

Abstract
Mental retardation/intellectual disability is a devastating neurodevelopmental disorder with serious impact on affected individuals and their families, as well as on health and social services. It occurs with a prevalence of ∼2%, is an etiologically heterogeneous condition, and is frequently the result of genetic aberrations. Autosomal-recessive forms of nonsyndromic MR (NS-ARMR) are believed to be common, yet only five genes have been identified. We have used homozygosity mapping to search for the gene responsible for NS-ARMR in a large Pakistani pedigree. Using Affymetrix 5.0 single nucleotide polymorphism (SNP) microarrays, we identified a 3.2 Mb region on 8q24 with a continuous run of 606 homozygous SNPs shared among all affected members of the family. Additional genotype data from microsatellite markers verified this, allowing us to calculate a two-point LOD score of 5.18. Within this region, we identified a truncating homozygous mutation, R475X, in exon 7 of the gene TRAPPC9. In a second large NS-ARMR/ID family, previously linked to 8q24 in a study of Iranian families, we identified a 4 bp deletion within exon 14 of TRAPPC9, also segregating with the phenotype and truncating the protein. This gene encodes NIK- and IKK-β-binding protein (NIBP), which is involved in the NF-κB signaling pathway and directly interacts with IKK-β and MAP3K14. Brain magnetic resonance imaging of affected individuals indicates the presence of mild cerebral white matter hypoplasia. Microcephaly is present in some but not all affected individuals. Thus, to our knowledge, this is the sixth gene for NS-ARMR to be discovered.
Main Text
Mental retardation/intellectual disability (MR/ID) is a neurodevelopmental disorder characterized by low intelligence quotient (IQ) and deficits in adaptive behaviors that interfere with everyday life and is estimated to affect 1%–3% of the population. A diagnosis of mental retardation requires an IQ of 70 or below and at least two deficits in adaptive behaviors, such as delayed language, social skills, or self-help skills.1 MR can present as the sole clinical feature (nonsyndromic) or can be present with additional clinical or dysmorphological features (syndromic). Due to the high male-to-female ratio, X-linked MR has been extensively studied, and over 80 causal genes have been cloned.2,3 However, it is likely that autosomal forms of MR are more common than X-linked MR, as only ∼4% of genes reside on the X chromosome. Despite this, little is known about the genetic basis of nonsyndromic autosomal-recessive mental retardation (NS-ARMR).
To date, only five genes have been identified for NS-ARMR. These include PRSS12 (MIM 606709), CRBN (MIM 609262), CC2D1A (MIM 610055), GRIK2 (MIM 138244), and TUSC3 (MIM 601385). These genes have a variety of functions and participate in multiple biochemical pathways. In addition, there are several known disease loci for NS-ARMR for which genes have not yet been identified.4
In the present study, a family from the Abbottabad area in the North-West Frontier Province of Pakistan was recruited through COMSATS Institute of Information Technology (Islamabad, Pakistan) after informed written consent was given (for affected individuals and children 16 or under, parental consent was given). Institutional research ethics board consent was given for the study through COMSATS and through the Centre for Addiction and Mental Health (Toronto). This family has seven living and one deceased affected members, across three generations and three branches, where almost all marriages are between first cousins (Figure 1). Six of the affected individuals are female, and one is male. In addition, an affected female from the grandparental generation (III:4) is now deceased. Informed written consent was obtained for each participating family member.
Figure 1.
