

Abstract
Select members of the bovine pancreatic ribonuclease A (RNase A) superfamily are potent cytotoxins. These cytotoxic ribonucleases enter the cytosol, where they degrade cellular RNA and cause cell death. Ribonuclease inhibitor (RI), a cytosolic protein, binds to members of the RNase A superfamily with inhibition constants that span 10 orders of magnitude. Here, we show that the affinity of a ribonuclease for RI plays an integral role in defining the potency of a cytotoxic ribonuclease. RNase A is not cytotoxic and binds RI with high affinity. Onconase, a cytotoxic RNase A homolog, binds RI with low affinity. To disrupt the RI-RNase A interaction, three RNase A residues (Asp-38, Gly-88, and Ala-109) that form multiple contacts with RI were replaced with arginine. Replacing Asp-38 and Ala-109 with an arginine residue has no effect on the RI–RNase interaction. In addition, these variants are not cytotoxic. In contrast, replacing Gly-88 with an arginine residue yields a ribonuclease (G88R RNase A) that retains catalytic activity in the presence of RI and is cytotoxic to a transformed cell line. Replacing Gly-88 with aspartate also yields a ribonuclease (G88D RNase A) with a decreased affinity for RI and cytotoxic activity. The cytotoxic potency of onconase, G88R RNase A, and G88D RNase A correlate with RI evasion. We conclude that ribonucleases that retain catalytic activity in the presence of RI are cytotoxins. This finding portends the development of a class of chemotherapeutic agents based on pancreatic ribonucleases.
Keywords: cancer/onconase/inhibitor
Bovine pancreatic ribonuclease A (RNase A; EC 3.1.27.5) catalyzes the cleavage of RNA (1). Select homologs of RNase A use this activity to effect diverse biological phenomena. Angiogenin (ANG) promotes the growth of new blood vessels (2). Bovine seminal ribonuclease (BS-RNase) displays immunosuppressive, embryotoxic, aspermatogenic, and antitumor activities (3). Onconase (ONC), a ribonuclease from the oocytes and early embryos of the Northern leopard frog (Rana pipiens), is a potent toxin to several transformed cell lines (4). Significantly, the biological activities of ANG, BS-RNase, and ONC depend on their ribonucleolytic activity. Though an efficient catalyst of RNA cleavage, RNase A does not display any pronounced biological action.
Members of the RNase A superfamily have divergent amino acid sequences but similar tertiary structures. The amino acid sequences of BS-RNase A and RNase A, which are from the same species, are 80% identical (5). ONC and human ANG, however, share only 30% and 33% amino acid identity, respectively, with RNase A (6, 7). The majority of nonconserved residues are in surface loops, which appear to play a significant role in the special biological activities of ANG, BS-RNase, and ONC (8, 9). The three-dimensional structure of the pancreatic-type ribonucleases is comprised of a central four-stranded antiparallel β-sheet, flanked by two α-helices. The active site is located in a cleft defined by a third N-terminal α-helix and one edge of the β-sheet. This structure is stabilized by three or four disulfide bonds.
Ribonuclease inhibitor (RI) is a 50-kDa protein that constitutes ≈0.01% of the cytosolic protein in mammalian cells (10–12). Mammalian RIs are highly conserved. For example, porcine RI (pRI) and human RI (hRI) share 77% amino acid sequence identity, with no insertions or deletions except for a four-residue extension on the N terminus of hRI. Both inhibitors contain 15 leucine-rich, β–α repeat units arranged symmetrically in a horseshoe. The β-strands form a solvent-exposed β-sheet that defines the inner circumference of RI. The α-helices define the outer surface of the inhibitor (13). RI forms a 1:1, noncovalent complex with target ribonucleases, including RNase A (14, 15) and ANG (14). Values of the inhibition constant (Ki) are in the fM range. The unusually low Ki values are a consequence of an association rate constant (kon) close to the diffusion limit and a dissociation rate constant (koff) near 10−7⋅s−1 (14–16).
Atomic details of RI–ribonuclease interactions are apparent from the three-dimensional structures of the crystalline pRI⋅RNase A and hRI⋅ANG complexes (17–19). To a first approximation, the two structures are similar. In both, one-third of the ribonuclease rests within the RI horseshoe and the remainder extends out of the plane defined by the inhibitor (Fig. 1A). This arrangement centers each enzyme’s active site on the C terminus of the inhibitor. The intermolecular contacts in the RI⋅ribonuclease complexes differ significantly, but this heterogenety is caused largely by the low sequence identity between RNase A and ANG.
Figure 1.

Molecular interactions between pRI (red) and RNase A (blue). This figure was created with the programs molscript (20) and raster3d (21) by using atomic coordinates derived by x-ray diffraction analysis (17). (A) Three-dimensional structure of the crystalline pRI⋅RNase A complex. The indicated residues were replaced in RNase A variants. (B) Contacts between RI and RNase A near Gly-88 of RNase A. The views in A and B are from opposite sides of the pRI⋅RNase A complex. Trp-259 of pRI corresponds to Trp-264 of hRI.
The inhibitory activity of RI is manifested in the cytosol. This location provides the reducing environment that is necessary to maintain RI activity. Mammalian RIs contain 30 or 32 reduced cysteine residues (22). Oxidation of a single cysteine residue causes rapid oxidation of the remaining cysteine residues and consequent inactivation of RI (23). All known RI ligands, including RNase A, are secreted ribonucleases. This observation and the cytosolic localization of RI supports the hypothesis that the inhibitor functions to preserve the integrity of cellular RNA should a secretory ribonuclease reach the cytosol (11, 24).
The protection offered to cells by RI is limited. The cytotoxic activities of BS-RNase and ONC appear to be a consequence of their abilities to escape inactivation by RI. BS-RNase is isolated as a homodimer, covalently linked by two disulfide bonds. As a dimer, BS-RNase is not inhibited by RI and is cytotoxic. RI becomes a potent inhibitor when BS-RNase is reduced to its monomeric form (8, 25–27). Though monomeric, ONC also evades tight binding by RI (28). ONC retains the elements of tertiary structure that characterize pancreatic-type ribonucleases, but its surface loops are truncated severely compared with their counterparts in RNase A and ANG. Further, the majority of RNase A and ANG residues that contact RI are replaced by dissimilar residues in ONC.
