

Abstract
Cilia and flagella are evolutionarily conserved structures that play various physiological roles in diverse cell types. Defects in motile cilia result in primary ciliary dyskinesia (PCD), the most prominent ciliopathy, characterized by the association of respiratory symptoms, male infertility, and, in nearly 50% of cases, situs inversus. So far, most identified disease-causing mutations involve genes encoding various ciliary components, such those belonging to the dynein arms that are essential for ciliary motion. Following a candidate-gene approach based on data from a mutant strain of the biflagellated alga Chlamydomonas reinhardtii carrying an ODA7 defect, we identified four families with a PCD phenotype characterized by the absence of both dynein arms and loss-of-function mutations in the human orthologous gene called LRRC50. Functional analyses performed in Chlamydomonas reinhardtii and in another flagellated protist, Trypanosoma brucei, support a key role for LRRC50, a member of the leucine-rich-repeat superfamily, in cytoplasmic preassembly of dynein arms.
Main Text
Cilia are evolutionarily conserved complex structures that protrude from the surface of most eukaryotic cells.1 These organelles can be classified according to the arrangement of their microtubule cytoskeleton core, called an axoneme.2 The axoneme consists of nine outer-doublet microtubules, connected by nexin links, surrounding a central pair of microtubules (i.e., “9+2” pattern) or not (i.e., “9+0” pattern). The 9+0 primary cilia are immotile, except in the embryonic node, where they are involved in left-right asymmetry.3 The 9+2 motile cilia, structurally identical to spermatozoan flagella, are involved in the transport of extracellular fluids, as in the respiratory tract, where they propel mucus and represent, therefore, the first line of airway defenses.2 Motile cilia are powered by outer dynein arms (ODAs) and inner dynein arms (IDAs), multiprotein ATPase complexes that are attached to the peripheral doublets and are essential for ciliary motion.2 In mammals, congenital defects in motile cilia, termed primary ciliary dyskinesia (PCD) (CILD1 [MIM 244400]), result in respiratory disease due to impaired mucociliary clearance.4 PCD is a genetically heterogeneous ciliopathy usually transmitted as an autosomal-recessive trait. Approximately half of all patients with pcd display situs inversus, symptomatic of Kartagener syndrome (CILD1 [MIM 244400]). In addition, most male patients are infertile. This complex phenotype, combining chronic airway infections, situs inversus, and male infertility, is explained by a single event: a defect in one of the numerous components common to the axonemes found in respiratory cilia and embryonic nodal cilia, as well as spermatozoan flagella. To date, although several genes have been implicated in PCD,5–9 most mutations so far identified were found in patients whose cilia lack only ODAs. In addition, the molecular basis of the most common ciliary phenotype, characterized by the absence of both dynein arms,10–12 remains essentially unexplored. This phenotype found in about 30% of the patients with PCD13 has been previously explained in only two families that were shown to carry mutations in KTU (CILD10 [MIM 612517]), a gene encoding a protein required for cytoplasmic preassembly of axonemal dyneins.14
Data obtained in the unicellular biflagellated alga Chlamydomonas reinhardtii (Cr), indicating that the product of the ODA7 gene (GeneID: 5715340) is needed for dynein preassembly and may participate in a structural link between inner- and outer-row dyneins,15 prompted us to explore whether PCD could result from mutations in the orthologous human gene, LRRC50, which encodes a member of the superfamily of leucine-rich-repeat (LRR)-containing proteins (Figure 1A).16 To this end, we first determined the human expression pattern of LRRC50 transcripts by quantitative RT-PCR. This analysis revealed that LRRC50 is expressed mainly in adult trachea and testis, a pattern typical of PCD-associated genes (Figure S1, available online).
Figure 1.

LRRC50 Molecular Defects in Patients with PCD
(A) Exonic organization of the human LRRC50 cDNA (top) and domain-organization model of the corresponding protein (bottom), on which are shown the mutations identified in the four families described in this study. The 12 exons are indicated by empty or hashed boxes, depicting translated or untranslated sequences, respectively. “CC,” coiled-coil domain; “Pro-rich,” proline-rich domain.
