abnormalities in the limbs (large epiphyses, wide metaphyses) only become visible after birth with the appearance of the secondary ossification centers. Second, Nkx3-2−/− mice have asplenia, a feature that has not been reported in patients with SMMD. Ultrasonographic evaluation of the abdomen in patient IV.3 revealed the presence of a normal spleen.
In summary, our findings define SMMD as a distinct entity within the group of rare skeletal dysplasias. The identification of homozygous inactivating mutations in NKX3-2 supports the autosomal-recessive inheritance of this condition and underscores the crucial role of this homeobox-containing protein in ossification of the human vertebral column. The presence of mega- and pseudoepiphyses with wide growth plates in tubular bones of SMMD patients confirms the more generalized role of NKX3-2 in endochondral ossification of both the axial and appendicular skeletal elements.
Acknowledgments
We are grateful to the patients for their collaboration. This work was supported by a grant (# G.0279.07) from the Research Foundation—Flanders (FWO). J.H. has a postdoctoral mandate at the FWO and G.M. is senior clinical investigator at the FWO. A.D. has a predoctoral grant (BOF) from the Ghent University.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl, http://www.ensembl.org/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
RTprimerDB, http://www.rtprimerdb.org/
Accession Numbers
The Ensembl accession number for the used NKX3-2 reference sequence is ENSG00000109705.
References
- 1.Hirsinger E., Jouve C., Dubrulle J., Pourquie O. Somite formation and patterning. Int. Rev. Cytol. 2000;198:1–65. doi: 10.1016/s0074-7696(00)98002-1. [DOI] [PubMed] [Google Scholar]
- 2.Tam P.P., Trainor P.A. Specification and segmentation of the paraxial mesoderm. Anat. Embryol. (Berl.) 1994;189:275–305. doi: 10.1007/BF00190586. [DOI] [PubMed] [Google Scholar]
- 3.Christ B., Ordahl C.P. Early stages of chick somite development. Anat. Embryol. (Berl.) 1995;191:381–396. doi: 10.1007/BF00304424. [DOI] [PubMed] [Google Scholar]
- 4.Bulman M.P., Kusumi K., Frayling T.M., McKeown C., Garrett C., Lander E.S., Krumlauf R., Hattersley A.T., Ellard S., Turnpenny P.D. Mutations in the human delta homologue, DLL3, cause axial skeletal defects in spondylocostal dysostosis. Nat. Genet. 2000;24:438–441. doi: 10.1038/74307. [DOI] [PubMed] [Google Scholar]
- 5.Mortier G.R., Wilkin D.J., Wilcox W.R., Rimoin D.L., Lachman R.S., Eyre D.R., Cohn D.H. A radiographic, morphologic, biochemical and molecular analysis of a case of achondrogenesis type II resulting from substitution for a glycine residue (Gly691→Arg) in the type II collagen trimer. Hum. Mol. Genet. 1995;4:285–288. doi: 10.1093/hmg/4.2.285. [DOI] [PubMed] [Google Scholar]
- 6.Silverman F.N., Reiley M.A. Spondylo-megaepiphyseal-metaphyseal dysplasia: A new bone dysplasia resembling cleidocranial dysplasia. Radiology. 1985;156:365–371. doi: 10.1148/radiology.156.2.3925497. [DOI] [PubMed] [Google Scholar]
- 7.Agarwal P.P., Srinivasan A., Sharma R., Kabra M., Gupta A.K. Spondylo-megaepiphyseal-metaphyseal dysplasia: An unusual bone dysplasia. Pediatr. Radiol. 2003;33:893–896. doi: 10.1007/s00247-003-1033-3. [DOI] [PubMed] [Google Scholar]
- 8.Superti-Furga A., Unger S. Nosology and classification of genetic skeletal disorders: 2006 revision. Am. J. Med. Genet. A. 2007;143:1–18. doi: 10.1002/ajmg.a.31483. [DOI] [PubMed] [Google Scholar]
- 9.Akazawa H., Komuro I., Sugitani Y., Yazaki Y., Nagai R., Noda T. Targeted disruption of the homeobox transcription factor Bapx1 results in lethal skeletal dysplasia with asplenia and gastroduodenal malformation. Genes Cells. 2000;5:499–513. doi: 10.1046/j.1365-2443.2000.00339.x. [DOI] [PubMed] [Google Scholar]
- 10.Tribioli C., Lufkin T. Molecular cloning, chromosomal mapping and developmental expression of BAPX1, a novel human homeobox-containing gene homologous to Drosophila bagpipe. Gene. 1997;203:225–233. doi: 10.1016/s0378-1119(97)00520-9. [DOI] [PubMed] [Google Scholar]
- 11.Collins J.S., Schwartz C.E. Detecting polymorphisms and mutations in candidate genes. Am. J. Hum. Genet. 2002;71:1251–1252. doi: 10.1086/344344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeng L., Kempf H., Murtaugh L.C., Sato M.E., Lassar A.B. Shh establishes an Nkx3.2/Sox9 autoregulatory loop that is maintained by BMP signals to induce somitic chondrogenesis. Genes Dev. 2002;16:1990–2005. doi: 10.1101/gad.1008002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshiura K.I., Murray J.C. Sequence and chromosomal assignment of human BAPX1, a bagpipe-related gene, to 4p16.1: A candidate gene for skeletal dysplasia. Genomics. 1997;45:425–428. doi: 10.1006/geno.1997.4926. [DOI] [PubMed] [Google Scholar]
