

Abstract
Retinal cones are depolarized in darkness, keeping voltage-gated Ca2+ channels open and sustaining exocytosis of synaptic vesicles. Light hyperpolarizes the membrane potential, closing Ca2+ channels and suppressing exocytosis. Here, we quantify the Ca2+ concentration in cone terminals, with Ca2+ indicator dyes. Two-photon ratiometric imaging of fura-2 shows that global Ca2+ averages ∼360 nM in darkness and falls to ∼190 nM in bright light. Depolarizing cones from their light to their dark membrane potential reveals hot spots of Ca2+ that co-label with a fluorescent probe for the synaptic ribbon protein ribeye, consistent with tight localization of Ca2+ channels near ribbons. Measurements with a low-affinity Ca2+ indicator show that the local Ca2+ concentration near the ribbon exceeds 4 μM in darkness. The high level of Ca2+ near the ribbon combined with previous estimates of the Ca2+ sensitivity of release leads to a predicted dark release rate that is much faster than observed, suggesting that the cone synapse operates in a maintained state of synaptic depression in darkness.
Keywords: Cone, Photoreceptor, Ca2+, Synaptic transmission, Exocytosis
Introduction
The biochemical signaling steps of rod and cone phototransduction are known in quantitative detail, but our understanding of how the light response is transmitted to postsynaptic neurons is more limited. We know that sustained depolarization in darkness activates L-type Ca2+ channels in photoreceptor terminals (Corey et al., 1984; Wilkinson & Barnes, 1996; Baldridge et al., 1998) and that intracellular Ca2+ is required for synaptic vesicle exocytosis, which occurs tonically in darkness (Cervetto & Piccolino, 1974). Intraterminal Ca2+ is known to decrease in the light (Johnson et al., 2007), but the concentrations of Ca2+ that underlie synaptic vesicle release in darkness and light have not been accurately quantified with ratiometric Ca2+ indicators.
Previous studies on isolated photoreceptors (Rieke & Schwartz, 1996) provided estimates of Ca2+ in terminals by using voltage clamp to hold the membrane potential near the expected dark and light voltages. In cone terminals, the steady-state Ca2+ concentration is a consequence of influx through ion channels [both L-type Ca2+ channels and cyclic nucleotide-gated (CNG) channels; Savchenko et al., 1997], balanced by cytoplasmic buffering and efflux through the plasma membrane Ca2+-ATPase (Morgans et al., 1998; Krizaj & Copenhagen, 1998). In rods, Ca2+-dependent Ca2+ release from intracellular stores also plays a role in setting the intracellular Ca2+ level (Krizaj et al., 1999, 2003; Cadetti et al., 2006; Suryanarayanan & Slaughter, 2006). We have shown previously that the dark Ca2+ concentration is maintained at a lower concentration in rods than in cones, accounting for the slower dark rate of exocytosis in rods than in cones (Sheng et al., 2007).
Here, we measure Ca2+ in cone synaptic terminals with the fluorescent ratiometric indicator dye, fura-2. To monitor Ca2+ in darkness, we use two-photon microscopy, in which infrared light excites dye fluorescence in cone terminals while minimally exciting phototransduction in outer segments. Infrared light penetrates deeply into tissue, enabling visualization of Ca2+ in cone terminals in the intact retina. To localize subcellular sites of Ca2+ entry, we examine individual cone terminals more closely in retinal slices with low-affinity nonratiometric Ca2+ dyes dialyzed into cells with a patch pipette.
Photoreceptor terminals have synaptic ribbons which play a role in tonic release, but the specific function of the ribbon is unclear (Prescott & Zenisek, 2005). Voltage-gated Ca2+ channels are found adjacent to ribbons (Nachman-Clewner et al., 1999; Morgans, 2001), consistent with local control of Ca2+-dependent exocytosis at the plasma membrane, but other Ca2+-dependent events may also occur at the ribbon (Heidelberger et al., 2005). Measuring and localizing light-induced changes in the Ca2+ concentration will help constrain the possible locations and affinities of Ca2+ sensors for vesicle replenishment and exocytosis, bringing us closer to understanding how neurotransmission is regulated at the ribbon synapse.
Materials and methods
Tissue preparation and dye loading
The retina from the lizard, Anolis segrei, was isolated with the retinal pigment epithelium attached (Choi et al., 2005a,b; Sheng et al., 2007) with procedures approved by the UC Berkeley Animal Care and Use Committee. The retina was prepared and imaged in saline containing (in mM) NaCl 149, KCl 4, CaCl2 1.5, MgCl2 1.5, HEPES 10, Glucose 10 (pH 7.4). First, a membrane-permeant Ca2+ indicator dye, either Oregon Green BAPTA-1-AM (OGB-1) or fura-2-AM, was applied at 100 μM for 2 h to load retinal cells. The saline contained 1% DMSO and 0.2% pluronic acid to enhance dye solubility and cell permeation. Dye-loaded retinas were then mounted flat on nitrocellulose filter paper with the inner retina facing the microscope objective. For dual imaging of synaptic vesicles and Ca2+, the retina was double labeled, first with FM 4-64 (Choi et al., 2005a) and then with fura-2-AM. Retinal preparation and labeling were carried out at 21°C in complete darkness.