We have directly investigated the relationship between RI inhibition of ribonucleases and ribonuclease cytotoxicity. Specifically, we reasoned that RNase A could be endowed with a cytotoxic activity by specifically decreasing its susceptibility to inactivation by RI. We created two RI-evasive RNase A variants by incorporating amino acid residues that introduce steric and electrostatic strain into the RI⋅RNase A complex. As anticipated, these variants are toxic to a transformed cell line. Our data indicate that ribonuclease cytotoxicity is a direct consequence of an enzyme’s ability to overcome inhibition by RI.
MATERIALS AND METHODS
Materials.
Escherichia coli strain BL21(DE3) and the pET22b(+) expression vector were from Novagen. K-562 cells were from the American Type Culture Collection. Enzymes used for DNA manipulation were from Promega or New England Biolabs. Ribosomal RNA (16S and 23S) was from Boehringer Mannheim. Poly(cytidylic acid) [poly(C)] was from Midland Certified Reagent (Midland, TX). [Methyl-3H]thymidine (6.7 Ci/mmol) was from DuPont/NEN. All other chemicals and reagents were of commercial reagent grade or better and were used without further purification.
DNA oligonucleotides for DNA sequencing and site-directed mutagenesis were from Integrated DNA Technologies (Coralville, IA). Reagents for DNA sequencing, including AmpliTaq DNA polymerase, FS, were from Applied Biosystems. DNA was sequenced by using an Applied Biosystems Automated DNA Sequencer at the University of Wisconsin’s Biotechnology Center.
Terrific broth (29) medium contained (in 1 liter) 12 g of Bacto tryptone (Difco), 24 g of Bacto yeast extract (Difco), 4 ml of glycerol, 2.31 g of KH2PO4, and 12.54 g of K2HPO4. It was prepared in distilled, deionized water and autoclaved. RPMI 1640 medium, fetal bovine serum, and penicillin-streptomycin were from Life Technologies (Gaithersburg, MD). PBS contained (in 1 liter) 0.20 g of KCl, 0.20 g of KH2PO4, 8.0 g of NaCl, and 2.16 g of Na2HPO4⋅7H2O.
UV absorbance measurements were made on a Cary Model 3 spectrophotometer (Varian, Palo Alto, CA) equipped with a Cary temperature controller. Circular dichroism (CD) spectra were collected on an Aviv Model 62A DS CD spectrophotometer (Lakewood, NJ) equipped with an Aviv temperature controller.
Design of Ribonuclease Variants.
Our goal was to alter RNase A so as to perturb only its interaction with RI. We selected target residues in RNase A based on the following criteria. First, the residue must either form a hydrogen bond with RI or make van der Waals contacts (<4 Å) with RI, as defined in the structure of the crystalline pRI⋅RNase A complex (Fig. 1A; refs. 17 and 18). Because of the high sequence identity between pRI and hRI, it is likely that such contacts are preserved in the hRI⋅RNase A complex. Second, the target residues must not be in the RNase A active site. Ribonucleolytic activity is requisite for ribonuclease cytotoxicity (7, 30, 31). Consequently, any amino acid change that diminishes catalytic activity is likely to also reduce cytotoxicity. Third, the target residues must be solvent exposed and confined to the surface loops of RNase A. Substitutions in the enzymic core could decrease the stability of the native three-dimensional structure. Three RNase A target residues were selected (Fig. 1A):
Asp-38 forms a salt bridge with Arg-453 of pRI. In addition, Asp-38 makes van der Waals contacts with Ile-455 of pRI.
Gly-88 lies within a hydrophobic region of pRI defined by Trp-257, Trp-259, and Trp-314 (Fig. 1B). Gly-88 makes one atom-to-atom contact with Trp-257 and six atom-to-atom contacts with Trp-259.
Ala-109 contributes to a hydrophobic pocket that is occupied by Tyr-433 of pRI. The residues make two atom-to-atom contacts in the pRI⋅RNase A crystal structure.
Initially, we replaced target residues in RNase A with arginine to yield D38R RNase A, G88R RNase A, and A109R RNase A. The arginine side chain is the second largest (van der Waals volume: 148 Å3) and most polar of all amino acid side chains (32). Its δ-guanido group is hydrated extensively when exposed to solvent, and this hydration shell significantly increases the effective size of the side chain. Thus, to form the inhibitor⋅enzyme complex, RI must accommodate the polarity and size of the arginine side chain. In an attempt to perturb further the interaction with RI, we combined the changes in single variants to yield two double variants: D38R/G88R RNase A and G88R/A109R RNase A.
We also replaced Gly-88 with an aspartate residue to yield G88D RNase A. The aspartate side chain is the second most polar amino acid side chain (32). Like the δ-guanido group of arginine, the β-carboxylate group of aspartate is hydrated extensively when solvent exposed. In contrast to arginine, the aspartate side chain is anionic and small (van der Waals volume: 91 Å3).
Finally, we replaced Trp-264 of hRI with an alanine residue. As first proposed by Crick (33), receptor-ligand interactions can resemble “knobs-into-holes.” Trp-264 is homologous to Trp-259 of pRI and is in the hydrophobic pocket that surrounds Gly-88 in the RI⋅RNase A complex (Fig. 1B). By making the W264A variant of hRI, we created a “hole” that could better accommodate an RNase A variant with a “knob” at position 88. The relative ability of W264A hRI to inhibit G88R RNase A and G88D RNase A thus could provide new information on the RI⋅RNase A complex.
Production of Ribonucleases.
Plasmid pBXR (34) directs the expression of RNase A in E. coli. Oligonucleotide-mediated site-directed mutagenesis (35) of plasmid pBXR was used to replace Asp-38, Gly-88, and Ala-109 with arginine residues, and to replace Gly-88 with an aspartate residue.
