

Abstract
Y-linked single-nucleotide polymorphisms (SNPs) have served as powerful tools for reconstructing the worldwide genealogy of human Y chromosomes and for illuminating patrilineal relationships among modern human populations. However, there has been no systematic, worldwide survey of sequence variation within the protein-coding genes of the Y chromosome. Here we report and analyze coding sequence variation among the 16 single-copy “X-degenerate” genes of the Y chromosome. We examined variation in these genes in 105 men representing worldwide diversity, resequencing in each man an average of 27 kb of coding DNA, 40 kb of intronic DNA, and, for comparison, 15 kb of DNA in single-copy Y-chromosomal pseudogenes. There is remarkably little variation in X-degenerate protein sequences: two chromosomes drawn at random differ on average by a single amino acid, with half of these differences arising from a single, conservative Asp→Glu mutation that occurred ∼50,000 years ago. Further analysis showed that nucleotide diversity and the proportion of variant sites are significantly lower for nonsynonymous sites than for synonymous sites, introns, or pseudogenes. These differences imply that natural selection has operated effectively in preserving the amino acid sequences of the Y chromosome's X-degenerate proteins during the last ∼100,000 years of human history. Thus our findings are at odds with prominent accounts of the human Y chromosome's imminent demise.
Main Text
Study of single-nucleotide polymorphisms (SNPs) in the male-specific region of the Y chromosome (the MSY; Figure 1) has differed markedly from studies of SNPs elsewhere in the human genome. Study of MSY SNPs has focused on refining the genealogical tree of human MSYs and improving its utility in understanding genetic relationships among populations. These efforts have yielded a robust and unambiguous genealogical tree of human Y chromosomes.1–3 This achievement was possible because, unlike the rest of the genome, the MSY does not participate in crossing over and reshuffling with a homolog but instead displays clonal, strictly patrilineal inheritance. Conversely, there has been little consideration of the possibility of natural selection acting on MSY SNPs, which would complicate the use of the SNP-based genealogy as a tool for understanding relationships among populations.
Figure 1.
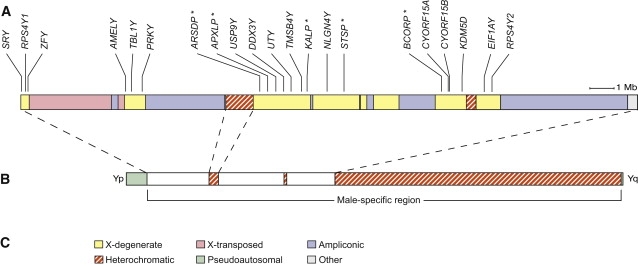
Single-Copy, X-Degenerate Genes and Pseudogenes in the MSY
(A) Schematic representation of euchromatic portions of the MSY (male-specific region of Y chromosome) highlighting locations of X-degenerate genes and pseudogenes studied. All of these genes and pseudogenes have formerly allelic homologs on the X chromosome. Asterisks indicate pseudogenes.
(B) The MSY does not undergo meiotic crossing over with a homolog. By contrast, the pseudoautosomal regions (green) undergo crossing over between the X and Y chromosomes at male meiosis and thus are shared by the two chromosomes. The MSY's X-degenerate genes and pseudogenes are distinct from the genes found in the pseudoautosomal regions.
(C) Sequence classes within the human Y chromosome. See 4 for details.
Nevertheless, the MSY contains 16 evolutionarily conserved, single-copy genes, and their patterns of nucleotide variation may illuminate selective constraints on the MSY and be important to future studies of MSY function in health and disease. These 16 genes are the X-degenerate genes of the human MSY (Figure 1),4 which are legacies of the common ancestry of the human Y and X chromosomes. Over long evolutionary time periods, these genes were part of regions shared by the Y and X chromosomes—the so-called pseudoautosomal regions. The X-degenerate genes eventually became recombinationally isolated in the MSY at points in time ranging from hundreds to tens of millions of years ago.4,5
We sought to answer several questions. Worldwide, what common coding differences exist among the X-degenerate genes of the human MSY? Are there differences between patterns of nucleotide variation that affect protein sequences versus those that do not? If so, do these patterns shed light on selective constraints or recent Y-chromosome degeneration?