The retina from the larval tiger salamander, Ambystoma tigrinum, was isolated with procedures approved by the University of Nebraska Medical Center Institutional Animal Care and Use Committee. Retinal slices were prepared (Rabl et al., 2005), and whole-cell voltage-clamp recordings were obtained with an Optopatch patch-clamp amplifier (Cairn Instruments, Faversham, Kent, UK) with 8–15 Mohm borosilicate glass patch electrodes. The pipette solution contained (in mM) 94 CsGluconate, 9.4 TEACl, 1.9 MgCl2, 9.4 MgATP, 0.5 GTP, 0.5 EGTA, 32.9 HEPES (pH 7.2). To label the synaptic ribbon, a rhodamine-conjugated ribeye peptide (Zenisek et al., 2004) was used at 50 μM.
Imaging
Anole cones were imaged with a Zeiss LSM two-photon microscope equipped with a tunable Mai-Tai laser (Spectraphysics, Mountain view, CA). The excitation wavelength was tuned to 800 nm for OGB-1 and 700 and 760 nm for ratiometric imaging of fura-2. To stimulate cone phototransduction, we used white light from a halogen lamp with intensity of 107 photons/μm2/s, attenuated with neutral density filters. Images were analyzed with Scion Image software (Scion Corporation, Frederick, MD).
The intraterminal Ca2+ concentration of anole cones was calculated from the ratio of emitted light evoked by two-photon dye excitation of fura-2 with 700- and 760-nm light, with standard equations (Grynkiewicz et al., 1985). Calibration constants were determined using a fura-2 calcium imaging calibration kit (Invitrogen) as described previously (Sheng et al., 2007), yielding a Kd of 234 nM (Fig. 1G). A similar midpoint ratio value was obtained in situ by recording from individual lizard cones and dialyzing them with highly buffered, known concentrations of Ca2+. For the midpoint ratio, the pipette solution had a free Ca2+ concentration of 180 nM and consisted of (in mM) 132 KCl, 2 NaCl, 10 EGTA, 3.5 CaCl2, 2 MgCl2, 20 HEPES (pH 7.1, N = 8 cells). The minimum and maximum ratios were obtained by dialyzing solutions containing 0 CaCl2 (N = 4) or 0 EGTA (N = 6).
Fig. 1.
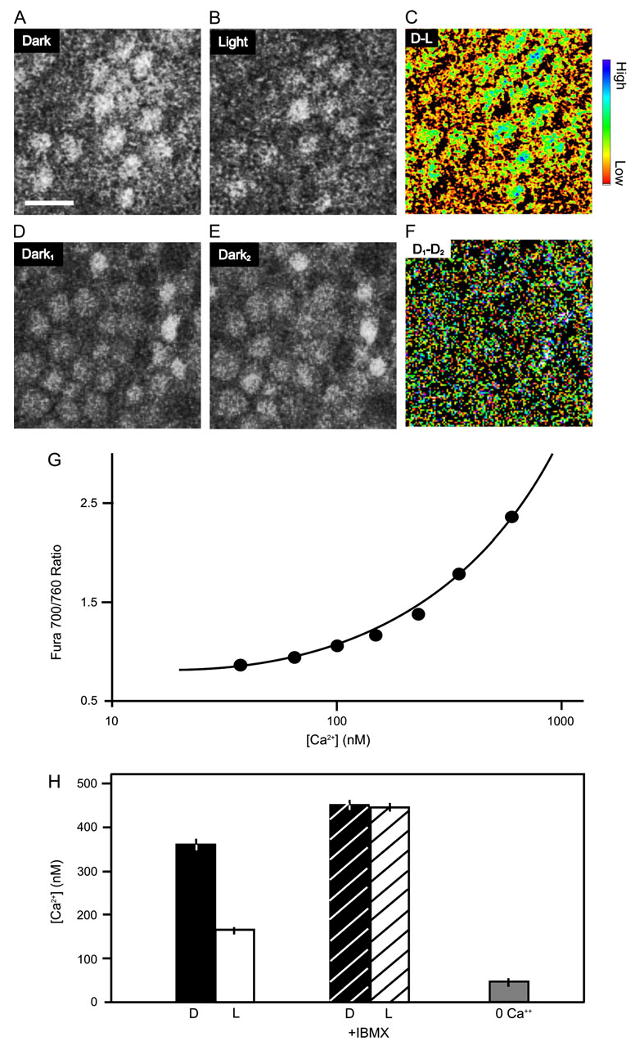
Light-elicited decrease in average Ca2+ in cone terminals. (A-D) Images from two-photon scans of OPL of a flat-mounted anole retina loaded with the Ca2+ dye OGB-1. To reveal the decrease in Ca2+ resulting from light, scans were taken from dark-adapted retina before (A) and after (B) 5-min illumination with white light. Pseudocolor difference image (scan in B − scan in A) highlights the drop in Ca2+ resulting from illumination (C) Scale bar in A is 10 μm. The blue end of the color scale represents a larger decrease in Ca2+; the red end of the scale represents a smaller decrease. To control for possible effects of scanning, control scans were taken before (D) and after (E) a continued period of 5 min in darkness. The difference image (scan in E − scan in D) shows no decrease in Ca2+ (F). Quantification of spatially averaged Ca2+ in cone terminals by two-photon imaging of fura-2. (G) The calibration curve was determined with a fura-2 calcium imaging calibration kit (Invitrogen) and fit with the standard equation (Grynkiewicz et al., 1985), yielding a best-fit Kd of 234 nM. For clarity, only the lower concentration portion of the sigmoidal calibration curve is shown (continuous line). (H) White light stimulation causes a 50% drop in internal Ca2+. Blocking rod and cone PDE with IBMX leads to a rise in Ca2+ above the dark level and eliminates the effect of light. Exposing the retina to Ca2+-free saline for 15 min causes Ca2+ to drop below the light level.