(B) Mutations identified in families D115, D33, D11, and D42. The expected consequences of these sequence abnormalities (circled), which are detailed in the text, are shown. The intragenic deletion (del) found in patient D42_II1 (P) involves exons 2 and 3 and flanking intronic sequences, as identified through long-range PCR amplifications (right; C stands for control) followed by direct sequencing (bottom). The evolutionary conservation of the deleted exons is shown (dashed frame).
We therefore screened for LRRC50 mutations in 24 patients (12 males and 12 females, from 23 independent families) with PCD related to an absence of both dynein arms. The diagnosis of PCD was confirmed by analysis of nasal or bronchial biopsies that showed ciliary immotility and, at the ultrastructural level, a total absence of dynein arms in all examined cilia. The PCD phenotype was associated with situs inversus in ten patients, and infertility was noted in five adult patients (two males and three females). With consent, blood samples for genetic studies were obtained from all patients and/or their parents. The PCD genetic study was approved by the local ethics committee (CPP Henri-Mondor, Créteil, France). We identified six distinct heterozygous defects consistent with a loss of function of LRRC50 in three unrelated patients, as well as a homozygous missense mutation in two siblings born to a consanguineous union. The phenotypic features of these patients—all born to healthy parents—are summarized in Table 1.
Table 1.
Phenotypic Features of Patients with Identified LRRC50 Mutations
| Patient | Gender | Situs inversus | Airway Disease | Fertility |
Cilia |
|
|---|---|---|---|---|---|---|
| CBFa(Hz) | TEMbDefects | |||||
| D11_II1c | M | no | bronchitis, sinusitis, otitis, bronchiectasis, lobectomy | not tested | 0 | absence of both dynein arms |
| D11_II2c | M | no | bronchitis, sinusitis, otitis, bronchiectasis, lobectomy | not tested | 0 | absence of both dynein arms |
| D33_II1 | F | yes | bronchitis, sinusitis, bronchiectasis | not tested | 0 | absence of both dynein arms |
| D42_II1 | F | yes | bronchitis, otitis, bronchiectasis | IVFd | 0 | absence of both dynein arms |
| D115_II2 | F | yes | bronchitis, sinusitis, bronchiectasis | sterility | 0 | absence of both dynein arms |
Ciliary beat frequency.
Transmission electron microscopy.
Born to a consanguineous union.
In vitro fertilization.
Patient II2 from family D115 carries compound-heterozygous mutations introducing premature stop codons (Figures 1A and 1B). The p.Tyr264X nonsense mutation in exon 6 (c.792C>A) is predicted to result in a severely truncated protein lacking almost two thirds of the protein, including the last LRR domain as well as the coiled-coil and proline-rich domains (Figure 1A). The second LRRC50 allele carries a 1 bp insertion in exon 4 (c.508dupG); this mutation results in a frameshift and a premature translation stop codon at position 179 (p.Glu170GlyfsX10). The patient from family D33 (Figures 1A and 1B) carries a 1 bp insertion in exon 1 (c.115dupT) and a 23 bp deletion in exon 8 (c.1300_1322del23bp); these two mutations are predicted to introduce reading frameshifts (p.Cys39LeufsX44 and p.Gly434ProfsX4) leading to premature stop codons, one very early, at residue 44, and the other one at residue 437 in the proline-rich domain. Compound-heterozygous defects were also found in patient II1 from family D42 (Figures 1A and 1B): a 4 bp insertion in exon 8 (c.1198_1199insTCGC) leading to a premature stop codon at position 405 (p.Pro400LeufsX6) and a 5376 bp intragenic deletion involving exons 2 and 3 and flanking intronic sequences (c.124+1536_353-2102del5376bp) that would lead to an in-frame deletion of 76 amino acids (p.Glu42_Lys117 del), including a region highly conserved throughout evolution (Figure 1B, bottom).