The MSYs intensively studied, clonal genealogy, and its relationship to human geography present opportunities for efficiently and comprehensively capturing coding variation. Within human genetics, this situation is paralleled only in the mitochondrial genome. We used information from the MSY genealogy in the selection of DNA samples for study, as we sought to maximize MSY diversity and the representation of recent MSY evolutionary history. We did this by selecting DNA samples from as many branches of the MSY genealogical tree as possible, and also by selecting, when possible, multiple samples from those branches that are populous or geographically widespread.
Specifically, we employed previously described SNPs to determine the MSY haplotypes of 473 candidate Y chromosomes. From among these we selected 105 chromosomes (Table S1 available online), representing 47 branches of the MSY genealogy, in which to resequence the X-degenerate genes. Both the initial MSY haplotyping and the subsequent resequencing were performed on genomic DNAs extracted from EBV-transformed lymphoblastoid cell lines or, in a few cases, from fibroblast cell cultures. The study was approved by the institutional review board of the Massachusetts Institute of Technology.
In the 105 selected samples, we resequenced the 16 single-copy MSY genes (Table 1) and 5 single-copy MSY pseudogenes (Table S2). GenBank accession numbers for the resequenced portions of these genes and pseudogenes are listed in Table S3. The referenced GenBank entries specify the PCR primer pairs and reaction conditions with which we amplified the resequenced portions of the genes and pseudogenes. We were able to design reliable assays for PCR amplification of all but 4 of the 185 X-degenerate coding exons. We carried out sequencing reactions with “BigDye” kits (Applied Biosystems) and read the sequencing-reaction products on an ABI3700 automated sequencer.
Table 1.
Genes Surveyed for DNA Sequence Variation
| Gene |
Numbers of Nucleotide Sites Surveyed |
||
|---|---|---|---|
| Synonymous | Nonsynonymous | Intron | |
| SRY | 174 | 429 | 0 |
| RPS4Y1 | 235 | 549 | 1,374 |
| ZFY | 636 | 1,634 | 1,624 |
| AMELY | 146 | 343 | 1,609 |
| TBL1Y | 396 | 1,006 | 3,775 |
| PRKY | 193 | 472 | 1,538 |
| USP9Y | 2,176 | 5,305 | 10,507 |
| DDX3Y | 582 | 1,395 | 2,954 |
| UTY | 1,185 | 2,853 | 6,519 |
| TMSB4Y | 36 | 93 | 185 |
| NLGN4Y | 307 | 775 | 1,282 |
| CYORF15A | 116 | 274 | 757 |
| CYORF15B | 155 | 385 | 1,007 |
| KDM5D | 1,235 | 2,847 | 4,131 |
| EIF1AY | 125 | 304 | 1,413 |
| RPS4Y2 | 236 | 550 | 1,654 |
| Total | 7,932a | 19,215a | 40,329 |
NOTE: Table S3 provides details of STSs (primer pairs) used to amplify exons and their intronic flanks.
Totals listed differ from column sums (7,933 and 19,214 nucleotides, respectively) because of rounding of nonintegral values.
We used the phred program (see Web Resources) for base calling and calculation of initial quality scores at each nucleotide position.6 We utilized the phrap program (see Web Resources) to assemble sequence reads together with the corresponding reference sequence. Where reads from neighboring PCR products overlapped, we assembled them together.
By using a neighborhood quality score, we stringently assessed whether each nucleotide in a given read had been sequenced accurately.7 We considered a nucleotide to have been sequenced accurately if it and all 20 nucleotides on each side had phred scores ≥20. In cases of overlapping reads from a single DNA sample, we relied upon the highest phred score of any of the reads from that sample. We confirmed variant sites by visually examining sequencing chromatograms from variant and reference alleles.
In analyzing the resultant set of high-accuracy DNA sequences, we utilized a perl program (available on request) to calculate nucleotide diversity at each nonsynonymous, synonymous, intron, or pseudogene site. We calculated nucleotide diversity at each site as the mean number of differences when comparing all pairs of chromosomes at that site.8 We calculated amino acid diversity in the analogous fashion. To calculate the number of nonsynonymous and synonymous sites examined, we counted as “nonsynonymous” all nondegenerate sites and two-thirds of all 2-fold degenerate sites. We counted as “synonymous” all 4-fold degenerate sites and one-third of all 2-fold degenerate sites. We excluded gene flanks and 5′ and 3′ UTRs from analysis because they were not uniformly well defined. We compared variant sites to dbSNP (build 129) and submitted all novel variants (Table S4).