Salamander cones were imaged with a spinning disk confocal microscope (Perkin Elmer Ultraview LCI) equipped with a cooled CCD camera (Orca ER, Hama-matsu Corp., Japan). Images were acquired at 60-ms intervals with single-frame durations of 48–56 ms. Pixel values were binned 2 × 2. Ca2+ indicator dyes were included in the patch pipette at 100 μM and were dialyzed into cells during whole-cell recording. We used three dyes: Oregon Green BAPTA-1 (OGB), Oregon Green BAPTA-6F (OGB-6F), and Oregon Green 488 BAPTA-5N (OGB-5N), with Kd values of 0.17, 3, and 20 μM, respectively, as reported by Molecular Probes (Eugene, OR). We estimated changes in Ca2+ with OGB-5N by using the following equation (Helmchen, 2000):
| (1) |
ΔF/F represents the fractional change in fluorescence resulting from a brief depolarizing step. (ΔF/F)max was determined from the maximal fluorescence change produced by a 500-ms depolarization to −10 mV. Variability in the amount of dye that enters each cell during whole-cell recording produces cell-to-cell differences in absolute fluorescence that prevented in situ determination of the Kd for OGB-5N. We therefore used the Kd value of 20 μM provided by Molecular Probes. There was no added Ca2+ in the pipette solution, and thus, the resting Ca2+ concentration ([Ca2+]rest) was assumed to be 10 nM, but varying this value from 1 to 100 nM had only a small effect (<100 nM) on the calculated value of Δ[Ca2+]i. To validate measurements obtained using eqn (1), we measured fluorescence changes produced by the three different Oregon Green dyes under identical stimulation conditions and obtained similar estimates of intraterminal Ca2+ concentration. This comparison was carried out in salamander rods because recordings are more stable and fluorescent hot spots larger and easier to measure than in cones (unpublished observations).
Results
Illumination causes a decrease in the cone terminal Ca2+ concentration
We first examined light-regulated changes in synaptic Ca2+ in flat mounts of the cone-only retina of the anole lizard. Cone terminals in the outer plexiform layer (OPL) were identified by their loading with the synaptic vesicle marker dye FM4-64 (Rea et al., 2004). The same terminals also label strongly with the nonratiometric Ca2+ indicator Oregon Green BAPTA-1 (OGB) (Fig. 1A). Light stimulation results in a decrease in dye fluorescence, indicating a drop in intraterminal Ca2+ concentration (Fig. 1B). A difference image from scans of the OPL before and after exposure to white light reveals a decrease in Ca2+ in nearly all the cone terminals (Fig. 1C). We controlled for possible effects of the laser scan by constructing a difference image from repeated scans in the dark. Repeated scanning alone caused no change in Ca2+ (Fig. 1D–1F).
To quantify the change in Ca2+ elicited by light stimulation, we used the ratiometric Ca2+ indicator fura-2. Scans of the OPL were obtained with 700- and 760-nm light to minimize photostimulation of cones, and pixel-by-pixel ratio values were converted into Ca2+ concentrations. To enable this conversion, we calibrated the Ca2+ dependence of the 700:760 nm fura-2 excitation ratio in test solutions (Fig. 1G). A complete in situ dye calibration in patch-clamped cones was not possible because high Ca2+ concentrations resulted in cell death. However, we did confirm that dialysis into cones of a solution containing 180 nM free Ca2+ produced an excitation ratio of 1.32 ± 0.04 (n = 8), in good agreement with the ratio predicted from the cell-free calibration (1.31). These measurements indicate that the average Ca2+ concentration in cone terminals was ∼359 ± 24 nM in darkness (n = 8) but dropped to ∼188 ± 10 nM after a 5-min exposure to bright white light (n = 8) (Fig. 1H). We observed that the decrease in Ca2+ elicited by light was slow to develop (>2 min), presumably resulting from slow-buffered diffusion of Ca2+ throughout the cytoplasm of the cone terminal. Hence, the steady-state concentration of Ca2+ in bright light is likely to be underestimated. Consistent with the notion that prolonged cessation of Ca2+ entry would lower Ca2+ further, application of Ca2+-free saline for 15 min resulted in a larger decrease in internal Ca2+ to ∼88 ± 7 nM (n = 4).
To investigate the role of phototransduction in mediating the effect of light on intraterminal Ca2+, we applied the phosphodiesterase (PDE) inhibitor isobutylmethylxanthine (IBMX). We found that IBMX eliminated the light-dark difference and also elevated the basal Ca2+ concentration to ∼450 nM. IBMX inhibition of PDE leads to a supranormal accumulation of cGMP, thereby opening CNG channels in cone terminals (Savchenko et al., 1997) and outer segments (Fesenko et al., 1985). Enhancement of the CNG current can lead to membrane depolarization and greater Ca2+ influx, which could contribute to elevating basal Ca2+ and blunting the light-dark difference in synaptic Ca2+ concentration.
Ca2+ entry is localized to synaptic ribbons
To localize sites of Ca2+ entry in cone terminals, we imaged Ca2+ changes resulting from depolarizing steps. Ca2+ indicator dyes were introduced through an inner segment-attached patch electrode. A long axon separates the terminal from the inner segment of anole cones, so to avoid possible space clamp errors, we used tiger salamander cones, which have a synaptic terminal incorporated directly into the inner segment.