A cDNA that codes for Met(−1)/M23L ONC was a generous gift of R. J. Youle (National Institute of Neurological Disorders and Stroke, National Institutes of Health) (28). This cDNA was modified to produce ONC that is identical to the native protein isolated from frog. The ONC cDNA was amplified with the PCR such that the product has a 5′ blunt end and a 3′ SalI site downstream from the termination codon. The PCR product was inserted into pET22B(+) by using the MscI and SalI sites. Site-directed mutagenesis was used to restore the methionine residue at position 23. The resulting plasmid, pONC, carries a cDNA coding for the wild-type ONC in-frame with the pelB signal sequence.
The RNase A variants and ONC were produced and purified by methods described previously (36), with the following modifications. A culture of E. coli strain BL21(DE3), transformed with the appropriate plasmid, was grown to an OD of 1.6–2.0 at 600 nm in terrific broth medium. Expression was induced by the addition (to 0.5 mM) of isopropyl-1-thio-β-d-galactopyranoside, and cells were collected 3–4 h after induction. After cell lysis with a French pressure cell, inclusion bodies were recovered by centrifugation and resuspended in a solution of 20 mM Tris⋅HCl buffer, pH 8.0, containing 7 M guanidine-HCl, 10 mM DTT, and 10 mM EDTA. The inclusion bodies were solubilized and denatured by stirring at room temperature under N2(g) for 2 h. The protein solution then was diluted 10-fold with 20 mM acetic acid (AcOH), centrifuged to remove precipitant, and dialyzed overnight versus 20 mM AcOH. Material that precipitated during dialysis was removed by centrifugation. Refolding of RNase A and ONC was initiated by diluting the supernatant rapidly to a protein concentration of ≈0.5 mg/ml in 0.10 M Tris-AcOH buffer, pH 8.0, containing 0.10 M NaCl, 3.0 mM reduced glutathione, and 0.6 mM oxidized glutathione. D38R RNase A, G88R RNase A, G88D RNase A, A109R RNase A, D38R/G88R RNase A, and G88R/A109R RNase A were refolded by rapid dilution to a protein concentration of ≈0.25 mg/ml in 0.10 M Tris-AcOH buffer, pH 8.0, containing 0.5 M l-arginine (to prevent protein aggregation), 3.0 mM reduced glutathione, and 0.6 mM oxidized glutathione at 10°C. Samples were concentrated by ultrafiltration with a YM10 membrane (10,000 Mr cut-off; Amicon) and applied to a Superdex G-75 gel filtration FPLC column (Pharmacia) in 50 mM sodium acetate buffer, pH 5.0, containing NaCl (0.10 M) and NaN3 (0.02% wt/vol). Protein from the major peak was collected, concentrated by ultrafiltration, dialyzed versus 50 mM Hepes-HCl buffer, pH 8.0, and applied to a Mono S cation exchange FPLC column (Pharmacia). Ribonucleases were eluted from the column with a linear gradient of NaCl (0.2–0.4 M) in 50 mM Hepes-HCl buffer, pH 8.0. Protein concentrations were determined by UV spectroscopy using extinction coefficients of ɛ278 = 0.72 mg ml−1⋅cm−1 for RNase A (37) and its variants and ɛ280 = 0.87 mg ml−1⋅cm−1 for ONC. The extinction coefficient for ONC was calculated with the method of Pace and coworkers (38).
Production of Ribonuclease Inhibitor.
A cDNA that codes for hRI was a generous gift of Promega. This cDNA was inserted into the pET22b(+) expression vector between the EcoRI and SphI sites to give plasmid pET–RI. Oligonucleotide-mediated site-directed mutagenesis (35) of plasmid pET–RI was used to replace Trp-264 with an alanine residue. A culture of E. coli strain BL21(DE3), transformed with the appropriate plasmid, was grown in terrific broth medium to OD of 1.0 at 600 nm. Expression then was induced by the addition (to 0.5 mM) of isopropyl-1-thio-β-d-galactopyranoside. After an additional 4 h of growth, cells were collected by centrifugation and resuspended in 10 mM potassium phosphate buffer, pH 7.5, containing 0.15 M NaCl, 1 mM EDTA, 1 mM phenylmethanesulfonyl fluoride, and 10 mM DTT. Cells were lysed by two passes through a French pressure cell. Insoluble debris was removed by centrifugation. The remaining steps of the purification, including RNase A–affinity chromatography, were performed as described (39, 40). After elution from the RNase A–affinity column, the hRI was transferred by ultrafiltration to 0.10 M Mes-NaOH buffer, pH 6.0, containing 0.10 M NaCl and 10 mM DTT. The resulting solution was stored in a sealed vial at 4°C. The molar concentration of active wild-type hRI or W264A hRI was measured by titration against a known concentration of RNase A (41), with remaining ribonucleolytic activity monitored by using poly(C) as a substrate (see below).
Thermal Stability Assays.
Because cytotoxicity is assessed by prolonged incubation of ribonucleases at physiological temperature, it is important to establish that the RNase A variants retain adequate thermal stability. Thermal stabilities of the variants were measured by monitoring the change in absorbance at 287 nm (A287) with increasing temperature (42). The temperature of the ribonuclease solutions (0.1–0.2 mg/ml in PBS) was increased from 25°C to 80°C in 1°C increments. The A287 was recorded after a 6-min equilibration at each temperature.
Attempting to measure the thermal stability of ONC with UV spectroscopy yielded uninterpretable thermal denaturation curves. Consequently, the thermal stability of ONC was determined by using CD spectroscopy to monitor the change in molar ellipticity at 204 nM ([Θ]204) with increasing temperature. The temperature of an ONC solution (0.2 mg/ml in PBS) was increased from 50°C to 104°C in 2°C increments. The [Θ]204 was recorded after a 2.5-min equilibration at each temperature.
UV and CD data were fitted to a two-state model for denaturation, and these fits were used to determine values of the midpoint of the thermal denaturation curve (Tm).
Ribonuclease Inhibitor Binding Assays.