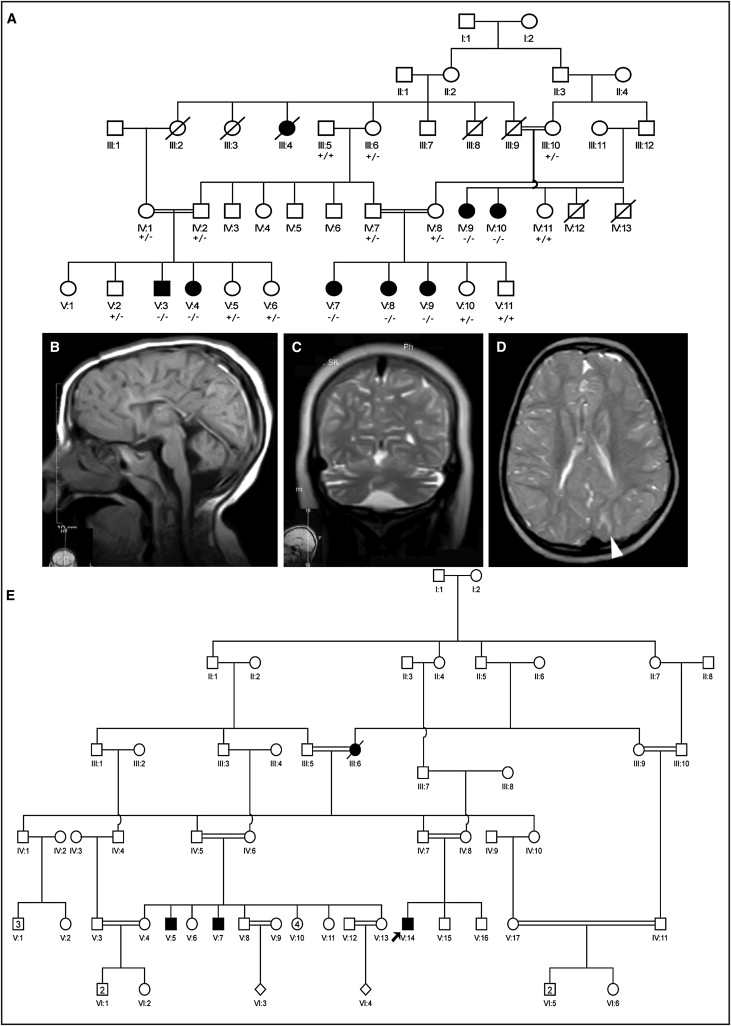
Pedigrees of Families and MRI Images
(A) Pedigree and MRI images for family MR-2019 are shown in (A–E). Pedigree for MR-2019 (A): affected individuals are connected by loops of consanguinity over five generations.
(B) MRI image from female affected V:9: sagittal T1-weighted image showing thin corpus callosum and a small cerebellar vermis with prominent folia. The brain stem is also diffusely thin.
(C) MRI image from female affected V:8: coronal T2-weighted image showing enlarged CSF spaces, suggesting cortical volume loss.
(D) MRI images from female affected V:9: axial T2-weighted image showing areas of T2 hyperintensity in subcortical white matter (arrowhead), enlarged sulci, and reduced white matter volume compatible with mildly diminished cerebral cortical and white matter volume.
(E) Pedigree for M001: affected individuals are connected by loops of consanguinity over five generations.
The assessment of level of MR in the Pakistani family was performed on three affected members (V:3, V:8, and V:9) (Figure 1) by a team of three experienced clinicians who come from the same culture and speak the local language of the family. The 3-year-old girl (V:9) did not walk independently and spoke fewer than 20 meaningful words. She could not feed herself and had not achieved continence during the day or night. No seizures were reported. The 6-year-old female (V:8) started walking at 4 years of age. She did not use full sentences. She was overweight but not obese (weight, 38 Kg; height, 116 cm; BMI, 28.2) and is reported to have clonic/tonic seizures, for which neuroleptic treatment has started. The 9-year-old male (V:3) started walking at 5 years of age. He was able to ask for food, but was unable to feed himself. He was able to convey the need to go to the toilet, but needed help with washing and cleaning. Mild kyphosis was observed, but no seizures were reported.
Although fully standardized IQ testing was not possible due to cultural and linguistic barriers, all affected individuals were assessed using the Vineland Adaptive Behavior Scales, Second Edition5 and Portage Guide to Early Intervention6 as the framework for the assessment. The information gathered was discussed within the team of clinicians, and consensus diagnoses of moderate to severe MR/ID were reached based on DSM IV criteria.1 Developmental trajectories did not differ between the affected individuals, and the degree of disability was similar across all the domains of functioning. No clinical features to indicate the presence of Autism Spectrum Disorder or mental illness (other than MR/ID) were present. They typically started to walk at 5 years of age. In spite of the availability of the opportunity, none of the affected individuals had acquired the ability to read and write.