The Ca2+ concentration in the nm-scale region (the “nanodomain”) adjacent to the inner mouth of an open Ca2+ channel is likely to exceed 100 μM (Matthews, 1996; Naraghi & Neher, 1997; Demuro & Parker, 2006). Consistent with this, we found that activating the Ca2+ current by applying depolarizing steps from −70 to −10 mV produced a localized change in Ca2+ that could be visualized with the low-affinity Ca2+ dye, OGB-5N, which has a Kd of 20 μM. Subtraction of the image obtained at −70 mV (Fig. 2A1) from that obtained at −10 mV (Fig. 2A2) yields a difference image (Fig. 2A3), revealing a “hot spot” of Ca2+ in the synaptic terminal region at the base of the cone. We converted fluorescence changes in a 2-μm2 region at the center of the hot spot into Ca2+ levels with eqn (1) (see Methods), yielding an estimate of 8.1 ± 1.1 μM (N = 6). For the experiment in Fig. 2, we plotted Ca2+ changes as a function of time. The graph at the left shows Ca2+ levels measured in the hot spot, and the graph at the right shows Ca2+ levels measured in an adjacent region (just to the upper right of the hot spot) and a region in the center of the soma. The graph shows that Ca2+ levels rise abruptly during the test step and fall quickly afterward. Because diffraction limits of light microscopy blur accurate localization of fluorescence, these measurements underestimate the Ca2+ concentration adjacent to the open pore of a voltage-gated Ca2+ channel. Unlike what is found in rods (Cadetti et al., 2006; Suryanarayanan & Slaughter, 2006), we did not observe Ca2+ waves in response to depolarizing steps of 200 ms or longer, even when we used OGB-1, a high-affinity indicator (data not shown). This is consistent with other observations suggesting little role for Ca2+-induced Ca2+ release in cones (Krizaj et al., 2003).
Fig. 2.
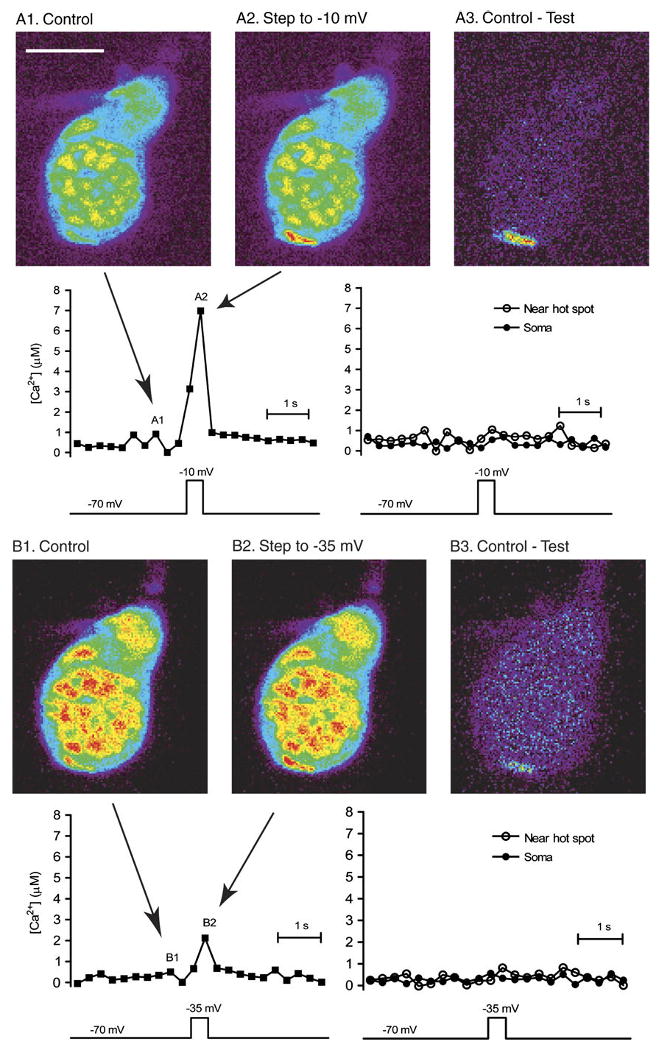
Depolarizing steps to −35 and −10 mV evokes localized Ca2+ increases in voltage-clamped cones that can be detected with the low-affinity Ca2+ dye, OGB-5N. The figure shows pseudocolor images from a single confocal section of a cone filled with OGB-5N. A1 is a control image obtained prior to the depolarizing test step. A2 shows the image obtained during the last 55 ms of a 200-ms depolarizing test step from −70 to −10 mV, which produced a localized fluorescence increase in the synaptic region at the base of the cone. The Ca2+ hot spot can be seen more clearly in the difference image in A3. The graphs show changes in Ca2+ over time for regions within the hot spot (left graph), just to the upper right of the hot spot (right graph), and in the soma (right graph). ΔF/F was converted to [Ca2+] using eqn (1). Images illustrated in the figure were acquired at time points indicated in the left graph. B1 shows a subsequent control image obtained in the same cone, B2 shows an image of the cone obtained during a depolarizing step from −70 to −35 mV, and B3 shows the difference image of the hot spot produced by this modest depolarization. OGB-5N was imaged with 488-nm excitation and 525-nm emission filters. As in panel A, the graphs show [Ca2+] as a function of time for the hot spot, an adjacent region, and the soma. Image acquisition time: 55 ms. Scale bar = 10 μm.
We next examined changes in Ca2+ evoked by depolarization to −35 mV, equivalent to the dark potential of cones. Only a small fraction of the voltage-gated Ca2+ conductance should be active at −35 mV (Thoreson et al., 2003). Nevertheless, steps to −35 mV elicited a clear increase in fluorescence (Fig. 2B), with Ca2+ hot spots reaching levels of several micromolars (4.6 ± 1.5 μM; N = 4). Thus, while Ca2+ levels in the terminal cytoplasm average ∼360 nM in darkness, local Ca2+ levels near the channels are much higher.