The RNase A variants were screened initially for ribonucleolytic activity in the presence of hRI by using an agarose gel-based assay (24, 43). Briefly, 10 ng of ribonuclease was added to a reaction mixture containing 50 mM Tris⋅HCl buffer, pH 7.4, containing DTT (10 mM), 16S- and 23S-rRNA (4 μg), and recombinant wild-type hRI (0, 20, or 40 units, where one unit of hRI is defined as the amount of hRI required to inhibit the ribonucleolytic activity of 5 ng of RNase A by 50%). Each mixture (10 μl) was incubated for 10 min at 37°C. The assays then were stopped by the addition of 2 μl of loading buffer, which was 10 mM Tris⋅HCl buffer, pH 7.5, containing 50 mM EDTA, glycerol (30% vol/vol), xylene cyanol FF (0.25% wt/vol), and bromophenol blue (0.25% wt/vol), and subjected to electrophoresis through a NuSieve agarose gel (1.5% wt/vol) containing ethidium bromide (0.4 μg/ml). This same assay was performed with W254A hRI (20 units).
Values of Ki for the hRI⋅G88R RNase A and the hRI⋅G88D RNase A interactions were determined by measuring the steady-state rate of poly(C) cleavage in the presence of hRI. Reactions were performed at 25°C in Mes-NaOH buffer, pH 6.0, containing 0.10 M NaCl, 50 pM enzyme, and 61–69 μM poly(C). Eight hRI concentrations (25 pM—2.5 nM) were used to determine the value of Ki′ = Ki(1 + S/Km), the apparent inhibition constant for the hRI⋅G88R RNase A complex. Seven hRI concentrations (49 pM—0.49 nM) were used to determine the value of Ki′ for the hRI⋅G88D RNase A complex. Values of Ki′ were calculated by fitting steady-state rates to an equation that describes tight-binding inhibitors (44):
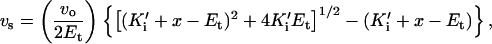 |
1 |
where vo is the rate of poly(C) cleavage in the absence of RI, vs is the steady-state rate, x is the concentration of active hRI, and Et is the concentration of ribonuclease.
Steady-State Kinetics Assays.
Polymeric RNA is hypochromic. The ability of a ribonuclease to catalyze the cleavage of poly(C) (ɛ = 6,200 M−1⋅cm−1 per nucleotide at 268 nm) was monitored by the increase in UV absorption (Δɛ = 2,380 M−1⋅cm−1 at 250 nm) (45). Assays were performed at 25°C in 0.10 M Mes-NaOH buffer, pH 6.0, containing NaCl (0.10 M), poly(C) (5 μM to 1.5 mM), and enzyme (2.5 nM for an RNase A variant; 1.5 μM for ONC). Initial velocity data were used to calculate values of kcat and kcat/Km with the program hypero (46).
Cytotoxicity Assays.
Cytotoxicity assays were conducted with K-562 cells, which are from a continuous human erythroleukemia line. K-562 cells were maintained at 37°C in a humidified atmosphere containing CO2 (g; 5% vol/vol). Culture medium was RPMI 1640 medium supplemented with fetal bovine serum (10% vol/vol), penicillin (100 units/ml), and streptomycin (100 μg/ml). Cytotoxicity was evaluated by measuring [methyl-3H]thymidine incorporation into newly synthesized DNA, as described previously (8, 31, 43, 47). Briefly, aliquots (95 μl) of cultured K-562 cells (5 × 104 cells/ml) were placed in a microtiterplate, and sterile solutions (5 μl) of ribonucleases in PBS were added to the aliquots. Cells were incubated in the presence of ribonucleases for 44 h, followed by a 4-h pulse with [methyl-3H]thymidine (0.20 μCi per well). Cells then were harvested onto glass fiber filters by using a PHD cell harvester (Cambridge Technology; Watertown, MA) and lysed by the passage of several milliliters of water through the filters. DNA and other cellular macromolecules are retained by the filter; small molecules, including unincorporated label, pass through the filters. After washing extensively with water, the filters were dried with methanol and counted by using a liquid scintillation counter. Results from the cytotoxicity assays were expressed as the percentage of [methyl-3H]thymidine incorporated into the DNA of PBS-treated control cells. Data represent the average of triplicate samples within an individual assay. All cytotoxicity assays were repeated at least three times.
RESULTS
Ribonucleases and Ribonuclease Inhibitor.
RNase A, the RNase A variants, and ONC were produced in E. coli. After purification, each ribonuclease migrated as a single species of the appropriate Mr during SDS/PAGE (data not shown), indicating that the pelB signal sequence had been removed by endogenous E. coli proteases. Wild-type RNase A, G88R RNase A, and G88D RNase A had identical CD spectra (data not shown), suggesting that they had a similar tertiary structure. Previous studies had demonstrated that our purification scheme effectively removes all endotoxin from ribonuclease preparations (9). The isolated yields for all ribonucleases were 5–50 mg per liter of E. coli culture.
ONC isolated from frog oocytes has an N-terminal pyroglutamyl residue that contributes to the structure of its active site (48). This N-terminal pyroglutamyl residue is produced by the spontaneous cyclization of an N-terminal glutamine residue. If ONC is produced with an N-terminal methionine residue [Met(−1)], then Gln-1 cannot cyclize to form a pyroglutamate. The resulting protein [Met(−1) ONC] retains only 2% of the ribonucleolytic activity of ONC and is ineffective as a cytotoxin, despite being folded properly (48). To overcome this limitation, a cDNA that codes for ONC was fused in-frame with the pelB signal sequence. As listed in Table 1, catalysis of poly(C) cleavage by this recombinant ONC occurred with steady-state kinetic parameters nearly identical to those reported for ONC isolated from frog oocytes (28). This result suggests that, after removal of the pelB signal sequence, Gln-1 did cyclize to form a pyroglutamyl residue and that the ONC described herein is identical to ONC from frog oocytes.
Table 1.
Ribonuclease thermal stabilities and steady-state kinetic parameters for catalysis of poly(C) cleavage
| Ribonuclease | Tm,* °C | kcat, s−1 | kcat/Km, 106 M−1⋅s−1 |
|---|---|---|---|
| RNase A | 63 | 510 ± 20† | 5.7 ± 0.5† |
| D38R RNase A | 59 | ND | ND |
| G88R RNase A | 60 | 790 ± 20 | 2.9 ± 0.3 |
| G88D RNase A | 64 | 370 ± 10 | 3.2 ± 0.2 |
| A109R RNase A | 64 | ND | ND |
| ONC | 90 | 0.20 ± 0.01 | 0.0014 ± 0.0001 |
Not determined.