The p.Leu175Arg missense mutation found in family D11 (Figures 1A and 1B) lies in the third LRR domain of the protein. Such LRR-containing domains occur in thousands of functionally diverse proteins from viruses to eukaryotes.16 LRR domains contain several repeats of an LRR motif of 20–30 amino acids and are believed to be involved in protein-protein interactions through their common curved architecture.16 Each repeat contains a highly conserved segment of 11 or 12 residues (with the consensus sequence LxxLxLxxNxL or LxxLxLxxCxxL, in which “x” can be any amino acid; “L” is Leu, Ile, Val, or Phe; “N” is Asn, Thr, Ser, or Cys; and “C” is Cys or Ser) and a variable segment that has allowed classification into one of multiple subfamilies.17,18 The repeats in LRRC50 orthologs (Figure 2A) most closely resemble those in the SDS22-like subfamily15 defined by the consensus sequence LxxLxLxxNxIxxIxxLxzxLxx,18 in which “z” indicates frequent deletions. The p.Leu175Arg mutation, which replaces an apolar and neutral amino acid by a polar and charged amino acid, involves a residue invariant in LRRC50 orthologs (Figure 2A). We subsequently used Chlamydomonas reinhardtii (Cr) to assess the functional consequences of this missense mutation. To model the structure of CrODA7, residues 31–200 of CrODA7 were fit to residues 1–171 of 1M9L, the Chlamydomonas ODA LC1 light-chain structure, by the Swiss Model server, and an image was generated with Accelrys ViewerLite (Accelerys, San Diego, CA, USA) with the use of POV-Ray V3.6 (Persistence of Vision Raytracer, Williamstown, Australia). This modeling (Figure 2B) confirmed that Leu92 (the equivalent of human Leu175) is one of the leucine residues that contribute to the hydrophobic core of the LRR arcs.16 These data, together with the fact that, in humans, the p.Leu175Arg mutation was identified in the heterozygous state in the two healthy parents (Figure 1B), strongly suggest that this missense mutation results in a complete loss of protein function. To confirm this hypothesis and see specifically whether the p.Leu175Arg mutation disrupts a conserved function, we mutated the equivalent residue in CrODA7 (Leu92) and expressed the modified proteins in oda7-1, a null-mutant strain carrying a 1.2 kb deletion spanning exon 1.15 To this end, a triple-HA coding cassette19 was amplified and cloned into an expression vector based on pGenD-ble,20 and ODA7 cDNA sequences encoding amino acids 1–423 were inserted for the creation of a C-terminal HA tag. For the expression vector, overlapping primers 5′-CGCATATGGTGGAATTCCGGATCCTGGCTAGCC-3′ and 5′-TTGTTAGACGTCCTGGCTAGCCAGGATCC-3′ were used to generate a polylinker containing NdeI, EcoRI, BamHI, NheI, AatII, and MfeI sites, which was digested with NdeI and MfeI and cloned between NdeI and EcoRI sites in place of the ble gene in pGenD-ble. The triple-HA coding region was amplified with primers 5′-AGAACTAGGAATTCCCCCGGGGAG-3′ and 5′-GCCGGCGAGTACTGCTAGCGGCG-3′ and was cloned between EcoRI and NheI sites (underlined) in the expression vector. Coding regions of ODA7, including residues 1–423 (out of 432), were amplified with primers 5′-CCACTGTACCATATGTGTGCTTG-3′ and 5′-CGACCGCTCGCGATATCCC-3′ and were inserted between NdeI and NruI (underlined) in the HA vector, generating a C-terminal, HA-tagged ODA7 protein. Point mutations were generated by in vitro mutagenesis. Plasmids were cotransformed along with ARG gene plasmid pJD67 into an arg7oda7 strain. Transformants were selected by PCR, and those expressing the HA-tagged transgene were identified by whole-cell immunoblotting with the anti-HA rat monoclonal antibody 3F10 (Figure S2). As shown in Figure 2C (panel 1), expression of wild-type (WT)-tagged ODA7-HA fully rescues the oda7-1 strain to WT motility, even though expression levels of the tagged protein remain lower than those of endogenous ODA7 in WT strains (Figure S2). Similar results were obtained after expression of an ODA7 protein carrying the p.Leu92Ala (L92A) mutation, whereas no phenotypic rescue was observed with an ODA7 protein carrying the p.Leu92Arg (L92R) or p.Leu92Asp (L92D) mutation (Figure 2C, panel 1).
Figure 2.

Function of LRRC50, as Inferred from Cross-Species Studies
(A) Evolutionary conservation of the third LRR domain (conserved residues defining the LRR consensus are in bold). Leucine 175 is shown in yellow.
(B) Predicted structure of the LRR region of CrODA7. Side chains are indicated for three rows of leucines that line the center of this beta barrel; Leu92 (the equivalent of human Leu175) is indicated (yellow arrow). Red, alpha-helix; blue, beta-sheet; green, turn; gray, random coil.
