

Abstract
Background
Primary graft dysfunction (PGD) in the immediate post-lung transplant period strongly increases the risk of chronic rejection (BOS). Here we hypothesized that PGD-induced inflammation augments alloimmunity, thereby predisposing to BOS.
Methods
PGD and BOS were diagnosed according to the established ISHLT criteria. Anti-HLA-alloantibodies were analyzed using Flow-PRA. Donor HLA-class II specific T-cells were analyzed using IFN-γ ELISPOT. Serum levels of 25 cytokines and chemokines were measured using LUMINEX.
Results
Of the 127 subjects, 29(22.8%) had no PGD (grade 0), 42 (33.2%) had PGD-1, 36 (28.3%) had PGD-2, and 20 (15.7%) had PGD-3. PGD+ve patients had elevated proinflammatory mediators MCP-1, IP-10, IL-1β, IL-2, IFN-γ, and IL-12 in the sera during the early post-transplant period. On serial analysis, PGD+ve patients revealed increased development of de novo anti-HLA-II (5 yr: 52.2% Vs PGD-ve 13.5%, p=0.008). However, no difference was found in anti-HLA-I alloantibody development (PGD+ve patients 48% Vs PGD-ve 39.6%, p=0.6). Furthermore, PGD+ve patients had increased frequency of donor HLA class-II specific CD4+ T-cells [(91.4±19.37)×10−6 Vs (23.6±15.93)x10−6, p=0.003].
Conclusion
PGD induces proinflammatory cytokines that can upregulate donor HLA II antigens on the allograft. Increased donor HLA II expression along with PGD-induced allograft inflammation promotes the development of donor specific alloimmunity. This provides an important mechanistic link between early post-transplant lung allograft injury and reported association with BOS.
Keywords: bronchiolitis obliterans, rejection
Introduction
Broncholitis Obliterans Syndrome (BOS) represents chronic lung allograft rejection and remains the predominant cause for poor long-term survival following lung transplantation. BOS develops in about 50% of human lung allograft recipients within three years and over 90% at nine years post transplantation (1). BOS is postulated to have a multifactorial etiology. Nevertheless, alloimmunity constitutes the predominant form of injury that contributes to the pathogenesis of BOS. Sundaresan et al (2) reported that development of HLA class-I antibodies was an independent predictor for the development of BOS. These antibodies precede the development of BOS by about 20 months (3). Jaramillo et al further demonstrated that anti-HLA class-I antibodies activate AEC inducing proliferation and apoptosis. The activated AEC produce growth factors that also lead to smooth muscle and fibro-proliferation, characteristic of BOS histopathology (4). Data from our laboratory (5) and others (6) has further demonstrated that development of HLA class-II antibodies is associated with increased risk of BOS. Similarly, both donor HLA class-I (7) and class-II (5, 8) alloreactive T-cells are increased in patients with BOS suggesting a role for cellular alloimmunity in the pathogenesis of BOS (9).
There is now accumulating evidence that early post-transplant events promote the development of BOS. In a previous study from our laboratory, we found that patients with BOS had elevated pro-inflammatory mediators including IP-10, MCP-1, IL-1β, IL-2, IL-12, and IL-15 during the early post-transplant interval (5). The increase in pro-inflammatory mediators was associated with development donor-specific HLA class-II alloimmunity. However, the cause of elevated pro-inflammatory cytokines in patients with BOS remained unclear. Recently, in a retrospective review of 334 adult lung transplant recipients Hachem et al found that primary lung allograft dysfunction (PGD) was associated with increased risk of BOS (RR 1.73-2.53), independent of acute rejection, lymphocytic bronchitis, and community-acquired respiratory viral infections (10). PGD has been proposed to produce inflammation and amplify the immunogenicity of the allograft. Here we hypothesized that PGD-induced inflammation upregulated MHC expression on the allograft leading to increased alloantigen presentation and production of anti-donor antibodies that are known to contribute to the immunopathogenesis of BOS.
Material and Methods
Study Subjects
Adult patients undergoing lung transplantation at Washington University Medical Center/ Barnes-Jewish Hospital were prospectively enrolled in the study between May 1990 and Jan 2005 after obtaining informed consent, in accordance with a protocol approved by the Institutional Review Board. Only those patients that consented to donate blood at post-transplant follow-up visits were included. The peripheral blood mononuclear cells (PBMCs) were isolated from heparinized blood by Ficoll-Hypaque density gradient centrifugation (Pharmacia, Sweden), and stored in the laboratory sample bank at −135°C until further use. The plasma separated from peripheral blood was stored at −70°C. Patients were excluded if they had HLA antibodies pre-transplant, developed hyperacute rejection, got re-transplanted, or expired within 6 months following transplantation. All patients were free of acute rejection or respiratory infection for at least one month prior to the time of analysis. The standard immunotherapy protocol consisted of cyclosporine, azathioprine, and prednisone. After BOS was diagnosed, the immunotherapy protocol was modified to FK506 (Tacrolimus), mycophenolate mofetil, and prednisone.