Tm values (±2 °C) of RNase A (T. A. Klink and R.T.R., unpublished results) and RNase A variants in PBS were determined by UV spectroscopy. The Tm value (±2 °C) of ONC in PBS was determined by CD spectroscopy.
From ref. 49.
Wild-type hRI and W264A hRI were produced in E. coli. After purification, each hRI migrated as a single species of the appropriate Mr during SDS/PAGE (data not shown). The inhibitory activities of the purified hRIs was quantitated by titrating a sample of RNase A with the inhibitor and recording remaining enzymatic activity. The isolated yields for the two hRIs were ≈1.2 mg (2.6 nmol) per 4 liters of E. coli culture.
Thermal Stability.
To effect a cytotoxic response, either in cell culture or in vivo, a ribonuclease must retain its native conformation for an extended time at physiological temperature. The RNase A amino acid substitutions described herein were designed to weaken the interaction between RNase A and RI without compromising the enzyme’s thermal stability. As listed in Table 1, Tm values of all single variants were within 4°C of RNase A. Consequently, a shift to physiological temperature would cause inconsequential changes to the enzymes’ tertiary structures.
Surprisingly, the Tm of ONC was determined to be 90°C. The remarkable stability of ONC is potentially because of a disulfide bond (Cys-87—Cys-104) that tethers the C terminus to a middle strand of the antiparallel β-sheet (50). High thermal stability had been reported for other members of the RNase A superfamily, a ribonuclease from the liver of R. catesbeiana and ribonucleases from the oocytes of R. catesbeiana and R. japonica (51). Like ONC, each of these ribonucleases has a C-terminal disulfide bond. Moreover, the ribonuclease from R. catesbeiana oocytes is toxic to several transformed cell lines (52).
Binding to Ribonuclease Inhibitor.
An agarose gel-based assay was used to screen for inhibition of the RNase A variants by hRI. As shown in Fig. 2, RNase A was highly sensitive to the effects of hRI. In the absence of hRI, RNase A cleaved rRNA efficiently, producing a faint smear of low Mr molecules. Addition of excess hRI protected the rRNA completely. D38R RNase A and A109R RNase A showed patterns nearly identical to that of RNase A, indicating that these variants, despite the arginine substitutions, remained highly sensitive to hRI. Thus, it appears that RNase A, hRI, or the RI⋅RNase A complex is able to accommodate an arginine residue at positions 38 or 109 and still preserve contacts that are necessary for tight binding.
Figure 2.

Agarose gel-based assay of ribonuclease inhibition by hRI. Inhibition was assessed by visualizing the ribonuclease-catalyzed degradation of 16S- and 23S-rRNA in the absence or presence of excess hRI. − designates experiments that lacked ribonuclease or hRI (or both). (A) Inhibition of a ribonuclease (0 or 10 ng) by wild-type hRI (0, 20, or 40 units). (B) Inhibition of RNase A varying at residue 88 (0 or 10 ng) by wild-type hRI or W264A hRI (0 or 20 units).
In the absence of hRI, G88R RNase A and G88D RNase A displayed enzymatic activity comparable to that of RNase A. In contrast to the native enzyme, addition of excess hRI to reactions containing the Gly-88 variants did not inhibit the cleavage of rRNA. Thus, replacing Gly-88 with an arginine or aspartate residue significantly reduced the susceptibility of RNase A to hRI-mediated inactivation. The additional arginine residue in the double variants, D38R/G88R RNase A and G88R/A109R RNase A, did not enhance resistance to hRI (data not shown). Based on these results, subsequent characterization of the RNase A variants focused on G88R RNase A and G88D RNase A.
In the absence of hRI, ONC catalyzed rRNA cleavage. But, the cleavage products were of higher Mr than those produced by RNase A. This result is consistent with ONC having a lower specific catalytic activity than RNase A. Like G88R RNase A and G88D RNase A, ONC catalyzed rRNA cleavage despite the presence of excess of hRI. Thus, ONC, G88R RNase A, and G88D RNase A are far less sensitive to the inhibitory activity of hRI than is RNase A. As listed in Table 2, the inhibition of G88R RNase A by hRI had Ki = 0.41 nM. RI inhibition of G88D RNase A was more pronounced, with Ki = 0.052 nM. These values are 104-fold and 103-fold greater, respectively, than that for the pRI⋅RNase A interaction.
Table 2.
RI constants by hRI and IC50 values for K-562 cell toxicity
To explore further the interactions between hRI and RNase A near Gly-88, we created a variant of hRI in which Trp-264 is replaced by an alanine residue to yield W264A hRI. As shown in Fig. 2B, wild-type hRI and W264A hRI inhibit the ribonucleolytic activity of wild-type RNase A to a similar extent. W264A hRI is, however, a much more potent inhibitor of G88R RNase A and G88D RNase A than is wild-type hRI. Apparently, RNase A and G88R and G88D variants bind to hRI in a similar orientation. Replacing Trp-264 with an alanine residue creates a pocket that is able to accommodate the side chain of an arginine or aspartate residue, thereby relieving the steric strain that weakens binding to wild-type hRI. Thus, the behavior of residue 88 of RNase A and residue 264 of RI is consistent with the “knobs-into-holes” model of receptor—ligand interaction (33).
Steady-State Kinetics.
Several studies had shown that the biological activities of ribonucleases, including ONC cytotoxicity, rely on the enzymes’ catalytic activity (31, 53). The results of the agarose gel-based assays showed that all of the ribonucleases were active catalysts (Fig. 2) and that G88R RNase A, G88D RNase A, and ONC evaded inhibition by hRI. We evaluated the ribonucleolytic activity of these latter enzymes in detail.
G88R RNase A and G88D RNase A were found to be efficient catalysts of RNA cleavage (Table 1). Their kcat values did not deviate significantly from that of RNase A, and their kcat/Km values were lower by only 2-fold. These high levels of ribonucleolytic activity indicate that the active-site structures and catalytic mechanisms of the RNase A variants were similar to those of RNase A. Moreover, because G88R RNase A and G88D RNase A are thermally stable, the enzymes likely maintain a high level of ribonucleolytic activity at physiological temperature.