We tested whether the sites of Ca2+ influx revealed with these dyes are close to the ribbon, as predicted by immunohistochemistry (Nachman-Clewner et al., 1999; Morgans, 2001; tom Dieck et al., 2005). To label synaptic ribbons, we used a Rhodamine-tagged ribbon-binding peptide (Rh-RBP) that specifically binds to the ribbon protein ribeye (Zenisek et al., 2004). Rh-RBP and the Ca2+-sensitive dye, OGB-6F, were introduced into cones through patch pipettes. Depolarization of the cone elicited a rise in OGB-6F fluorescence, indicating a localized rise in Ca2+. The regions exhibiting the largest change in Ca2+ are again highlighted by subtracting the −70 mV image (Fig. 3A) from the −10 mV image (Fig. 3B). The resulting difference image shows three hot spots of Ca2+ (Fig. 3C). Consistent with the results using OGB-5N (Fig. 2), Ca2+ levels measured with OGB-6F in the hot spot rose to micromolar levels (Fig. 3D). Visualization of Rh-RBP in the same cell showed selective labeling of three spots at the base of the cone (Fig. 3E), where synaptic ribbons are located. Merging the OGB-6F and Rh-RBP images shows that the Ca2+ hot spots overlap with the ribbon labeling (Fig. 3F). Similar co-localization was seen in 11 photoreceptors (six rods and five cones). This suggests that high levels of Ca2+ are attained at the ribbon, the principle site of synaptic vesicle exocytosis.
Fig. 3.
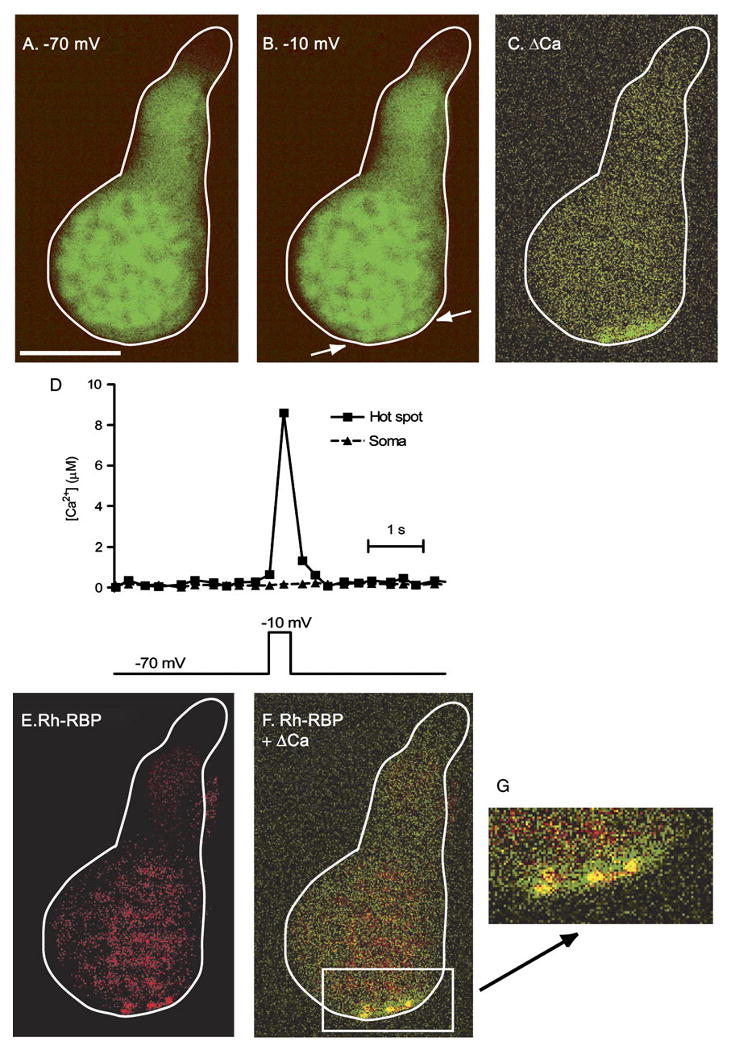
Ca2+ hot spots co-localize with synaptic ribbons. Cone loaded via a patch pipette with the low-affinity Ca2+ indicator dye OGB-6F and the ribbon-specific peptide, Rh-RBP. Fluorescent images were taken at −70 mV (A) and during depolarization to −10 mV (B). The voltage clamp stimulus and times at which images A and B were obtained are illustrated below the images. Difference image (C) shows regions at the base of the terminal exhibiting increased fluorescence (green pixels), indicating a higher Ca2+ concentration. Note the three Ca2+ “hot spots.” OGB-6F was imaged with 488-nm excitation and 525-nm emission filters. Duration of each frame during Ca2+ imaging was 48 ms. (D) The graph shows changes in Ca2+ within the hot spot and soma over time. ΔF/F was converted to [Ca2+] using eqn (1) and a Kd of 3 μM for OGB-6F. (E) Binding of Rh-RBP to synaptic ribbons in the same cone was visualized by exciting the dye with 568-nm excitation and 607-nm emission filters. Note the three spots of bright labeling (red pixels), presumably from three distinct synaptic ribbons. (F) Overlap of the change in OGB-6F fluorescence and Rh-RBP binding (yellow pixels), showing co-localization of Ca2+ hot spots and synaptic ribbons (panel C + panel E). (G) Magnification of the region containing Ca2+ hot spots. Scale bar =10 μm.