Definitions
BOS was diagnosed according to the ISHLT criteria (11) based on the percentage decline in forced expiratory volume in 1 second (FEV1) compared to baseline and graded as follows: 1=80–66% of baseline value, 2=65–51% of baseline value, and 3=50% or less of baseline value. Other causes of decreased lung function such as infection and bronchial anastomotic stricture were ruled out.
PGD was diagnosed immediately following transplant on arrival of the patient to the intensive care unit according to the established definition of the ISHLT (12). Grade 0 is characterized by a partial pressure of arterial oxygen to the fraction of inspired oxygen (PaO2/FiO2) ratio > 300 mm Hg and a clear chest radiograph; Grade 1 by a PaO2/FiO2 ratio > 300 mm Hg and radiographic infiltrates consistent with pulmonary edema; Grade 2 by a PaO2/FiO2 ratio = 200-300 mm Hg and pulmonary infiltrates; and Grade 3 by a PaO2/FiO2 ratio < 200 mm Hg with pulmonary infiltrates. The absence of other potential causes of lung allograft dysfunction such as hyperacute rejection, venous anastomotic complications, cardiogenic pulmonary edema, and pneumonia are implicit in this definition.
Assays and reagents
Flow-PRA analysis for the detection of HLA antibodies was done using flow-cytometry as per the manufacturer’s protocol (One Lambda Inc, CA). The percent PRA is determined by the percent of microparticles that are bound by the antibodies in the serum. PRA ≥ 2.9% for HLA class-I and 2.4% for HLA class-II was considered positive. Serum levels of IL-1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12, IL-13, IL-15, IL-17, Eotaxin, IP-10, MIG, MCP-1, MIP-1α, MIP-1β, RANTES, TNF-α, IFN-α, IFN-γ, GM-CSF, IL-1Rα, and IL-2R were analyzed using the solid phase sandwich multiplex bead LUMINEX immunoassays (Biosource International Inc, CA) according to the manufacturer’s protocols. To detect donor specific anti-HLA class II CD4+ T-cell alloreactivity, we used peptides corresponding to the to the β-chain hypervariable region of the mismatched HLA-DR*0101, HLA-DR*0301, HLA-DR*0701, and HLA-DR*1501 (Research Genetics Inc., Huntsville, AL) (8). These techniques have been described in detail in our previous publication (5).
ELISPOT
ELISPOT assay for IFN-γ was performed as previously described (13). ELISPOT is a recent technique to analyze the frequency of antigen-specific T-cells in a given sample. Briefly, multiscreen 96-well filtration plates (Millipore, MA) were coated with 5.0μg/ml of capture human cytokine-specific mAb (BD Biosciences) at 4 °C overnight. The plates were then blocked with 1% BSA for 2h and washed with PBS. Subsequently, 3×105 PBMCs were cultured in triplicate in the presence of donor HLA-DR peptides (10μg/ml) and irradiated feeder autologous PBMCs (APCs) (1:1 ratio). After 48 h, the plates were washed and then, 2.0μg/ml of a biotinylated human cytokine-specific mAb (BD Biosciences) in PBS/BSA/Tween-20 was added to the wells. After an overnight incubation at 4°C, the plates were washed (3x) and horseradish peroxidase-labeled streptavidin (BD Biosciences), diluted 1:2000 in PBS/BSA/Tween-20, was added to the wells. After 2h, the assay was developed by 3-amino-9-ethylcarbazole substrate reagent (BD Biosciences) for 5–10 min. The plates were washed with tap water to stop the reaction and air-dried. The spots were analyzed in an ImmunoSpot Series I analyzer (Cellular Technology, OH).
Statistical analysis
Continuous data was checked for normality using Shapiro-Wilk test. Non-normal data were transformed with a log transformation. Type 1 error was controlled when performing multiple t-tests using Dunn-Sidak correction. Tabular data were compared using the Fisher’s exact test for 2 × 2 tables and chi-square for 2 × n tables. For more than two group comparisons and analyzing multiple dependent variables, MANOVA was used. The frequency of alloreactive T-cells was compared between PGD0 and PGD1-3 patients using two-tailed t-test. Serial development of HLA antibodies on Flow-PRA was analyzed using the Kaplan-Meir analysis and the groups were compared using logrank test.