ONC was found to be a relatively inefficient catalyst of RNA cleavage. The values of kcat and kcat/Km for ONC were 104-fold lower than those of RNase A (Table 1). This lower catalytic activity is caused, in part, by differences in substrate preference. When assayed for the cleavage of a polymeric substrate, ONC shows a 25-fold preference for poly(uridylic acid) over poly(C) (28). RNase A activity, however, is maximal when assayed for the cleavage of poly(C) (54).
Cytotoxicity.
Our hypothesis is that overcoming inhibition by RI is central to ribonuclease cytotoxicity. Accordingly, the increased Ki values for G88R RNase A and G88D RNase A interactions should be manifested as a cytotoxic activity. The ability of G88R RNase A and G88D RNase A to kill mammalian cells was assessed by measuring [methyl-3H]thymidine incorporation into cellular DNA after a 44-h incubation with the ribonucleases. As shown in Fig. 3 and listed in Table 2, ONC is a potent cytotoxin with IC50 ≅ 0.5 μM. This value is similar to that reported by Youle and coworkers, who measured [35S]methionine incorporation into cellular proteins to assess ONC toxicity to K-562 cells (55). Like ONC, G88R RNase A has a pronounced effect on K-562 cell viability. The IC50 value of G88R RNase A (IC50 ≅ 7 μM) was ≈10-fold greater than that of ONC. The additional arginine residue in D38R/G88R RNase A and G88R/A109R RNase A did not enhance the cytotoxic potency of G88R RNase A (data not shown). Like the G88R enzyme, G88D RNase A inhibited K-562 cell viability, with an IC50 value (IC50 = 30 μM) that was ≈5-fold greater than that of G88R RNase A. At concentrations used in this assay, RNase A had no measurable effect on K-562 cell viability. The IC50 values determined for ONC, G88R RNase A, and G88D RNase A varied by ≤30% in three distinct assays (data not shown).
Figure 3.

Effect of ribonucleases on the proliferation in culture of K-562 cells. Proliferation was measured by incorporation of [methyl-3H]thymidine into cellular DNA after a 44-h incubation with the ribonucleases. Values reported are the mean from three cultures and are expressed as a percentage of the control, which is the mean from cultures lacking exogenous ribonuclease. The standard error of each value is <11%.
DISCUSSION
Attributes of a Cytotoxic Ribonuclease.
A ribonuclease must have several properties to be cytotoxic. First, its structure must be stable at physiological temperature. Otherwise, the ribonuclease would be an inactive catalyst as well as a target for proteolysis. The values of Tm for G88R RNase A, G88D RNase A, and ONC indicate that each of these enzymes retains its native structure during a cytotoxicity assay (Table 1).
Second, a ribonuclease must be an active catalyst. Cytotoxic ribonucleases effect cell death by catalyzing the cleavage of cellular RNA (24, 53, 56). G88R RNase A and G88D RNase A are equally efficient catalysts of RNA cleavage (Table 1). Yet, G88R RNase A is a significantly more potent cytotoxin than is G88D RNase A. Moreover, both RNase A variants are more efficient catalysts than is ONC. Yet, ONC is more cytotoxic than is either G88R RNase A or G88D RNase A. This result indicates that ribonucleolytic activity alone is not responsible for the different cytotoxic potencies of ribonucleases.
Third, a ribonuclease must evade RI. As the inhibition constant (Ki) for a ribonuclease decreases, the IC50 value for cytotoxicity increases (Table 2). Should a tight-binding ribonuclease such as RNase A reach the cytosol, it would be inactivated rapidly by endogenous RI. In contrast, cytotoxic ribonucleases, such as ONC, G88R RNase A, and G88D RNase A, resist inactivation by RI. These ribonucleases then catalyze the degradation of cellular RNA, ultimately causing cell death.
An important conclusion from our data is that wild-type RNase A has all of the properties necessary to be cytotoxic, except for resistance to RI. In other words, ONC is not unique; other monomeric homologs of RNase A can be endowed with potent cytotoxic activity. Other data support this conclusion. For example, monomeric BS-RNase, which is 80% identical to RNase A, is not cytotoxic. Carboxymethylation of Cys-31 in monomeric C32S BS-RNase (to give MCM31) or Cys-32 in monomeric C31S BS-RNase (to give MCM32) diminishes affinity for RI (43). Moreover, MCM31 and MCM32 are aspermatogenic to mice.
Replacing residues 1–9 of human pancreatic ribonuclease with the corresponding residues of ONC decreases the Ki of RI by 28-fold (28). In the presence of retinoic acid, this hybrid ribonuclease is toxic to U251 cells. Under the same conditions, human pancreatic ribonuclease does not affect cell viability. These results add further support to the proposal that ribonuclease susceptibility to RI defines the cytotoxic potential of a ribonuclease.
The cytotoxic potency of a ribonuclease correlates with the presence of ribonucleolytic activity in the cytosol. RI constitutes ≈0.01% of the cytosolic protein in a typical mammalian cell (10, 11). By using this value and data on the volume and composition of the cytosol (57), we estimate that the cytosolic concentration of RI is near 10−6 M. Thus, any ribonuclease with Ki > 10−6 M would be largely free (not bound by RI) in the cytosol. The Ki value of ONC is in that range (28), but those of G88R RNase A and G88D RNase A are not (Table 2). The RNase A variants could compensate for being more susceptible to RI by being better catalysts of RNA degradation (Table 1). Alternatively, K-562 cells could have an unusually low concentration of RI. As a consequence, the Ki values for G88R RNase A and G88D RNase A could approach the RI concentration. Nonetheless, the concentration of cytosolic RI in K-562 cells is sufficient to fully protect these cells from high doses of RNase A (Fig. 3).
Prospectus.
Ribonucleases have much potential as chemotherapeutics. For example, ONC presently is undergoing phase III human clinical trials for the treatment of malignant mesothelioma. Here, we have used rational design to weaken the interaction between RI and RNase A. The consequence of the amino acid substitutions at Gly-88 is a pronounced cytotoxicity to a human leukemic cell line. Because RNase A is a mammalian protein and ONC is an amphibian protein, a chemotherapeutic based on RNase A is likely to be less immunogenic and thus more efficacious than is ONC. Finally, appropriate substitutions to the surface loop containing residue 88 in human pancreatic ribonuclease could endow that enzyme with potent cytotoxicity.