- 14.Heathcote K., Braybrook C., Abushaban L., Guy M., Khetyar M.E., Patton M.A., Carter N.D., Scambler P.J., Syrris P. Common arterial trunk associated with a homeodomain mutation of NKX2.6. Hum. Mol. Genet. 2005;14:585–593. doi: 10.1093/hmg/ddi055. [DOI] [PubMed] [Google Scholar]
- 15.Goldmuntz E., Geiger E., Benson D.W. NKX2.5 mutations in patients with tetralogy of fallot. Circulation. 2001;104:2565–2568. doi: 10.1161/hc4601.098427. [DOI] [PubMed] [Google Scholar]
- 16.Schott J.J., Benson D.W., Basson C.T., Pease W., Silberbach G.M., Moak J.P., Maron B.J., Seidman C.E., Seidman J.G. Congenital heart disease caused by mutations in the transcription factor NKX2–5. Science. 1998;281:108–111. doi: 10.1126/science.281.5373.108. [DOI] [PubMed] [Google Scholar]
- 17.Weir B.A., Woo M.S., Getz G., Perner S., Ding L., Beroukhim R., Lin W.M., Province M.A., Kraja A., Johnson L.A. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–898. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schorderet D.F., Nichini O., Boisset G., Polok B., Tiab L., Mayeur H., Raji B., de la Houssaye G., Abitbol M.M., Munier F.L. Mutation in the human homeobox gene NKX5–3 causes an oculo-auricular syndrome. Am. J. Hum. Genet. 2008;82:1178–1184. doi: 10.1016/j.ajhg.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krude H., Schutz B., Biebermann H., von Moers A., Schnabel D., Neitzel H., Tonnies H., Weise D., Lafferty A., Schwarz S. Choreoathetosis, hypothyroidism, and pulmonary alterations due to human NKX2–1 haploinsufficiency. J. Clin. Invest. 2002;109:475–480. doi: 10.1172/JCI14341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Breedveld G.J., van Dongen J.W., Danesino C., Guala A., Percy A.K., Dure L.S., Harper P., Lazarou L.P., van der Linde H., Joosse M. Mutations in TITF-1 are associated with benign hereditary chorea. Hum. Mol. Genet. 2002;11:971–979. doi: 10.1093/hmg/11.8.971. [DOI] [PubMed] [Google Scholar]
- 21.Dentice M., Cordeddu V., Rosica A., Ferrara A.M., Santarpia L., Salvatore D., Chiovato L., Perri A., Moschini L., Fazzini C. Missense mutation in the transcription factor NKX2–5: A novel molecular event in the pathogenesis of thyroid dysgenesis. J. Clin. Endocrinol. Metab. 2006;91:1428–1433. doi: 10.1210/jc.2005-1350. [DOI] [PubMed] [Google Scholar]
- 22.Azpiazu N., Frasch M. tinman and bagpipe: Two homeo box genes that determine cell fates in the dorsal mesoderm of Drosophila. Genes Dev. 1993;7:1325–1340. doi: 10.1101/gad.7.7b.1325. [DOI] [PubMed] [Google Scholar]
- 23.Lettice L.A., Purdie L.A., Carlson G.J., Kilanowski F., Dorin J., Hill R.E. The mouse bagpipe gene controls development of axial skeleton, skull, and spleen. Proc. Natl. Acad. Sci. USA. 1999;96:9695–9700. doi: 10.1073/pnas.96.17.9695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tribioli C., Lufkin T. The murine Bapx1 homeobox gene plays a critical role in embryonic development of the axial skeleton and spleen. Development. 1999;126:5699–5711. doi: 10.1242/dev.126.24.5699. [DOI] [PubMed] [Google Scholar]
- 25.Murtaugh L.C., Chyung J.H., Lassar A.B. Sonic hedgehog promotes somitic chondrogenesis by altering the cellular response to BMP signaling. Genes Dev. 1999;13:225–237. doi: 10.1101/gad.13.2.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murtaugh L.C., Zeng L., Chyung J.H., Lassar A.B. The chick transcriptional repressor Nkx3.2 acts downstream of Shh to promote BMP-dependent axial chondrogenesis. Dev. Cell. 2001;1:411–422. doi: 10.1016/s1534-5807(01)00039-9. [DOI] [PubMed] [Google Scholar]
- 27.Lengner C.J., Hassan M.Q., Serra R.W., Lepper C., van Wijnen A.J., Stein J.L., Lian J.B., Stein G.S. Nkx3.2-mediated repression of Runx2 promotes chondrogenic differentiation. J. Biol. Chem. 2005;280:15872–15879. doi: 10.1074/jbc.M411144200. [DOI] [PubMed] [Google Scholar]
- 28.Rodrigo I., Hill R.E., Balling R., Munsterberg A., Imai K. Pax1 and Pax9 activate Bapx1 to induce chondrogenic differentiation in the sclerotome. Development. 2003;130:473–482. doi: 10.1242/dev.00240. [DOI] [PubMed] [Google Scholar]
- 29.Provot S., Kempf H., Murtaugh L.C., Chung U.I., Kim D.W., Chyung J., Kronenberg H.M., Lassar A.B. Nkx3.2/Bapx1 acts as a negative regulator of chondrocyte maturation. Development. 2006;133:651–662. doi: 10.1242/dev.02258. [DOI] [PubMed] [Google Scholar]
- 30.Vandesompele J., De Preter K., Pattyn F., Poppe B., Van Roy N., De Paepe A., Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-7-research0034. RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.