It has been reported that the Ca2+ indicator dye Fluo-3 can accumulate on ribbons in hair cell terminals (Issa & Hudspeth, 1996), which could confound identification of local regions having a high Ca2+ concentration. However, OGB-6F fluorescence is weak at the hot spot region when the cell is hyperpolarized (Fig. 3A), indicating no local accumulation of the dye. Moreover, elevating Ca2+ homogenously by flash photolysis of caged Ca2+ (DM-nitrophen) did not produce hot spots (data not shown).
Discussion
Optical measurement of Ca2+ in light and darkness
The signaling steps that link photoreceptor light responses to suppression of neurotransmitter release have been studied chiefly with electrophysiological techniques. Thus, light-induced hyperpolarization (Baylor & Fuortes, 1970), the consequent decrease in voltage-gated Ca2+ current (Corey et al., 1984), and the resulting change in membrane capacitance resulting from Ca2+-dependent exocytosis (Rieke & Schwartz, 1996; Thoreson et al., 2004) were all revealed by electrical recording methods. Only recently has optical imaging been applied to measuring the decrease in synaptic vesicle exocytosis in response to light (Choi et al., 2005a). Here, we used optical methods to visualize the one crucial signaling step between phototransduction and synaptic release that has remained unmeasured, the light-triggered decrease in synaptic Ca2+ concentration.
There are several technical challenges in optically measuring synaptic Ca2+ while keeping cones dark-adapted. The first challenge is minimizing the inadvertent photoisomerization of cone opsins while eliciting fluorescence from Ca2+ indicator dyes. Our solution is to use two-photon microscopy, which excites the dyes with long-wavelength light (>700 nm), largely outside the spectral range of photoreceptor activation. Even so, we are limited to capturing brief and infrequent “snapshots” of Ca2+, as more prolonged or repeated (>5 s) illumination with these wavelengths does indeed elicit a photoresponse of sufficient magnitude to alter synaptic function (Sheng et al., 2007).
A second challenge is localizing Ca2+ signals with high spatial accuracy. Given our illumination parameters, diffraction limits direct microscopic resolution to ∼300 nm (Stelzer, 2000). This blurring of the observed Ca2+ gradient limits the ability to accurately measure highly localized Ca2+ changes, leading to underestimation of their values (Augustine et al., 2003). Nevertheless, our measurements indicate that at a minimum, steady-state levels of Ca2+ at the base of the synaptic ribbon reach several micromolars in darkness. We expect that the local Ca2+ concentration probably reaches much higher levels at release sites, which appear to be located within nanometers of Ca2+ channels.
Roles of Ca2+ in synaptic transmission
In neurons that fire action potentials, voltage-gated Ca2+ channels are tightly localized near sites of synaptic vesicle fusion. This co-localization enables the high Ca2+ concentration in nanodomains near the mouth of an open channel (>100 μM) to saturate the release machinery on neighboring primed vesicles (Augustine et al., 2003; Schneggenburger & Neher, 2005). This arrangement ensures rapid and reliable phasic release of vesicles to a very brief stimulus, namely the action potential.
Photoreceptors, in contrast, generate graded voltage signals and release neurotransmitter in a tonic manner. Photoreceptor synapses have several biochemical and morphological specializations not found in conventional synapses, but how these specializations contribute to tonic release is not well understood. The Ca2+ sensor for neurotransmitter release in rods and cones has an unusually high affinity, enabling release at submicromolar Ca2+ levels (Rieke & Schwartz, 1996; Thoreson et al., 2004; Sheng et al., 2007). This would seem to alleviate the need for high local Ca2+ and therefore close association between Ca2+ channels and Ca2+-sensitive release sites. In fact, it has been suggested that the sensor for neurotransmitter release is so far removed from Ca2+ channels so that vesicle fusion events are not tightly coordinated with individual channel opening events (Rieke & Schwartz, 1996). The present results revealed a twofold change in intraterminal Ca2+ levels between light and dark. A twofold change in Ca2+ levels should produce only a twofold change in synaptic release from cones (Thoreson et al., 2004; Sheng et al., 2007), but release rates vary 25-fold between light and dark (Choi et al., 2005a). These results therefore suggest that larger, local Ca2+ changes are more directly responsible for regulating release than average intraterminal Ca2+ levels.
We found that Ca2+ influx through voltage-gated Ca2+ channels in cones produced hot spots that tightly co-localize with synaptic ribbons, similar to results obtained in hair cells and retinal bipolar cells (Issa & Hudspeth, 1996; Zenisek et al., 2003, 2004). Furthermore, our results showed that micromolar dark Ca2+ concentrations are achieved near the base of ribbons and probably much higher concentrations near individual open Ca2+ channels. Our functional imaging results are also consistent with morphological evidence for Ca2+ channel localization near ribbons. Freeze-fracture Electron Microscopy studies show rows of intramembranous particles, proposed to be Ca2+ channels, at the base of synaptic ribbons (Raviola & Gilula, 1975), and more recent immuno-EM studies show labeling for α1 Ca2+ channel subunits clustered near synaptic ribbons (Nachman-Clewner et al., 1999; Morgans, 2001; tom Dieck et al., 2005).