(C) CrODA7 is primarily cytoplasmic, and requires Leu92 for its function. C1: Expression of HA-tagged ODA7 proteins in the oda7-1 mutant background is shown by a blot of whole cell protein probed with anti-HA. Tubulin is shown as a loading control. The beat frequency (Hz) of each strain, measured by strobe analysis,33 is shown below each lane. C2: The relative abundance of ODA7 is shown by blots of stoichiometric amounts of whole cells (WC) compared with deflagellated cell bodies (CB) and flagella (FL). More ODA7 is present in cell bodies than in a 50-fold excess of flagella. Other proteins for comparison include a chloroplast protein (cyt f) that localizes, therefore, only to the cytoplasm, an intraflagellar transport protein (IFT46) that is present in both the cytoplasm and the flagella, and an ODA subunit (IC2) that localizes mainly to the flagella.14 Data for blots of cell fractions probed with anti-IFT46, anti-cyt f, and anti-IC2 appeared in a previous publication,14 and an identical blot prepared from the same samples was probed with polyclonal anti-ODA7.15 C3: Rescue of ODA assembly is shown by a blot of flagellar proteins probed with anti-IC2. Dynein assembly is supported by the L92 (WT) and the L92A, but not the L92R-tagged product; very low levels of IC2 are present in flagella of the L92D strain (see text). Inner-row dynein subunit IC140 is shown as a loading control.
(D) TbODA7 localizes to the cell body and is required for ODA formation. Upper panel: left, GFP signal for GFP:TbODA7; right, flagellum labeled with an anti-PFR2 marker antibody (red). Lower panel: left, DAPI staining merged with the phase-contrast picture; right, TbDNAI1, a component of ODAs, is still produced in TbODA7RNAi-induced cells but fails to be correctly incorporated into the flagellar skeleton. T, total; S, soluble fraction; C, cytoskeleton (detergent-resistant fraction). The PFR2 protein (probed with monoclonal antibody L13D6,34 1:50) and BiP35 (probed with rabbit polyclonal antibody, 1:1000) are shown as loading and fractionation controls of the cytoskeleton and soluble fractions, respectively. One representative experiment of four independent experiments is shown.
Although the absence of ODA7 prevents preassembly of axonemal dynein in the cytoplasm,21 missense mutations such as L92R or L92D might act as dominant negatives instead and block dynein function in flagella. Previous studies showed that ODA7 associates with both inner- and outer-row dyneins in flagella15 but did not look at the subcellular distribution of ODA7. Immunoblot analysis of stoichiometrically loaded cell fractions (Figure 2C, panel 2) revealed abundant ODA7 in deflagellated cell bodies, but none was detected in flagella. The ODA7 protein was, however, detectable when a 50-fold excess of the flagellar fraction was loaded. We conclude that over 99% of ODA7 resides in the cytoplasm, rather than in the flagellar compartment (Figure 2C, panel 2). This result is consistent with the hypothesis that the major effect of a lack of ODA7 is the disruption of dynein preassembly in the cytoplasm.21 Immunoblots of flagellar fractions showed that modifying Leu92 to an alanine does not affect the ability of ODA7-HA to support assembly of ODAs, whereas introducing an arginine or an aspartate residue at this position blocks dynein assembly (Figure 2C, panel 3). Very low levels of IC2 were present in flagella of the L92D strain, suggesting that either the amount of dynein assembled was below the threshold needed for the production of measurable beat frequency differences or the dynein that assembled was unable to contribute to force generation. Taken together, these results suggest that replacement of the neutral Leu92 residue with a charged residue disrupts function of ODA7, but a negative charge (aspartate) may be marginally less disruptive than a positive charge (arginine).