By using these analytic tools, we examined, in each of the 105 selected Y chromosomes, an average of 27,147 coding nucleotides in 181 exons, 40,329 intronic nucleotides, and 15,251 pseudogene nucleotides (Table 1; Table S2). In this sequence, we detected a total of 126 single-nucleotide variants (Table S4). We were able to place each of these 126 variants on one or another unique branch of the genealogical tree of human Y chromosomes (Figure 2; Figures S1–S4).1–3 Thus each of the variants observed is likely the result of a single mutational event.
Figure 2.
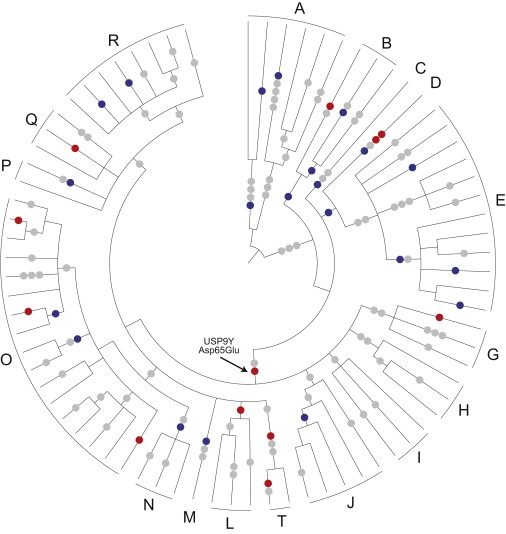
Genealogical Tree of Single-Copy Coding Sequence Variation in the MSY
The tree is shown in radial fashion with the root at the center and haplogroups (A through E, G through J, L through R, and T) indicated around the circumference. On the branches of the tree are shown the inferred genealogical locations of nonsynonymous nucleotide substitutions (red circles), synonymous nucleotide substitutions within coding sequence (blue circles), and substitutions in introns or pseudogenes (gray circles). The arrow indicates the most widespread nonsynonymous substitution (USP9Y Asp65Glu). See Figures S1–S4 for detailed haplotypes of the 105 Y chromosomes examined and for identifiers of the variants observed.
The 126 single-nucleotide variants that we detected in X-degenerate genes and pseudogenes result in very little diversity in the encoded proteins (Table 2). Only 12 of the 126 variants result in amino acid substitutions. Among the 105 chromosomes studied, amino acid diversity in the X-degenerate proteome is only 0.89 residues. Thus, on average, the sets of X-degenerate proteins from two Y chromosomes drawn at random from the study sample differ by a single amino acid substitution. Furthermore, a single variant accounts for almost half of that diversity. This variant corresponds to an aspartic acid to glutamic acid substitution at residue 65 of USP9Y (MIM 400005). We infer directionality (Asp→Glu) from the corresponding sequence in chimpanzee and from the context in the human MSY genealogy. In addition, based on the position of this mutation in the MSY genealogy, we infer that it occurred about 50,000 years ago (95% CI 38,700 to 55,700).3 It is among the oldest of the 12 observed protein-coding changes and is ancestral to 8 of the 11 other protein-coding changes. None of the three comparably ancient protein-coding changes is demonstrably closer to the root of the MSY genealogy (Figure 2; Figure S1). Examination of this Asp→Glu substitution in broader evolutionary context suggests that it may be of little functional consequence: among the 12 most similar USP9 proteins in mammals and birds, 11 have glutamic acid at the homologous residue (Figure S5). Only the mouse USP9Y protein has aspartic acid, as in the ancestral sequence of human USP9Y.
Table 2.