Photographs of the affected individuals were examined for dysmorphic features by an experienced clinical geneticist (R.W.). Neurological assessment was performed by a consultant neurologist (M. Azam) trained in the UK and currently working in Islamabad. He performed a thorough physical and neurological clinical examination of patients V:3, V:8, and V:9, including structural magnetic resonance imaging (MRI), with multiplanar imaging done through T1/T2WI sagittal, axial, and coronal sequences. Blood biochemistry was tested at the Islamabad Diagnostic Centre, including testing for liver function, renal function, electrolytes, and hematology.
Assessment for dysmorphological features proved negative, save that measurement of occipitofrontal head circumference (OFC) was low, at around the fifth centile, for two affected girls, but borderline microcephaly for three others (2–3 SDs below the mean) and normal for one female (V:7) (Table 1). The height and weight were within normal range for the local population. Neurological examination was normal in all affected individuals. The size of the spleen and liver were also normal, and there were no cardiac or respiratory abnormalities. Blood hematology and biochemistry markers were within the normal range, except creatine phospokinase levels were elevated (V:3, 218 U/l; V:8, 227 U/l; V:9, 402 U/l).
Table 1.
Occipitofrontal Head Circumference Measurements for Affected Members from Family MR-2019
| Pedigree ID | Status | Sex | Age | OFC (cm) | OFC SD. |
|---|---|---|---|---|---|
| V:3 | Affected | M | 9 | 49 | −2.5 |
| V:4 | Affected | F | 11 | 50.5 | 5th centile |
| V:7 | Affected | F | 14 | 54 | normal |
| V:8 | Affected | F | 6 | 49 | 5th centile |
| V:9 | Affected | F | 3 | 46.5 | −2 |
| lV:9 | Affected | F | 34 | not available | not available |
| lV:10 | Affected | F | 30 | 52 | −2 |
MRI analysis on three affected individuals (V:3, V:8, and V:9) (Figure 1) showed diminished cerebral white matter volume, with sulcal enlargement, thinning of the corpus callosum, and mildly reduced cerebellar volume (Figures 1B–1D). In addition, several areas of T2 hyperintensity were present in the subcortical white matter (Figure 1D). The overall interpretation of the MRI scans was diminished volume of the supra- and infratentorial cortex and underlying white matter. Although this likely represents cortical atrophy with secondary degeneration of white matter, serial imaging showing progressive volume loss would be required to prove atrophy.
DNA from seven affected family members and one unaffected family member was analyzed using Affymetrix Genome-Wide Human single nucleotide polymorphism (SNP) Array 5.0 (Affymetrix; Santa Clara, CA). Microarray analysis was performed at the London Regional Genomics Centre (LRGC) (University of Western Ontario; London, ON, Canada). The data were then analyzed using dChip software for extended runs of homozygosity. We identified a 3.2 Mb region of autozygosity in the family at locus 8q24, from 139,465,102 to 142,726,810 (UCSC Genome Browser), consisting of a run of 606 consecutive homozygous SNPs. This region overlaps with a 6.8 Mb locus identified in an Iranian NS-ARMR pedigree.4 After confirming autozygosity between affected members, microsatellite genotype analysis was performed for all participating family members. The microsatellite markers used were D8S256, D8S1837, D8S1743, and D8S1704.
The 3.2 Mb region contains 12 genes, only one of which has been previously implicated in MR. This gene, KCNK9 (MIM 605874), has been shown to be causal in recently identified Birk-Barel Syndrome7 (MIM 612292). We excluded KCNK9 by direct sequencing. To select candidate MR genes in the locus, we looked for genes that are expressed at high levels in the brain or that function in neurogenesis and development. We also used the UGET algorithm, which uses banked Affymetrix gene-chip data to identify genes within the autozygous region that showed high correlation of expression with known disease genes.8 All primers used were designed using the Primer3 program. PCR reactions were performed under standard conditions. We sequenced the selected candidate genes, namely PTK2 [MIM 600758] and TRAPPC9 (NIBP [MIM 611966]). We identified the nucleotide substitution c.1422C → T in exon 7 of TRAPPC9 (GenBank Accession #NM_031466), which results in the nonsense mutation R475X (Figure 2).