Additional studies were performed in another flagellated protist, Trypanosoma brucei (Tb). The Tb ortholog of LRRC50, encoded by gene Tb11.01.5550 (GeneID: 3665162), was identified by reciprocal BLASTP analysis; it shares 36% identity and 53% similarity with the human LRRC50 protein. As shown in Figure 2D, TbODA7, like CrODA7, is located primarily in the cell body. To analyze the phenotype resulting from RNAi-induced silencing of ODA7 in Tb, a specific sequence corresponding to a 677 bp internal fragment of TbODA7 was selected with the RNAit algorithm22 and cloned into the pZJM vector.23 This plasmid contains two tetracycline-inducible T7 promoters facing each other. The forward primer was 5′-GATCAAGCTTCTTCAATGTCGTGGGTTCCT-3′ (HindIII site underlined), and 5′-CTAGCTCGAGTCCCTGTTTTCCTCCATACG-3′ served as reverse primer (XhoI site underlined) representing nucleotides 88–764 of the TbODA7 coding sequence. Upon transformation in 29-13 cells that express the T7 RNA polymerase and the tetracycline repressor,24 this plasmid construct allows tetracycline-inducible expression of complementary single-stranded RNA corresponding to 677 bp of TbODA7. TbODA7 RNAi silencing led to an abnormal motility phenotype resulting in cell sedimentation (Figure S3A), a result consistent with the reduction of ODAs in 53% of axonemes documented by transmission electron microscopy (TEM) (Figures S3B and S3C). The fate of DNAI1, an ODA component,25 was examined upon treatment with 1% Nonidet P40 in PEM for 2 min and separation of a detergent-resistant fraction (containing the cytoskeleton and the flagellum) and the soluble fraction (containing the cytoplasm) by high-speed centrifugation. A mouse anti-TbDNAI1 antibody was raised against a region encoded by nucleotides 142–757 of the TbDNAI1 coding sequence. As shown in Figure 2D, in control cells, the majority of TbDNAI1 pool was found to be resistant to detergent extraction and remained associated with the cytoskeleton. The densitometric analysis of four independent experiments, performed with the Quantity One software (Biorad), revealed that only 9% ± 4% of the total TbDNAI1 was present in the soluble fraction. Knockdown of TbODA7 did not affect the total amount of DNAI1 but led to its striking shift to the detergent-soluble fraction that contained 44% ± 9% of TbDNAI1 (Figure 2D). This difference is statistically significant in an unpaired Student's t test for data with equal variance (p < 10−3). This demonstrates that some dynein-arm components are properly synthesized but remain in the cytoplasm.
Overall, these data show that LRRC50 loss-of-function mutations result in a PCD phenotype characterized by an absence of both dynein arms. Assuming that disruption of both rows of dynein arms is found in about 30% of patients,13 the overall disease contribution of LRRC50 can be estimated to range from 4% to 5%. In this study, three patients had a complete situs inversus, consistent with the Lrrc50 expression in the zebrafish node equivalent (Kupfer's vesicle) and with left-right asymmetry defects in zebrafish LRRC50 mutants.26 Notably, two female patients displayed fertility problems. Although the number of patients may be too small for definite conclusions to be drawn, it is tempting to speculate that LRRC50 mutations may cause predisposition to fertility problems. From a general viewpoint, as for male patients with PCD, infertility is readily explained by a defect common to the axonemes found on respiratory cells and sperm cells. For female patients, the link between PCD and infertility is less obvious, but it is supported by several observations made in female patients with Kartagener syndrome, in which the diagnosis of PCD is unquestionable,27 as is the case for the two female patients with fertility problems and identified LRRC50 mutations. In all cases, cilia from nasal epithelial cells were found to be completely immotile. Strikingly, different phenotypes are found in other species with similar mutations. In Chlamydomonas reinhardtii and Trypanosoma brucei, the absence of ODA7 results in flagellar hypomotility, with a selective lack of ODAs and no detectable IDA abnormality. Given the fact that it is often difficult to visualize IDAs, we also analyzed the patients’ cilia by means of computational TEM.28 As shown in Figure 3, no IDAs could be seen, a result pointing to slight differences in axonemal molecular composition and/or LRRC50 functions among species. Consistent with this data, cilia from zebrafish carrying a homozygous premature-stop codon in Lrrc50 were shown to lack either ODAs or both dynein arms,29 leaving open the possibility that LRRC50 may also be involved in other ultrastructural phenotypes such as isolated ODA or IDA defects. Zebrafish with different Lrrc50 mutations have also been shown to develop pronephric cysts,26,29 whereas none of our patients developed symptoms suggestive of a kidney disease; a similar observation was made in the case of Ktu/KTU mutations,14 a situation that may reflect the different embryologic origin of kidneys in fish and mammals30 and the fact that zebrafish pronephros contains motile cilia, which generate fluid flow,31 whereas the normal human kidney does not.32 As recently shown for KTU,14 LRRC50/ODA7 is a cytoplasmic protein that participates in construction of dynein arms, therefore suggesting that quite sophisticated machinery could be involved in this process. Such proteins represent a novel set of candidates for PCD.