Results
Clinical profile of LT patients
The study included 127 adult lung transplant patients. The mean age of this cohort was 57.40±11.96 years, male to female 62:65, primary lung pathology: COPD (62.9%), Alpha-1 antitrypsin (9.5%), Cystic Fibrosis (14.9%), Idiopathic Pulmonary Fibrosis (7.9%), Bronchiectasis (2.4%), and others (2.4%). The patients predominantly underwent bilateral lung transplantation (90.5%). The mean ischemia time for the right and left lung allografts was 278.1±44.3 and 301.0±28.3 minutes, respectively. The patients had a mean of 0.81±0.78 acute rejection episodes during the study period. Mean HLA allele mismatch was 2.54±1.6 for class-I and 0.84±0.8 for class-II antigens. Of the study subjects, 42 (33.2%) had PGD grade 1, 36 (28.3%) had PGD 2, 20 (15.7%) had PGD 3, while 29 (22.8%) had no PGD (grade 0). The clinical and demographic profile of the patients is shown in Table 1. There were no significant differences in lung pathology, allograft ischemia time, acute rejection, and HLA class-I and class-II mismatch between the groups (Table 1).
Table 1.
Clinical and demographic profile of study subjects
| All n=127 |
PGD1-3 n=98(77.2%) |
No PGD n=29(22.8%) |
PGD 1 n=42(33.2%) |
PGD 2 n=36(28.3%) |
PGD 3 n=20(15.7%) |
p | |
|---|---|---|---|---|---|---|---|
| AGE | 57.40 ± 11.96 | 57.38 ± 11.88 | 57.47± 12.46 | 56.39 ± 12.73 | 57.57 ± 9.88 | 59.14 ± 13.60 | NS |
| GENDER | |||||||
| M | 62 (48.8%) | 52 (53.1%) | 10 (34.5%) | 22 (52.4%) | 18 (50%) | 12 (60%) | NS |
| F | 65 (51.2%) | 46 (46.9%) | 19 (65.5%) | 20 (47.6%) | 18 (50%) | 8 (40%) | |
| TRANSPLANT | |||||||
| Single | 12 (9.5%) | 11 (11.2%) | 1 (3.5%) | 3 (7.1%) | 5 (13.9%) | 3 (15%) | |
| Bilateral | 115 (90.5%) | 87 (88.8%) | 28 (96.5%) | 39 (92.9%) | 31 (86.1%) | 17 (85%) | NS |
| PATHOLOGY | |||||||
| COPD | 80 (62.9%) | 61 (62.2%) | 19 (65.5%) | 29 (69.1%) | 20 (55.6%) | 12 (60%) | NS |
| A1A | 12 (9.5%) | 9 (9.2%) | 3 (10.4%) | 3 (7.1%) | 4 (11.1%) | 2 (10%) | |
| CF | 19 (14.9%) | 15 (15.2%) | 4 (13.9%) | 6 (14.3%) | 6 (16.7%) | 3 (15%) | |
| IPF | 10 (7.9%) | 9 (9.2%) | 1 (3.4%) | 4 (9.5%) | 3 (8.3%) | 2 (10%) | |
| Bronchiectasis | 3 (2.4%) | 2 (2.1%) | 1 (3.4%) | 0 | 1 (2.8%) | 1 (5%) | |
| Others | 3 (2.4%) | 2 (2.1%) | 1 (3.4%) | 0 | 2 (5.5%) | 0 | |
|
ISCHEMIA (mins) |
|||||||
| R | 278.1 ± 44.3 | 286.1 ± 64.3 | 271.1 ± 34.3 | 277.3 ± 66.3 | 302.1 ± 57.7 | 292.5 ± 64.9 | NS |
| L | 301.0 ± 28.3 | 313.0 ± 48.6 | 296.0 ± 22.5 | 311.0 ± 33.3 | 321.0 ± 44.6 | 299.2 ± 55.3 | NS |
|
ACUTE REJECTION |
0.81 ± 0.78 | 0.73 ± 0.45 | 0.88 ± 0.98 | 0.63 ± 0.39 | 0.81 ± 0.57 | 0.79 ± 0.66 | NS |
| RACE | |||||||
| Caucasian | 120 (94.5%) | 91 (92.9%) | 29 (100%) | 40 (95.2%) | 35 (97.2%) | 16 (80%) | |
| AA | 6 (4.7%) | 6 (6.1%) | 0 | 2 (4.8%) | 1 (2.8%) | 3 (15%) | |
| Others | 1 (0.8%) | 1 (1.0%) | 0 | 0 | 0 | 1 (5%) | NS |
|
HLA I mismatch |
2.54 ± 1.6 | 2.51 ± 1.7 | 2.66 ± 1.4 | 2.54 ± 1.3 | 2.47 ± 1.9 | 2.49 ± 1.6 | NS |
|
HLA II mismatch |
0.84 ± 0.8 | 0.84 ± 0.7 | 0.86 ± 0.8 | 0.85 ± 0.7 | 0.80 ± 0.6 | 0.83 ± 0.7 | NS |
Upregulation of cytokines in the patients with PGD