Figure 2.
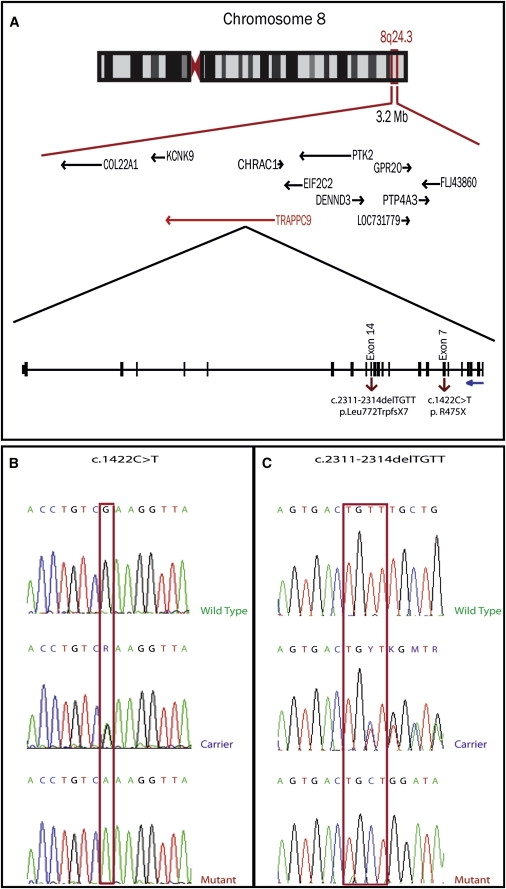
Disease Locus and TRAPPC9 Mutation
(A) Shared homozygous region in Family MR-2019 on chromosome 8q24.23–q24.3.
(B) Electropherograms for MR-2019 indicate the sequence change in exon 7, with wild-type, heterozygous carrier, and homozygous affected, leading to the amino acid change: Arginine 475 Stop.
(C) Electropherograms for M001 indicate a four base pair deletion in exon 14, resulting in a frameshift mutation that leads to the mutation p.Leu772TrpfsX7 and truncates the protein at residue 779.
The mutation is inherited in an autosomal-recessive fashion: all parents of affected members are heterozygous for the mutation, and all family members who are homozygous for the mutation express the phenotype. We screened 290 Pakistani controls (580 chromosomes) for the mutation by PCR amplification of exon 7 followed by TaqI restriction endonuclease. The TaqI cut site is destroyed by the mutation, so normal individuals should show digested PCR product on agarose gel. None of the controls had the mutated allele. Two-point parametric linkage analysis using easyLINKAGE Plus v5.02 gave a LOD score of 5.18 for marker D8S1704, situated at chr8:141,720,577–141,720,617 (UCSC hg18).
To assess the effect of this mutation on expression, we performed a quantitative RT-PCR experiment. This analysis of TRAPPC9 cDNA was performed using lymphoblast cDNA from two affected individuals from the family (V:3 and V:7) and three unrelated healthy controls, with the gene HPRT [MIM 308000] as the internal control housekeeping gene. Primers for the real-time PCR experiment were designed to include the end of TRAPPC9 exon 2 and the beginning of exon 3, thus spanning the second intron of the gene. We used SYBR Green Dye I chemistry (4309155, Applied Biosystems; Carlsbad, CA) using the Applied Biosystems 7300 Real-Time PCR System. The relative standard curve method was used to analyze the expression of TRAPPC9 in affected individuals and controls, and the experimental data were analyzed with the 7500 Software v2.0.1. Results indicated that, in individuals homozygous for 475X, the TRAPPC9 mRNA is expressed at only a fraction of the amount for controls with wild-type 475R (Figure 3). This suggests that the mutation results in a significant degree of nonsense-mediated mRNA decay, and hence, if there is any residual function in the truncated NIK- and IKK-β-binding protein (NIBP), expression levels would be very low.
Figure 3.