Figure 3.
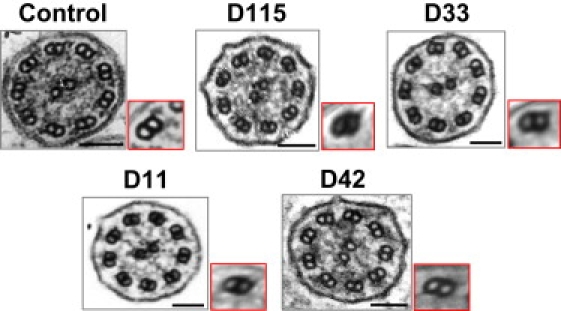
Electron Micrographs of Respiratory Cilia from Patients with LRRC50 Mutations, Showing the Absence of Both Dynein Arms
For each patient, the ten best-defined axonemal sections were selected, and the corresponding micrographs were submitted to computational TEM (composite images framed in red) for the improvement of IDA visualization, as described previously.28 Scale bars represent 0.1 μm.
Acknowledgments
We are grateful to the patients and their family members, whose cooperation made this study possible, and we thank all referring physicians. This work was supported by grants from the Legs Poix from the Chancellerie des Universités, the Assistance Publique-Hôpitaux de Paris (PHRC AOM06053, P060245), and the Agence Nationale pour la Recherche (ANR-05-MRAR-022-01). D.R.M. was supported by NIH-GM44228. Work on T. brucei was funded by Institut Pasteur and CNRS.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl Genome Browser, http://www.ensembl.org/
GenBank, http://www.ncbi.nlm.nih.gov/Genbank/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Swiss Model server, http://swissmodel.expasy.org/
T. brucei GeneDB database, http://www.genedb.org/genedb/tryp/blast.jsp
References
- 1.Haimo L.T., Rosenbaum J.L. Cilia, flagella, and microtubules. J. Cell Biol. 1981;91:125s–130s. doi: 10.1083/jcb.91.3.125s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Satir P., Christensen S.T. Overview of structure and function of mammalian cilia. Annu. Rev. Physiol. 2007;69:377–400. doi: 10.1146/annurev.physiol.69.040705.141236. [DOI] [PubMed] [Google Scholar]
- 3.Basu B., Brueckner M. Cilia multifunctional organelles at the center of vertebrate left-right asymmetry. Curr. Top. Dev. Biol. 2008;85:151–174. doi: 10.1016/S0070-2153(08)00806-5. [DOI] [PubMed] [Google Scholar]
- 4.Afzelius B.A. A human syndrome caused by immotile cilia. Science. 1976;193:317–319. doi: 10.1126/science.1084576. [DOI] [PubMed] [Google Scholar]
- 5.Duriez B., Duquesnoy P., Escudier E., Bridoux A.M., Escalier D., Rayet I., Marcos E., Vojtek A.M., Bercher J.F., Amselem S. A common variant in combination with a nonsense mutation in a member of the thioredoxin family causes primary ciliary dyskinesia. Proc. Natl. Acad. Sci. USA. 2007;104:3336–3341. doi: 10.1073/pnas.0611405104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fliegauf M., Benzing T., Omran H. When cilia go bad: cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 2007;8:880–893. doi: 10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- 7.Loges N.T., Olbrich H., Fenske L., Mussaffi H., Horvath J., Fliegauf M., Kuhl H., Baktai G., Peterffy E., Chodhari R. DNAI2 mutations cause primary ciliary dyskinesia with defects in the outer dynein arm. Am. J. Hum. Genet. 2008;83:547–558. doi: 10.1016/j.ajhg.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sharma N., Berbari N.F., Yoder B.K. Ciliary dysfunction in developmental abnormalities and diseases. Curr. Top. Dev. Biol. 2008;85:371–427. doi: 10.1016/S0070-2153(08)00813-2. [DOI] [PubMed] [Google Scholar]