Amino Acid Substitutions and Diversities
| Gene | Nucleotide Substitution | Exon | Amino Acid Substitution | Amino Acid Diversity |
|---|---|---|---|---|
| ZFY | A391G | 5 | Met131Val | 0.0370 |
| AMELY | G208A | 5 | Val70Met | 0.0317 |
| AMELY | G497A | 5 | Arg166Gln | 0.1051 |
| USP9Y | T195G | 4 | Asp65Glu | 0.4297 |
| USP9Y | C631T | 7 | Arg211Cys | 0.0404 |
| USP9Y | G3178A | 23 | Ala1060Thr | 0.0381 |
| DDX3Y | G487A | 6 | Asp163Asn | 0.0200 |
| DDX3Y | G1313C | 13 | Ser438Thr | 0.0217 |
| CYORF15A | A317C | 3 | Asn106Thr | 0.0230 |
| CYORF15A | C326T | 3 | Ser109Leu | 0.0887 |
| KDM5D | G3296A | 23 | Ser1099Asn | 0.0208 |
| KDM5D | G4433A | 27 | Arg1478Gln | 0.0312 |
| Aggregate | 0.89 | |||
We then asked whether patterns of variation are different for nonsynonymous nucleotide substitutions as compared to synonymous substitutions or substitutions in introns or pseudogenes. We found that nonsynonymous nucleotide diversity (0.5 × 10−4) is significantly lower than diversity at synonymous sites (1.5 × 10−4), in introns (1.2 × 10−4), and in pseudogenes (1.0 × 10−4) (Table 3; Table S5). Similarly, the proportion of nonsynonymous sites that vary is significantly lower than the proportions of synonymous, intron, and pseudogene sites that vary (Table 3; Table S5). By all these measures, in our systematic, worldwide survey of variation in MSY single-copy genes, nucleotide variants that alter protein sequence are significantly underrepresented relative to variants that do not alter protein sequence.
Table 3.
Both the Proportion of Variant Nucleotide Sites and Nucleotide Diversity Are Much Lower at Nonsynonymous Sites than at Synonymous Sites, in Introns, and in Pseudogenes
| Nonsynonymous | Synonymous | Intron | Pseudogene | Synonymous, Intron, and Pseudogene | |
|---|---|---|---|---|---|
| Variant nucleotide sites | 12 | 21 | 64 | 29 | 114 |
| Invariant nucleotide sites | 19,203 | 7,911 | 40,265 | 15,222 | 63,398 |
| Total nucleotide sites | 19,215 | 7,932 | 40,329 | 15,251 | 63,513 |
| Proportion of Variant Sites | 6.25 × 10−4 | 2.65 × 10−3 | 1.59 × 10−3 | 1.90 × 10−3 | 1.79 × 10−3 |
| p Values of Differences in Proportions of Variant Sites (Fisher's Exact Test, Two Sided) | |||||
| Nonsynonymous versus … | 5.4 × 10−5 | 1.9 × 10−3 | 7.8 × 10−4 | 1.2 × 10−4 | |
| Synonymous versus … | 0.055 | 0.30 | |||
| Intron versus … | 0.42 | ||||
| Nucleotide Diversity | 4.62 × 10−5 | 1.50 × 10−4 | 1.22 × 10−4 | 9.8 × 10−5 | 1.20 × 10−4 |
| p Values of Differences in Nucleotide Diversitiesa | |||||
| Nonsynonymous versus … | 1.4 × 10−5 | 2.1 × 10−3 | 6.3 × 10−4 | 2.7 × 10−4 | |
| Synonymous versus … | 0.039 | 0.25 | |||
| Intron versus … | 0.42 | ||||
p values calculated by two-sided Wilcoxon rank-sum tests comparing distributions of nucleotide diversities over all sites (wilcox.test function in R statistical computing environment; see Web Resources).
Several aspects of these findings merit discussion. First, in resequencing the Y chromosome's 16 single-copy genes in 105 men representing worldwide MSY diversity, we found little variability in predicted protein sequences. This low variability includes the sex-determining gene SRY (MIM 480000), in which we detected a single, synonymous substitution (Table S4). We also observed significantly less amino-acid-changing nucleotide variation than synonymous coding variation, intronic variation, or variation in decayed pseudogenes; this finding holds regardless of whether variation is assessed as mean nucleotide diversity or as the proportion of variant sites. These observations led us to conclude that most nonsynonymous mutations have been culled by natural selection while neutral mutations have more commonly persisted.