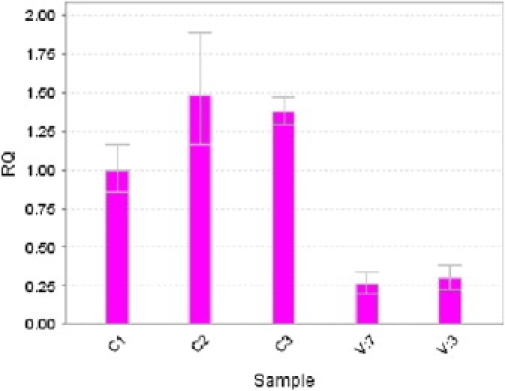
qPCR Assay of TRAPPC9 mRNA
Quantitative measurement of TRAPPC9 mRNA from lymphoblasts, measured using real-time PCR. The level of expression between individuals carrying a mutation in TRAPPC9 and those with the wild-type gene is clearly different, suggesting nonsense-mediated mRNA decay associated with the mutation. Subjects C1 and C2 are age- and sex-matched healthy controls. Affected individuals are V:7 and V:3. mRNA levels were normalized against the housekeeping gene HPRT1. Error bars indicate maximum and minimum values.
We obtained DNA from family members of the Iranian pedigree M001 from the Najmabadi et al. study.4 This large NS-ARMR family mapped to a 6.8 Mb critical interval on 8q24 that included TRAPPC9. Sequence analysis identified a 4 bp deletion: c.2311–2314 delTGTT, resulting in frameshift and premature truncation: p.Leu772TrpfsX7. This deletion segregates in a recessive manner in the family. Affected members of this family have severe NS-ARMR/ID and are ambulatory but nonverbal, with no history of seizures and no dysmorphic or unusual facial features. OFC head size was measured in three adult male affected individuals, aged 27, 33, and 37 years, as 54, 53, and 52.5 cm, respectively (between 1 and 3 SD below the mean).
We have identified a gene for autosomal-recessive mental retardation, TRAPPC9, by autozygosity mapping in a large consanguineous Pakistani family, with confirmation in an Iranian family. In the Pakistani family, although the phenotype appears to be nonsyndromic MR, mild cerebral white matter hypoplasia and small head circumference are present in most of the patients (Table 1). These head sizes, in comparison to age/sex means,9 would not be classified as true microcephaly, for which OFC would be >4 SD below the mean (see review by Woods et al.).10 The head growth trajectories for these subjects is not available; thus, it cannot be determined whether head size developed normally up to a certain age and then growth slowed or whether head growth was delayed from birth. The relevance of these clinical features to TRAPPC9 mutations can be confirmed only through the identification and evaluation of additional families.
TRAPPC9 is a 23 exon gene that encodes the 1148–1246 amino acid protein, NIBP. NIBP is highly conserved across evolution (protein sequence identity between human and mouse, 92%; chicken, 87%; zebrafish, 85%; pufferfish, 75%; sea urchin, 42%; bee, 35%); however, no known functional domains are present. COILS predicts the presence of coiled coil from amino acid residue 1062–1076. PSORT II predicts no signal peptides, no nuclear or organelle targeting sequences, and no ER retention signals and predicts NIBP to be a cytoplasmic protein.
NIBP contains one known conserved region, originally identified in Saccharomyces cerevisiae, called Trs120.11 It is believed to function as part of a large multisubunit tethering complex, TRAPPII, trafficking vesicles/proteins from the Golgi to the plasma membrane.11,12 Mutants of Trs120 in yeast cause defective trafficking from early endosome to late Golgi, suggesting a role for TRAPPII in sorting/recycling of proteins from the plasma membrane.13
In humans, multiple NIBP isoforms are expressed at high levels in the muscle and kidney and to a lesser extent in the brain, heart, and placenta.14 Only isoform 1 is present in the brain, where it is expressed in the cell bodies and processes of neurons.14 Knockdown of NIBP has been shown to reduce TNFα-induced NF-κB activation, prevent nerve growth factor-induced neuronal differentiation, and decrease Bcl-xL gene expression in PC12 cells.14 We speculate that the loss of NIBP function through truncating mutations in the Pakistani and Iranian families presented here might cause disruption of neuronal differentiation, which could result in the cerebral white matter hypoplasia observed through MRI. NIBP also directly interacts with IKK-β and MAP3K14 and is involved in both classical and alternative activation of the NF-κB signaling pathway.14
The activity of TRAPPC9/NIBP in the NF-κB pathway14 is of interest because another gene involved in nonsyndromic MR/ID, CC2D1A, was identified as an activator of the NF-κB signaling pathway,15 suggesting that disruption of this pathway might also be relevant to the etiology of MR/ID.