Acknowledgments
We thank Dr. A. D. Attie for use of his tissue culture facilities, Dr. B. Kobe for atomic coordinates of the pRI⋅RNase A complex, Dr. R. J. Youle for ONC cDNA, Promega for hRI cDNA, and M. C. Hebert and B. R. Kelemen for critical reading of the manuscript. CD spectra were obtained at the Biophysics Instrumentation Facility, which is supported by Grant BIR-9512577 (National Science Foundation). P.A.L. was supported by Molecular Biosciences Training Grant T32 GM07215 (National Institutes of Health). L.W.S. was supported by National Institutes of Health Postdoctoral Fellowship CA69750.
ABBREVIATIONS
- ANG
angiogenin
- BS-RNase
bovine seminal ribonuclease
- CD
circular dichroism
- hRI
human ribonuclease inhibitor
- ONC
onconase
- poly(C)
poly(cytidylic acid)
- pRI
porcine ribonuclease inhibitor
- RI
ribonuclease inhibitor
- RNase A
bovine pancreatic ribonuclease A
- Tm
midpoint of the thermal denaturation curve
References
- 1.Raines R T. Chem Rev. 1998;98:1045–1065. doi: 10.1021/cr960427h. [DOI] [PubMed] [Google Scholar]
- 2.Riordan J F. In: Ribonucleases: Structures and Functions. D’Alessio G, Riordan J F, editors. New York: Academic; 1997. pp. 445–489. [Google Scholar]
- 3.D’Alessio G, Di Donato A, Mazzarella L, Piccoli R. In: Ribonucleases: Structures and Functions. D’Alessio G, Riordan J F, editors. New York: Academic; 1997. pp. 383–423. [Google Scholar]
- 4.Youle R J, D’Alessio G. In: Ribonucleases: Structures and Functions. D’Alessio G, Riordan J F, editors. New York: Academic; 1997. pp. 491–514. [Google Scholar]
- 5.Suzuki H, Parente A, Farina B, Greco L, La Montagna R, Leone E. Biol Chem Hoppe Seyler. 1987;368:1305–1312. doi: 10.1515/bchm3.1987.368.2.1305. [DOI] [PubMed] [Google Scholar]
- 6.Fett J W, Strydom D J, Lobb R R, Alderman E M, Bethune J L, Riordan J F, Vallee B L. Biochemistry. 1985;24:5480–5486. doi: 10.1021/bi00341a030. [DOI] [PubMed] [Google Scholar]
- 7.Ardelt W, Mikulski S M, Shogen K. J Biol Chem. 1991;266:245–251. [PubMed] [Google Scholar]
- 8.Kim J-S, Souček J, Matoušek J, Raines R T. J Biol Chem. 1995;270:10525–10530. doi: 10.1074/jbc.270.18.10525. [DOI] [PubMed] [Google Scholar]
- 9.Raines R T, Toscano M P, Nierengarten D M, Ha J H, Auerbach R. J Biol Chem. 1995;270:17180–17184. doi: 10.1074/jbc.270.29.17180. [DOI] [PubMed] [Google Scholar]
- 10.Blackburn P, Moore S. Enzymes. 1982;XV:317–433. [Google Scholar]
- 11.Lee F S, Vallee B L. Prog Nucleic Acid Res. 1993;44:1–30. doi: 10.1016/s0079-6603(08)60215-9. [DOI] [PubMed] [Google Scholar]
- 12.Hofsteenge J. In: Ribonucleases: Structures and Functions. D’Alessio G, Riordan J, editors. New York: Academic; 1997. pp. 621–658. [Google Scholar]
- 13.Kobe B, Deisenhofer J. Nature (London) 1993;366:751–756. doi: 10.1038/366751a0. [DOI] [PubMed] [Google Scholar]
- 14.Lee F S, Shapiro R, Vallee B L. Biochemistry. 1989;28:225–230. doi: 10.1021/bi00427a031. [DOI] [PubMed] [Google Scholar]
- 15.Vicentini A M, Kieffer B, Matthies R, Meyhack B, Hemmings B A, Stone S R, Hofsteenge J. Biochemistry. 1990;29:8827–8834. doi: 10.1021/bi00489a046. [DOI] [PubMed] [Google Scholar]
- 16.Lee F S, Auld D S, Vallee B L. Biochemistry. 1989;28:219–224. doi: 10.1021/bi00427a030. [DOI] [PubMed] [Google Scholar]
- 17.Kobe B, Deisenhofer J. Nature (London) 1995;374:183–186. doi: 10.1038/374183a0. [DOI] [PubMed] [Google Scholar]
- 18.Kobe B, Deisenhofer J. J Mol Biol. 1996;264:1028–1043. doi: 10.1006/jmbi.1996.0694. [DOI] [PubMed] [Google Scholar]
- 19.Papageorgiou A C, Shapiro R, Acharya K R. EMBO J. 1997;16:5162–5177. doi: 10.1093/emboj/16.17.5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 21.Merritt E A, Murphy M E P. Acta Crystallogr D. 1994;50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]
- 22.Kawanomoto M, Motojima K, Sasaki M, Hattori H, Goto S. Biochem Biophys Acta. 1992;1129:335–338. doi: 10.1016/0167-4781(92)90513-y. [DOI] [PubMed] [Google Scholar]
- 23.Blázquez M, Fominaya J M, Hofsteenge J. J Biol Chem. 1996;271:18638–18642. doi: 10.1074/jbc.271.31.18638. [DOI] [PubMed] [Google Scholar]
- 24.Wu Y-N, Mikulski S M, Ardelt W, Rybak S M, Youle R J. J Biol Chem. 1993;268:10686–10693. [PubMed] [Google Scholar]
- 25.Murthy B S, Sirdeshmukh R. Biochem J. 1992;281:343–348. doi: 10.1042/bj2810343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cafaro V, De Lorenzo C, Piccoli R, Bracale A, Mastronicola M R, Di Donato A, D’Alessio G. FEBS Lett. 1995;359:31–34. doi: 10.1016/0014-5793(94)01450-f. [DOI] [PubMed] [Google Scholar]