How does the pattern of variation in the X-degenerate genes of the human Y chromosome compare to that of genes elsewhere in the genome? In our Y-chromosome data, the relative proportion of nonsynonymous to synonymous variant sites is 0.24 (1/1601 versus 1/378; Table 3), and the relative proportion of nonsynonymous to intronic variant sites is 0.39 (1/1601 versus 1/630). Roughly analogous values for a collection of 75 non-Y-linked human genes are similar: 0.38 and 0.54, respectively.9 Thus, there is presently no evidence that purifying selection is weaker or less effective in the Y chromosome's X-degenerate genes than in the human genome as a whole.
The intronic nucleotide diversity among the Y chromosomes studied here was substantially lower than in the rest of the genome: 1.2 × 10−4 versus 10.5 × 10−4 (standard deviation 2.7 × 10−4) for a sample of introns elsewhere in the genome, mostly in autosomes.9 Our finding of reduced intronic nucleotide diversity among Y chromosomes is in close agreement with an earlier study of four X-degenerate genes in a smaller collection of Y chromosomes.10 Part of this reduction in diversity among Y chromosomes is expected because of the lower population size of Y chromosomes compared to autosomes; the number of Y chromosomes in a population is one fourth the number of each autosome. From this, population-genetic theory predicts that MSY diversity would be proportionately reduced to one fourth of autosomal diversity.8 The additional deficit in diversity relative to the rest of the genome may be due to higher variance in reproductive success among men than among women,8 which would further reduce the effective population size of Y chromosomes.
The results reported here shed new light on an important question: how representative or typical is the sequenced human Y chromosome? Previous work showed that the sequenced MSY is representative with respect to copy number variation and is not an outlier with respect to large inversions.11 The results reported here demonstrate that it is also quite representative in terms of its X-degenerate proteome, bolstering evidence that the reference Y chromosome sequence is indeed representative.
The question of whether the reference Y chromosome is representative also arises in the context of Y chromosome evolution and the gene decay that it prominently features.3,4 If Y chromosomes were unconstrained by selection and decayed rapidly over evolutionary time periods, then different branches of the human MSY genealogy might show different degrees or kinds of decay. Indeed, after reports that no X-degenerate gene was lost in the human Y lineage since its divergence from the chimpanzee Y chromosome,12 the question arose as to whether this result might be highly dependent on the specific human Y chromosome selected for comparison with chimpanzee.13
In particular, some human Y chromosomes completely lack the X-degenerate genes AMELY (MIM 410000), TBL1Y (MIM 400033), and PRKY (MIM 40000) because of a contiguous deletion,14 with most such chromosomes mapping to branch J/-M241 of the MSY genealogy (branch J chromosomes with the derived allele for M241).15,16 A Y chromosome from this branch was not available to us at the outset of this study, and evidence to date indicates that this branch is rare, comprising only 0.23% of Y chromosomes even in India, where it is comparatively widespread.17 The deletion in this branch appears to be the consequence of a single, ancestral, unequal crossover between direct repeats that flank these three genes.14,16 This deletion has been studied intensively because a test for the AMELY gene has been used to detect male DNA in forensic studies, even though, by this test, DNA from a 46,XY man lacking AMELY appears to be from a woman.
Because this particular deletion was first detected and then attained prominence through a chance intersection with forensics, one might speculate that it represents one of many similar events among human Y chromosomes, with the others having escaped attention. However, we uncovered no evidence of other wholesale deletions of X-degenerate genes in the 105 chromosomes tested, although we selected them to represent as much diversity as possible among the samples available to us. Indeed, we found little variation in amino acid sequence among the X-degenerate genes. We conclude that, with respect to X-degenerate gene content, the chromosomes deleted for AMELY, TBL1Y, and PRKY are exceptional while the reference sequence is representative.