Identifying the genetic cause of MR in an individual or family could have significant benefits. For some genetic forms of MR, a simple metabolic therapy could be readily available, whereas for others, treatment could be anticipated in the near future. Additional benefits include anticipating comorbid medical problems and assisting with family planning. Also, in families where consanguineous marriages are practiced, this information might be used to advise the family how to prevent the disease recurring by avoiding marriage between mutation carriers.16
The identification of a mutation that eliminates NIBP—a component of the TRAPPII trafficking complex—suggests a potentially novel pathway involved in the brain dysfunction underlying MR. It is of interest that TRAPPC10, which encodes the human ortholog of the TRAPPII component Trs130, has been implicated in holoprosencephaly with epilepsy.17 Another TRAPP component, synbindin, encoded by TRAPPC4 (MIM 610971), binds with syndecan-2 (encoded by SDC2 [MIM 142460]) in neuronal dendritic spines.18 SDC2 has previously been identified at the 8q22.1 breakpoint of a translocation t(X;8)(p22.13;q22.1) in a patient with autism and multiple exostoses,19 and SDC2 transcription might be dysregulated by position effect. We speculate that mutations in other components of the TRAPPII complex that function in neurons might also cause NS-ARMR or other neuropsychiatric phenotypes. Indeed, several other components of vesicular trafficking pathways are already known to be associated with MR/ID, including VPS13B (Cohen syndrome; MIM 607817), GDI1 (NS-XLMR; MIM 300104), and GAP/TSC2 (tuberous sclerosis 2; MIM 191092).
Acknowledgments
We would like to express our deepest gratitude to the families involved for their participation in this study. Funding support for this work was through an Early Researcher Award from the Ontario provincial government to J.B.V. and also through a NARSAD Independent Investigator Award to J.B.V.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
UCSC Genome Browser, http://genome.ucsc.edu/
UCLA Gene Expression Tool (UGET), http://genome.ucla.edu/projects/UGET
References
- 1.Diagnostic and Statistical Manual of Mental Disorders . Fourth Edition. American Psychiatric Association; Arlington, VA: 2005. Text Revision (DSM-IV-TR) [Google Scholar]
- 2.Chiurazzi P., Schwartz C.E., Gecz J., Neri G. XLMR genes: Update 2007. Eur. J. Hum. Genet. 2008;16:422–434. doi: 10.1038/sj.ejhg.5201994. [DOI] [PubMed] [Google Scholar]
- 3.Ropers H.H., Hamel B.C. X-linked mental retardation. Nat. Rev. Genet. 2005;6:46–57. doi: 10.1038/nrg1501. [DOI] [PubMed] [Google Scholar]
- 4.Najmabadi H., Motazacker M.M., Garshasbi M., Kahrizi K., Tzschach A., Chen W., Behjati F., Hadavi V., Nieh S.E., Abedini S.S. Homozygosity mapping in consanguineous families reveals extreme heterogeneity of non-syndromic autosomal recessive mental retardation and identifies 8 novel gene loci. Hum. Genet. 2007;121:43–48. doi: 10.1007/s00439-006-0292-0. [DOI] [PubMed] [Google Scholar]
- 5.Sparrow S.S., Balla D.A., Cicchetti D.V. AGS Publishing; Circle Pines, MN: 2005. Vineland Adaptive Behavior Scales, Second Edition, Survey Forms Manual. [Google Scholar]
- 6.Shearer M.S., Shearer D.E. The Portage Project: A Model for Early Childhood Education. Exceptional Children. 1972;39:210–217. doi: 10.1177/001440297203900304. [DOI] [PubMed] [Google Scholar]