- 27.Murthy B S, Lorenzo C D, Piccoli R, D’Alessio G, Sirdeshmukh R. Biochemistry. 1996;35:3880–3885. doi: 10.1021/bi952429m. [DOI] [PubMed] [Google Scholar]
- 28.Boix E, Wu Y-N, Vasandani V M, Saxena S K, Ardelt W, Ladner J, Youle R J. J Mol Biol. 1996;257:992–1007. doi: 10.1006/jmbi.1996.0218. [DOI] [PubMed] [Google Scholar]
- 29.Tartof K D, Hobbs C A. Bethesda Res Lab Focus. 1987;9:12. [Google Scholar]
- 30.Sorrentino S, Glitz D G, Hamann K J, Loegering D A, Checkel J L, Gleich G J. J Biol Chem. 1992;267:14859–14865. [PubMed] [Google Scholar]
- 31.Kim J-S, Souček J, Matoušek J, Raines R T. Biochem J. 1995;308:547–550. doi: 10.1042/bj3080547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Radzicka A, Wolfenden R. Biochemistry. 1988;27:1664–1670. doi: 10.1021/bi00412a047. [DOI] [PubMed] [Google Scholar]
- 33.Crick F H C. Nature (London) 1952;170:882–883. doi: 10.1038/170882b0. [DOI] [PubMed] [Google Scholar]
- 34.delCardayré S B, Ribó M, Yokel E M, Quirk D J, Rutter W J, Raines R T. Protein Eng. 1995;8:261–273. doi: 10.1093/protein/8.3.261. [DOI] [PubMed] [Google Scholar]
- 35.Kunkel T A, Roberts J D, Zakour R A. Methods Enzymol. 1987;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- 36.Kim J-S, Raines R T. J Biol Chem. 1993;268:17392–17396. [PubMed] [Google Scholar]
- 37.Sela M, Anfinsen C B, Harrington W F. Biochem Biophys Acta. 1957;26:502–512. doi: 10.1016/0006-3002(57)90096-3. [DOI] [PubMed] [Google Scholar]
- 38.Pace C N, Vajdos F, Fee L, Grimsley G, Gray T. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blackburn P. J Biol Chem. 1979;254:12484–12487. [PubMed] [Google Scholar]
- 40.Lee F S, Vallee B L. Biochem Biophys Res Commun. 1989;160:115–120. doi: 10.1016/0006-291x(89)91628-8. [DOI] [PubMed] [Google Scholar]
- 41.Neumann U, Hofsteenge J. Protein Sci. 1994;3:248–256. doi: 10.1002/pro.5560030209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eberhardt E S, Wittmayer P K, Templer B M, Raines R T. Protein Sci. 1996;5:1697–1703. doi: 10.1002/pro.5560050823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matoušek J, Kim J-S, Souček J, Riha J, Ribó M, Leland P A, Raines R T. Comp Biochem Physiol. 1997;118:881–888. doi: 10.1016/S0305-0491(97)00278-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stone S R, Hofsteenge J. Biochemistry. 1986;25:4622–4628. doi: 10.1021/bi00364a025. [DOI] [PubMed] [Google Scholar]
- 45.delCardayré S B, Raines R T. Biochemistry. 1994;33:6031–6037. doi: 10.1021/bi00186a001. [DOI] [PubMed] [Google Scholar]
- 46.Cleland W W. Methods Enzymol. 1979;63:103–138. doi: 10.1016/0076-6879(79)63008-2. [DOI] [PubMed] [Google Scholar]
- 47.Kim J-S, Souček J, Matoušek J, Raines R T. J Biol Chem. 1995;270:31097–31102. doi: 10.1074/jbc.270.52.31097. [DOI] [PubMed] [Google Scholar]
- 48.Mikulski S M, Grossman A M, Carter P W, Shogen K, Kostanzi J J. Int J Oncol. 1993;3:57–64. doi: 10.3892/ijo.3.1.57. [DOI] [PubMed] [Google Scholar]
- 49.Fisher, B. M., Ha, J.-H. & Raines, R. T. (1998) Biochemistry, in press. [DOI] [PubMed]
- 50.Mosimann S C, Ardelt W, James M N G. J Mol Biol. 1994;236:1141–1153. doi: 10.1016/0022-2836(94)90017-5. [DOI] [PubMed] [Google Scholar]
- 51.Okabe Y, Katayama N, Iwama M, Watanabe H, Ohgi K, Irie M, Nitta K, Kawauchi H, Takayanagi Y, Oyama F, et al. J Biochem (Tokyo) 1991;109:786–790. doi: 10.1093/oxfordjournals.jbchem.a123457. [DOI] [PubMed] [Google Scholar]
- 52.Liao Y-D, Huang H-C, Chan H-J, Kuo S-J. Protein Exp Purif. 1996;7:194–202. doi: 10.1006/prep.1996.0027. [DOI] [PubMed] [Google Scholar]
- 53.Wu W-N, Saxena S, Ardelt W, Gadina M, Mikulski S M, De Lorenzo C, D’Alessio G, Youle R J. J Biol Chem. 1995;270:17476–17481. doi: 10.1074/jbc.270.29.17476. [DOI] [PubMed] [Google Scholar]
- 54.Sorrentino S, Libonati M. Arch Biochem Biophys. 1994;312:340–348. doi: 10.1006/abbi.1994.1318. [DOI] [PubMed] [Google Scholar]
- 55.Newton D L, Walbridge S, Mikulski S M, Ardelt W, Shogen K, Ackerman S J, Rybak S M, Youle R J. J Neuorsci. 1994;14:538–544. doi: 10.1523/JNEUROSCI.14-02-00538.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mastronicola M R, Piccoli R, D’Alessio G. Eur J Biochem. 1995;230:242–249. doi: 10.1111/j.1432-1033.1995.tb20557.x. [DOI] [PubMed] [Google Scholar]
- 57.Lodish H, Baltimore D, Berk A, Zipursky S L, Matsudaira P, Darnell J. Molecular Cell Biology. New York: Scientific American; 1995. [Google Scholar]