Our measurements indicate that the average Ca2+ concentration at hot spots in darkness is at least 4 μM and likely to be much higher within 50 nm of voltage-gated Ca2+ channels. Studies using paired recordings from cones and bipolar cells show that depolarization to the dark potential of cones triggers a rapid burst of exocytosis with an instantaneous vesicle fusion rate of several thousand vesicles per second (DeVries & Schwartz, 1999; Rabl et al., 2005). However, measurements of FM1-43 release indicate that cones sustain a tonic release rate of only 250 vesicles per second in darkness (Choi et al., 2005a). This discrepancy between abrupt and tonic release rates would be predicted if release sites were quite distant from Ca2+ channels. However, our results show that Ca2+ channels are clustered close to the synaptic ribbon and average intraterminal Ca2+ changes are too small to account for light–dark changes in release rates. A more likely explanation is that the cone terminal is in a continual state of synaptic depression in darkness. The high release rates stimulated by membrane depolarization cannot be maintained for more than a few milliseconds because release sites are rapidly depleted of vesicles (Rabl et al., 2006). Thus, release from cones declines to slower tonic rates as they remain depolarized in darkness, despite the presence of locally high Ca2+ levels at the ribbon synapse.
Acknowledgments
We thank Katalin Rabl for help with salamander imaging experiments, David Zenisek for providing Rh-RBP, and Robert S. Zucker for helpful discussions. This work was supported by National Institutes of Health grants EY15514 to R.H.K. and EY10542 to W.B.T. and a sabbatical research grant to W.B.T. from Research to Prevent Blindness.
References
- Augustine GJ, Santamaria F, Tanaka K. Local calcium signaling in neurons. Neuron. 2003;40:331–346. doi: 10.1016/s0896-6273(03)00639-1. [DOI] [PubMed] [Google Scholar]
- Baldridge WH, Kurennyi DE, Barnes S. Calcium-sensitive calcium influx in photoreceptor inner segments. J Neurophysiol. 1998;79:3012–3018. doi: 10.1152/jn.1998.79.6.3012. [DOI] [PubMed] [Google Scholar]
- Baylor DA, Fuortes MG. Electrical responses of single cones in the retina of the turtle. J Physiol. 1970;207:77–92. doi: 10.1113/jphysiol.1970.sp009049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadetti L, Bryson EJ, Ciccone CA, Rabl K, Thoreson WB. Calcium-induced calcium release in rod photoreceptor terminals boosts synaptic transmission during maintained depolarization. Eur J Neurosci. 2006;23:2983–2990. doi: 10.1111/j.1460-9568.2006.04845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervetto L, Piccolino M. Synaptic transmission between photoreceptors and horizontal cells in the turtle retina. Science. 1974;183:417–419. doi: 10.1126/science.183.4123.417. [DOI] [PubMed] [Google Scholar]
- Choi SY, Borghuis BG, Rea R, Levitan ES, Sterling P, Kramer RH. Encoding light intensity by the cone photoreceptor synapse. Neuron. 2005a;48:555–562. doi: 10.1016/j.neuron.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Choi SY, Sheng Z, Kramer RH. Imaging light-modulated release of synaptic vesicles in the intact retina: Retinal physiology at the dawn of the post-electrode era. Vision Res. 2005b;45:3487–3495. doi: 10.1016/j.visres.2005.08.011. [DOI] [PubMed] [Google Scholar]
- Corey DP, Dubinsky JM, Schwartz EA. The calcium current in inner segments of rods from the salamander (Ambystoma tigrinum) retina. J Physiol. 1984;354:557–575. doi: 10.1113/jphysiol.1984.sp015393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuro A, Parker I. Imaging single-channel calcium microdomains. Cell Calcium. 2006;40:413–422. doi: 10.1016/j.ceca.2006.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVries SH, Schwartz EA. Kainate receptors mediate synaptic transmission between cones and ‘Off’ bipolar cells in a mammalian retina. Nature. 1999;397:157–160. doi: 10.1038/16462. [DOI] [PubMed] [Google Scholar]
- Fesenko EE, Kolesnikov SS, Lyubarsky AL. Induction by cyclic GMP of cationic conductance in plasma membrane of retinal rod outer segment. Nature. 1985;313:310–313. doi: 10.1038/313310a0. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Heidelberger R, Thoreson WB, Witkovsky P. Synaptic transmission at retinal ribbon synapses. Prog Retin Eye Res. 2005;24:682–720. doi: 10.1016/j.preteyeres.2005.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmchen F. Calibration of fluorescent calcium indicators. In: Yuste R, Lanni F, Konnerth A, editors. Imaging Neurons: A Laboratory Manual. chap 32. New York: Cold Spring Harbor Laboratory Press; 2000. pp. 1–32. [Google Scholar]
- Issa NP, Hudspeth AJ. Characterization of fluo-3 labeling of dense bodies at the hair cell's presynaptic active zone. J Neurocytol. 1996;25:257–266. doi: 10.1007/BF02284801. [DOI] [PubMed] [Google Scholar]
- Johnson JE, Jr, Perkins GA, Giddabasappa A, Chaney S, Xiao W, White AD, Brown JM, Waggoner J, Ellisman MH, Fox DA. Spatiotemporal regulation of ATP and Ca2+ dynamics in vertebrate rod and cone ribbon synapses. Mol Vis. 2007;13:887–919. [PMC free article] [PubMed] [Google Scholar]