- 9.Castleman V.H., Romio L., Chodhari R., Hirst R.A., de Castro S.C., Parker K.A., Ybot-Gonzalez P., Emes R.D., Wilson S.W., Wallis C. Mutations in radial spoke head protein genes RSPH9 and RSPH4A cause primary ciliary dyskinesia with central-microtubular-pair abnormalities. Am. J. Hum. Genet. 2009;84:197–209. doi: 10.1016/j.ajhg.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chilvers M.A., Rutman A., O'Callaghan C. Ciliary beat pattern is associated with specific ultrastructural defects in primary ciliary dyskinesia. J. Allergy Clin. Immunol. 2003;112:518–524. doi: 10.1016/S0091-6749(03)01799-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jorissen M., Willems T., Van der Schueren B., Verbeken E., De Boeck K. Ultrastructural expression of primary ciliary dyskinesia after ciliogenesis in culture. Acta Otorhinolaryngol. Belg. 2000;54:343–356. [PubMed] [Google Scholar]
- 12.Noone P.G., Leigh M.W., Sannuti A., Minnix S.L., Carson J.L., Hazucha M., Zariwala M.A., Knowles M.R. Primary ciliary dyskinesia: diagnostic and phenotypic features. Am. J. Respir. Crit. Care Med. 2004;169:459–467. doi: 10.1164/rccm.200303-365OC. [DOI] [PubMed] [Google Scholar]
- 13.Papon J.F., Coste A., Roudot-Thoraval F., Boucherat M., Roger G., Tamalet A., Vojtek A.M., Amselem S., Escudier E. A 20-year experience of electron microscopy in the diagnosis of primary ciliary dyskinesia. Eur. Respir. J. 2009 doi: 10.1183/09031936.00046209. Published online October 19, 2009. [DOI] [PubMed] [Google Scholar]
- 14.Omran H., Kobayashi D., Olbrich H., Tsukahara T., Loges N.T., Hagiwara H., Zhang Q., Leblond G., O'Toole E., Hara C. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature. 2008;456:611–616. doi: 10.1038/nature07471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Freshour J., Yokoyama R., Mitchell D.R. Chlamydomonas flagellar outer row dynein assembly protein ODA7 interacts with both outer row and I1 inner row dyneins. J. Biol. Chem. 2007;282:5404–5412. doi: 10.1074/jbc.M607509200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bella J., Hindle K.L., McEwan P.A., Lovell S.C. The leucine-rich repeat structure. Cell. Mol. Life Sci. 2008;65:2307–2333. doi: 10.1007/s00018-008-8019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobe B., Kajava A.V. The leucine-rich repeat as a protein recognition motif. Curr. Opin. Struct. Biol. 2001;11:725–732. doi: 10.1016/s0959-440x(01)00266-4. [DOI] [PubMed] [Google Scholar]
- 18.Matsushima N., Tachi N., Kuroki Y., Enkhbayar P., Osaki M., Kamiya M., Kretsinger R.H. Structural analysis of leucine-rich-repeat variants in proteins associated with human diseases. Cell. Mol. Life Sci. 2005;62:2771–2791. doi: 10.1007/s00018-005-5187-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silflow C.D., LaVoie M., Tam L.W., Tousey S., Sanders M., Wu W., Borodovsky M., Lefebvre P.A. The Vfl1 Protein in Chlamydomonas localizes in a rotationally asymmetric pattern at the distal ends of the basal bodies. J. Cell Biol. 2001;153:63–74. doi: 10.1083/jcb.153.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer N., Rochaix J.D. The flanking regions of PsaD drive efficient gene expression in the nucleus of the green alga Chlamydomonas reinhardtii. Mol. Genet. Genomics. 2001;265:888–894. doi: 10.1007/s004380100485. [DOI] [PubMed] [Google Scholar]
- 21.Fowkes M.E., Mitchell D.R. The role of preassembled cytoplasmic complexes in assembly of flagellar dynein subunits. Mol. Biol. Cell. 1998;9:2337–2347. doi: 10.1091/mbc.9.9.2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Redmond S., Vadivelu J., Field M.C. RNAit: an automated web-based tool for the selection of RNAi targets in Trypanosoma brucei. Mol. Biochem. Parasitol. 2003;128:115–118. doi: 10.1016/s0166-6851(03)00045-8. [DOI] [PubMed] [Google Scholar]
- 23.Wang Z., Morris J.C., Drew M.E., Englund P.T. Inhibition of Trypanosoma brucei gene expression by RNA interference using an integratable vector with opposing T7 promoters. J. Biol. Chem. 2000;275:40174–40179. doi: 10.1074/jbc.M008405200. [DOI] [PubMed] [Google Scholar]