Prior to this study, MSY SNPs were analyzed primarily in reconstructing patrilineal relationships among modern human populations, with little heed to the SNPs' possible functional significance. Indeed, the conclusions of many such population studies have rested on the assumption that all MSY SNPs—as well as any structural polymorphisms in the Y chromosomes marked by these SNPs—are selectively neutral. Together with previous findings,18 our current data contradict this simplifying assumption. The MSY does not undergo sexual recombination with a homologous chromosome, so it is subject to natural selection as an indivisible unit. Even if the particular MSY SNPs employed in a population study are functionally inconsequential, they may have been coupled to detrimental or beneficial SNPs or structural variants elsewhere in the MSY. Previous studies have demonstrated that structural polymorphism in the MSY affects sperm production and male fertility.18 Similarly, our present findings imply that selection on coding SNPs has significantly affected the evolutionary trajectory and population frequencies of variant Y chromosomes during the past 100,000 years of human history. Taken together, these studies of structural polymorphism and coding sequence variation in the MSY highlight the role of natural selection in human MSY lineages. This new awareness means that we can no longer assume selective neutrality in the MSY when drawing conclusions from population genetic studies.
The results reported here also shed light on models of genetic decay in the human Y chromosome. Some geneticists have predicted gene loss to be so precipitous that it might lead to the complete demise of all human Y chromosomes in 10,000,000 years' time, if not sooner.19,20 To be sure, there is incontrovertible evidence of dramatic gene loss during the Y chromosome's evolution over periods of tens and hundreds of millions of years.4,5 Furthermore, during the 6,000,000 years since divergence of the chimpanzee and human lineages, the chimpanzee Y chromosome has lost the function of four X-degenerate genes, possibly as a result of increased specialization for spermatogenesis.12 By contrast, the human Y chromosome has not lost any X-degenerate genes during the same 6,000,000 years.12 Our present findings show that, in addition, X-degenerate gene content in the overwhelming majority of human Y lineages has changed little since the last common ancestor of modern human Y chromosomes, ∼100,000 years ago. Indeed, the results reported here imply that purifying selection has been effective in stabilizing and maintaining the amino acid sequences of the human MSY's X-degenerate proteins during this period. In combination with previous studies,12 our findings conclusively refute models of precipitous genetic decay in human Y-chromosome lineages.
Acknowledgments
We thank Laura Brown, Gail Farino, Vicki Frazzoni, and Loreall Pooler for technical assistance; Christine Disteche, Alan Donnenfeld, Nathan Ellis, Trefor Jenkins, Michael Hammer, Robert Oates, Pasquale Patrizio, Sherman Silber, Jean Weissenbach, and Brian Whitmire for human samples and cell lines; Andrew Clark for consultation on population genetic analysis; Michael Hammer and Peter Underhill for advice and guidance in genealogical studies; and D. Winston Bellott, Michelle Carmell, Greg Dokshin, Mark Gill, Jennifer Hughes, Jacob Mueller, Tatyana Pyntikova, and Shirleen Soh for comments on the manuscript. Supported by the National Institutes of Health and the Howard Hughes Medical Institute.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
GenBank (nucleotide sequences), http://www.ncbi.nlm.nih.gov/sites/entrez?db=nucleotide
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
phred and phrap programs, http://www.phrap.org/phredphradconsed.html
R statistical computing environment, http://www.r-project.org/
References
- 1.Underhill P.A., Shen P., Lin A.A., Jin L., Passarino G., Yang W.H., Kauffman E., Bonne-Tamir B., Bertranpetit J., Francalacci P. Y chromosome sequence variation and the history of human populations. Nat. Genet. 2000;26:358–361. doi: 10.1038/81685. [DOI] [PubMed] [Google Scholar]
- 2.Jobling M., Tyler-Smith C. The human Y chromosome: An evolutionary marker comes of age. Nat. Rev. Genet. 2003;4:598–612. doi: 10.1038/nrg1124. [DOI] [PubMed] [Google Scholar]