- 7.Barel O., Shalev S.A., Ofir R., Cohen A., Zlotogora J., Shorer Z., Mazor G., Finer G., Khateeb S., Zilberberg N. Maternally inherited birk barel mental retardation dysmorphism syndrome caused by a mutation in the genomically imprinted potassium channel KCNK9. Am. J. Hum. Genet. 2008;83:193–199. doi: 10.1016/j.ajhg.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Day A., Carlson M.R., Dong J., O'Connor B.D., Nelson S.F. Celsius: a community resource for Affymetrix microarray data. Genome Biol. 2007;8:R112. doi: 10.1186/gb-2007-8-6-r112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nellhaus G. Head circumference from birth to eighteen years. Practical composite international and interracial graphs. Pediatrics. 1968;41:106–114. [PubMed] [Google Scholar]
- 10.Woods C.G., Bond J., Enard W. Autosomal recessive primary microcephaly (MCPH): a review of clinical, molecular and evolutionary findings. Am. J. Hum. Genet. 2005;76:717–728. doi: 10.1086/429930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sacher M., Barrowman J., Schieltz D., Yates J.R., 3rd, Ferro-Novick S. Identification and characterization of five new subunits of TRAPP. Eur. J. Cell Biol. 2000;79:71–80. doi: 10.1078/S0171-9335(04)70009-6. [DOI] [PubMed] [Google Scholar]
- 12.Cox R., Chen S.H., Yoo E., Segev N. Conservation of the TRAPPII-specific subunits of a Ypt/Rab exchanger complex. BMC Evol. Biol. 2007;7:12. doi: 10.1186/1471-2148-7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cai H., Zhang Y., Pypaert M., Walker L., Ferro-Novick S. Mutants in trs120 disrupt traffic from the early endosome to the late Golgi. J. Cell Biol. 2005;171:823–833. doi: 10.1083/jcb.200505145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu W.H., Pendergast J.S., Mo X.M., Brambilla R., Bracchi-Ricard V., Li F., Walters W.M., Blits B., He L., Shaal S.M. NIBP, a novel NIK and IKK(beta)-binding protein that enhances NF-(kappa)B activation. J. Biol. Chem. 2005;280:29233–29241. doi: 10.1074/jbc.M501670200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsuda A., Suzuki Y., Honda G., Muramatsu S., Matsuzaki O., Nagano Y., Doi T., Shimotohno K., Harada T., Nishida E. Large-scale identification and characterization of human genes that activate NF-kappaB and MAPK signaling pathways. Oncogene. 2003;22:3307–3318. doi: 10.1038/sj.onc.1206406. [DOI] [PubMed] [Google Scholar]
- 16.Modell B., Darr A. Genetic counselling and customary consanguineous marriage. Nat. Rev. Genet. 2002;2:225–229. doi: 10.1038/nrg754. [DOI] [PubMed] [Google Scholar]
- 17.Yamakawa K., Mitchell S., Hubert R., Chen X.N., Colbern S., Huo Y.K., Gadomski C., Kim U.J., Korenberg J.R. Isolation and characterization of a candidate gene for progressive myoclonus epilepsy on 21q22.3. Hum. Mol. Genet. 1995;4:709–716. doi: 10.1093/hmg/4.4.709. [DOI] [PubMed] [Google Scholar]
- 18.Ethell I.M., Hagihara K., Miura Y., Irie F., Yamaguchi Y. Synbindin, a novel syndecan-2-binding protein in neuronal dendritic spines. J. Cell Biol. 2000;151:53–67. doi: 10.1083/jcb.151.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishikawa-Brush Y., Powell J.F., Bolton P., Miller A.P., Francis F., Willard H.F., Lehrach H., Monaco A.P. Autism and multiple exostoses associated with an X;8 translocation occurring within the GRPR gene and 3-prime to the SDC2 gene. Hum. Mol. Genet. 1997;6:1241–1250. doi: 10.1093/hmg/6.8.1241. [DOI] [PubMed] [Google Scholar]
- 20.Roche A.F., Mukherjee D., Guo S., Moore W.M. Head circumference reference data: birth to 18 years. Pediatrics. 1987;79:706–712. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.