- Krizaj D, Bao JX, Schmitz Y, Witkovsky P, Copenhagen DR. Caffeine-sensitive calcium stores regulate synaptic transmission from retinal rod photoreceptors. J Neurosci. 1999;19:7249–7261. doi: 10.1523/JNEUROSCI.19-17-07249.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krizaj D, Copenhagen DR. Compartmentalization of calcium extrusion mechanisms in the outer and inner segments of photoreceptors. Neuron. 1998;21:249–256. doi: 10.1016/s0896-6273(00)80531-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krizaj D, Lai FA, Copenhagen DR. Ryanodine stores and calcium regulation in the inner segments of salamander rods and cones. J Physiol. 2003;547:761–774. doi: 10.1113/jphysiol.2002.035683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews G. Synaptic exocytosis and endocytosis: Capacitance measurements. Curr Opin Neurobiol. 1996;6:358–364. doi: 10.1016/s0959-4388(96)80120-6. [DOI] [PubMed] [Google Scholar]
- Morgans CW. Localization of the δ1F calcium channel subunit in the rat retina. Invest Ophthalmol Vis Sci. 2001;42:2414–2418. [PubMed] [Google Scholar]
- Morgans CW, El Far O, Berntson A, Wässle H, Taylor WR. Calcium extrusion from mammalian photoreceptor terminals. J Neurosci. 1998;18:2467–2474. doi: 10.1523/JNEUROSCI.18-07-02467.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachman-Clewner M, St. Jules R, Townes-Anderson E. L-type calcium channels in the photoreceptor ribbon synapse: Localization and role in plasticity. J Comp Neurol. 1999;415:1–16. [PubMed] [Google Scholar]
- Naraghi M, Neher E. Linearized buffered Ca2+ diffusion in microdomains and its implications for calculation of [Ca2+] at the mouth of a calcium channel. J Neurosci. 1997;17:6961–6973. doi: 10.1523/JNEUROSCI.17-18-06961.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott ED, Zenisek D. Recent progress towards understanding the synaptic ribbon. Curr Opin Neurobiol. 2005;15:43143–43146. doi: 10.1016/j.conb.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Rabl K, Cadetti L, Thoreson WB. Kinetics of exocytosis is faster in cones than rods. J Neurosci. 2005;25:4633–4640. doi: 10.1523/JNEUROSCI.4298-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabl K, Cadetti L, Thoreson WB. Paired-pulse depression at photoreceptor synapses. J Neurosci. 2006;26:2555–2563. doi: 10.1523/JNEUROSCI.3667-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raviola E, Gilula NB. Intramembrane organization of specialized contacts in the outer plexiform layer of the retina. J Cell Biol. 1975;75:192–222. doi: 10.1083/jcb.65.1.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea R, Li J, Dharia A, Levitan ES, Sterling P, Kramer RH. Streamlined synaptic vesicle cycle in cone photoreceptor terminals. Neuron. 2004;4:755–766. doi: 10.1016/s0896-6273(04)00088-1. [DOI] [PubMed] [Google Scholar]
- Rieke F, Schwartz E. Asynchronous transmitter release: Control of exocytosis and endocytosis at the salamander rod synapse. J Physiol. 1996;493:1–8. doi: 10.1113/jphysiol.1996.sp021360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savchenko A, Barnes S, Kramer RH. Cyclic-nucleotide-gated channels mediate synaptic feedback by nitric oxide. Nature. 1997;390:694–698. doi: 10.1038/37803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, Neher E. Presynaptic calcium and control of vesicle fusion. Curr Opin Neurobiol. 2005;15:266–274. doi: 10.1016/j.conb.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Sheng Z, Choi SY, Dharia A, Li J, Sterling P, Kramer RH. Synaptic Ca2+ in darkness is lower in rods than cones, causing slower tonic release of vesicles. J Neurosci. 2007;27:5033–5042. doi: 10.1523/JNEUROSCI.5386-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzer EH. Practical limits to resolution in fluorescence light microscopy. In: Yuste R, Lanni F, Konnerth A, editors. Imaging Neurons: A Laboratory Manual. New York: Cold Spring Harbor Laboratory Press; 2000. pp. 12.1–12.9. [Google Scholar]
- Suryanarayanan A, Slaughter MM. Synaptic transmission mediated by internal calcium stores in rod photoreceptors. J Neurosci. 2006;26:1759–1766. doi: 10.1523/JNEUROSCI.3895-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreson WB, Rabl K, Townes-Anderson E, Heidelberger R. A highly Ca2+-sensitive pool of vesicles contributes to linearity at the rod photoreceptor ribbon synapse. Neuron. 2004;42:595–605. doi: 10.1016/s0896-6273(04)00254-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreson WB, Tranchina D, Witkovsky P. Kinetics of synaptic transfer from rods and cones to horizontal cells in the salamander retina. Neuroscience. 2003;122:785–798. doi: 10.1016/j.neuroscience.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Tom Dieck S, Altrock WD, Kessels MM, Qualmann B, Regus H, Brauner D, Fejtova A, Bracko O, Gundelfinger ED, Brandstätter JH. Molecular dissection of the photoreceptor ribbon synapse: Physical interaction of Bassoon and RIBEYE is essential for the assembly of the ribbon complex. J Cell Biol. 2005;168:825–836. doi: 10.1083/jcb.200408157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson MF, Barnes S. The dihydropyridine-sensitive calcium channel subtype in cone photoreceptors. J Gen Physiol. 1996;197:621–630. doi: 10.1085/jgp.107.5.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenisek D, Davila V, Wan L, Almers W. Imaging calcium entry sites and ribbon structures in two presynaptic cells. J Neurosci. 2003;23:2538–2548. doi: 10.1523/JNEUROSCI.23-07-02538.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenisek D, Horst NK, Merrifield C, Sterling P, Matthews G. Visualizing synaptic ribbons in the living cell. J Neurosci. 2004;24:9752–9759. doi: 10.1523/JNEUROSCI.2886-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]