- 3.Karafet T.M., Mendez F.L., Meilerman M.B., Underhill P.A., Zegura S.L., Hammer M.F. New binary polymorphisms reshape and increase resolution of the human Y chromosomal haplogroup tree. Genome Res. 2008;18:830–838. doi: 10.1101/gr.7172008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Skaletsky H., Kuroda-Kawaguchi T., Minx P.J., Cordum H.S., Hillier L., Brown L.G., Repping S., Pyntikova T., Ali J., Bieri T. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature. 2003;423:825–837. doi: 10.1038/nature01722. [DOI] [PubMed] [Google Scholar]
- 5.Ross M.T., Grafham D.V., Coffey A.J., Scherer S., McLay K., Muzny D., Platzer M., Howell G.R., Burrows C., Bird C.P. The DNA sequence of the human X chromosome. Nature. 2005;434:325–337. doi: 10.1038/nature03440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ewing B., Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- 7.Altshuler D., Pollara V.J., Cowles C.R., Van Etten W.J., Baldwin J., Linton L., Lander E.S. An SNP map of the human genome generated by reduced representation shotgun sequencing. Nature. 2000;407:513–516. doi: 10.1038/35035083. [DOI] [PubMed] [Google Scholar]
- 8.Hartl D.L., Clark A.G. Sinauer Associates, Inc.; Sunderland, MA: 2007. Principles of Population Genetics. [Google Scholar]
- 9.Halushka M.K., Fan J.B., Bentley K., Hsie L., Shen N., Weder A., Cooper R., Lipshutz R., Chakravarti A. Patterns of single-nucleotide polymorphisms in candidate genes for blood-pressure homeostasis. Nat. Genet. 1999;22:239–247. doi: 10.1038/10297. [DOI] [PubMed] [Google Scholar]
- 10.Shen P., Wang F., Underhill P.A., Franco C., Yang W.H., Roxas A., Sung R., Lin A.A., Hyman R.W., Vollrath D. Population genetic implications from sequence variation in four Y chromosome genes. Proc. Natl. Acad. Sci. USA. 2000;97:7354–7359. doi: 10.1073/pnas.97.13.7354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Repping S., van Daalen S.K., Brown L.G., Korver C.M., Lange J., Marszalek J.D., Pyntikova T., van der Veen F., Skaletsky H., Page D.C. High mutation rates have driven extensive structural polymorphism among human Y chromosomes. Nat. Genet. 2006;38:463–467. doi: 10.1038/ng1754. [DOI] [PubMed] [Google Scholar]
- 12.Hughes J.F., Skaletsky H., Pyntikova T., Minx P.J., Graves T., Rozen S., Wilson R.K., Page D.C. Conservation of Y-linked genes during human evolution revealed by comparative sequencing in chimpanzee. Nature. 2005;437:100–103. doi: 10.1038/nature04101. [DOI] [PubMed] [Google Scholar]
- 13.Tyler-Smith C., Howe K., Santos F.R. The rise and fall of the ape Y chromosome? Nat. Genet. 2006;38:141–143. doi: 10.1038/ng0206-141. [DOI] [PubMed] [Google Scholar]
- 14.Santos F.R., Pandya A., Tyler-Smith C. Reliability of DNA-based sex tests. Nat. Genet. 1998;18:103. doi: 10.1038/ng0298-103. [DOI] [PubMed] [Google Scholar]
- 15.Cadenas A.M., Regueiro M., Gayden T., Singh N., Zhivotovsky L.A., Underhill P.A., Herrera R.J. Male amelogenin dropouts: Phylogenetic context, origins and implications. Forensic Sci. Int. 2007;166:155–163. doi: 10.1016/j.forsciint.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 16.Jobling M.A., Lo I.C., Turner D.J., Bowden G.R., Lee A.C., Xue Y., Carvalho-Silva D., Hurles M.E., Adams S.M., Chang Y.M. Structural variation on the short arm of the human Y chromosome: Recurrent multigene deletions encompassing Amelogenin Y. Hum. Mol. Genet. 2007;16:307–316. doi: 10.1093/hmg/ddl465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kashyap V.K., Sahoo S., Sitalaximi T., Trivedi R. Deletions in the Y-derived amelogenin gene fragment in the Indian population. BMC Med. Genet. 2006;7:37. doi: 10.1186/1471-2350-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Repping S., Skaletsky H., Brown L., van Daalen S.K.M., Korver C.M., Pyntikova T., Kuroda-Kawaguchi T., de Vries J.W.A., Oates R.D., Silber S. Polymorphism for a 1.6-Mb deletion of the human Y chromosome persists through balance between recurrent mutation and haploid selection. Nat. Genet. 2003;35:247–251. doi: 10.1038/ng1250. [DOI] [PubMed] [Google Scholar]
- 19.Aitken R.J., Marshall Graves J.A. The future of sex. Nature. 2002;415:963. doi: 10.1038/415963a. [DOI] [PubMed] [Google Scholar]
- 20.Sykes B. W. W. Norton & Company; New York: 2004. Adam's Curse. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.