- 24.Wirtz E., Leal S., Ochatt C., Cross G.A. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 1999;99:89–101. doi: 10.1016/s0166-6851(99)00002-x. [DOI] [PubMed] [Google Scholar]
- 25.Branche C., Kohl L., Toutirais G., Buisson J., Cosson J., Bastin P. Conserved and specific functions of axoneme components in trypanosome motility. J. Cell Sci. 2006;119:3443–3455. doi: 10.1242/jcs.03078. [DOI] [PubMed] [Google Scholar]
- 26.van Rooijen E., Giles R.H., Voest E.E., van Rooijen C., Schulte-Merker S., van Eeden F.J. LRRC50, a conserved ciliary protein implicated in polycystic kidney disease. J. Am. Soc. Nephrol. 2008;19:1128–1138. doi: 10.1681/ASN.2007080917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin T.K., Lee R.K., Su J.T., Liu W.Y., Lin M.H., Hwu Y.M. A successful pregnancy with in vitro fertilization and embryo transfer in an infertile woman with Kartagener's syndrome: a case report. J. Assist. Reprod. Genet. 1998;15:625–627. doi: 10.1023/A:1020341629516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Escudier E., Couprie M., Duriez B., Roudot-Thoraval F., Millepied M.C., Pruliere-Escabasse V., Labatte L., Coste A. Computer-assisted analysis helps detect inner dynein arm abnormalities. Am. J. Respir. Crit. Care Med. 2002;166:1257–1262. doi: 10.1164/rccm.2111070. [DOI] [PubMed] [Google Scholar]
- 29.Sullivan-Brown J., Schottenfeld J., Okabe N., Hostetter C.L., Serluca F.C., Thiberge S.Y., Burdine R.D. Zebrafish mutations affecting cilia motility share similar cystic phenotypes and suggest a mechanism of cyst formation that differs from pkd2 morphants. Dev. Biol. 2008;314:261–275. doi: 10.1016/j.ydbio.2007.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drummond I.A., Majumdar A., Hentschel H., Elger M., Solnica-Krezel L., Schier A.F., Neuhauss S.C., Stemple D.L., Zwartkruis F., Rangini Z. Early development of the zebrafish pronephros and analysis of mutations affecting pronephric function. Development. 1998;125:4655–4667. doi: 10.1242/dev.125.23.4655. [DOI] [PubMed] [Google Scholar]
- 31.Kramer-Zucker A.G., Olale F., Haycraft C.J., Yoder B.K., Schier A.F., Drummond I.A. Cilia-driven fluid flow in the zebrafish pronephros, brain and Kupffer's vesicle is required for normal organogenesis. Development. 2005;132:1907–1921. doi: 10.1242/dev.01772. [DOI] [PubMed] [Google Scholar]
- 32.Ong A.C., Wagner B. Detection of proximal tubular motile cilia in a patient with renal sarcoidosis associated with hypercalcemia. Am. J. Kidney Dis. 2005;45:1096–1099. doi: 10.1053/j.ajkd.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 33.Mitchell D.R., Kang Y. Identification of oda6 as a Chlamydomonas dynein mutant by rescue with the wild-type gene. J. Cell Biol. 1991;113:835–842. doi: 10.1083/jcb.113.4.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kohl L., Sherwin T., Gull K. Assembly of the paraflagellar rod and the flagellum attachment zone complex during the Trypanosoma brucei cell cycle. J. Eukaryot. Microbiol. 1999;46:105–109. doi: 10.1111/j.1550-7408.1999.tb04592.x. [DOI] [PubMed] [Google Scholar]
- 35.Bangs J.D., Uyetake L., Brickman M.J., Balber A.E., Boothroyd J.C. Molecular cloning and cellular localization of a BiP homologue in Trypanosoma brucei. Divergent ER retention signals in a lower eukaryote. J. Cell Sci. 1993;105:1101–1113. doi: 10.1242/jcs.105.4.1101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.