To test the hypothesis that PGD induces inflammation, we analyzed serum levels of 25 clinically relevant cytokines and chemokines in the study subjects. Since PGD increases the risk of BOS (10), regardless of grade, we classified patients with PGD grades 1-3 together. Randomly selected serum samples from 20 patients in the PGD0 and 40 patients in PGD1-3 groups were analyzed. Patients were free of acute rejection and respiratory infections at the time of analysis. Furthermore, there was no difference in the type of immunosuppression used in this cohort. As shown in figure 1A, the time interval at which the samples were analyzed post-transplant in the two groups were comparable (PGD0: 3.5±1.4 months Vs PGD1-3: 3.1±1.4 months, p=0.33). Patients with PGD had significantly elevated levels of pro-inflammatory mediators including IP-10 (PGD0: 67±12 Vs PGD1-3 251±21 pg/ml, p=0.03), MCP-1 (PGD0: 212±32 Vs PGD1-3: 1221±56 pg/ml, p=0.01), IL-1β (PGD0: 189±23 Vs PGD1-3: 1124±213 pg/ml, p=0.01), IL-2 (PGD0: 98±23 Vs PGD1-3: 398±121 pg/ml, p=0.03), IFN-γ (PGD0: 56±25 Vs PGD1-3: 312±101, p=0.02) and IL-12 (PGD0: 287±33 Vs PGD1-3: 612±112, p=0.03) (Figure 1B). Although there was a trend of increased pro-inflammatory cytokines with higher grades of PGD, the differences were not statistically significant in this cohort (data not shown). There was no difference in the levels of IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-13, IL-15, IL-17, Eotaxin, MIG, MIP-1α, MIP-1β, RANTES, TNF-α, IFN-α, GM-CSF, IL-1Rα, and IL-2R (data not shown).
Figure 1.

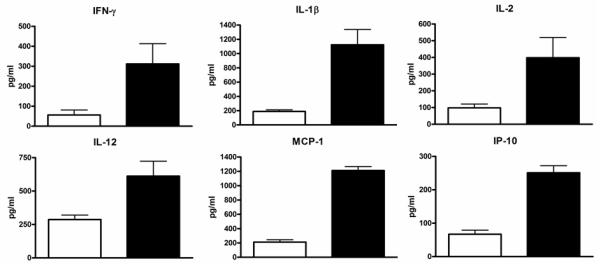
Upregulation of cytokines in the patients with PGD. Serum levels of 25 cytokines and chemokines were analyzed during the early post-transplant period in patients with and without PGD. A) Sampling time points for cytokine analysis were similar between the study groups (p=0.33). B) Pro-inflammatory chemokines IP-10 and MCP-1 and cytokines IFN-γ, IL-1β, IL-2, and IL-12 were found to be elevated in patients with PGD (black bars) compared to those without PGD (white bars). The difference in the serum levels of cytokines were statistically significant (p<0.05 for all). All values are expressed as pg/ml.
Development of alloimmunity in PGD patients
The study subjects were prospectively followed at 1-2 monthly intervals and the development of HLA class-I as well as HLA class-II antibodies analyzed using Flow-PRA. Patients were censored in case of death or development of BOS. The patients included in the study had no detectable HLA antibodies prior to transplantation. As is evident from Figure 2, development of de novo HLA antibodies, both class-I and II, was found to be similar in patients with PGD, regardless of PGD grade. Therefore, PGD grades 1, 2, and 3 were analyzed together. Patients with no PGD demonstrated a significantly decreased incidence of HLA class-II antibodies at 5 years post-transplant (PGD0: 13.5% Vs PGD1-3: 52.2%, p=0.008). However, there was no significant difference in the development of HLA class-I Abs at 5 years (PGD0: 39.6% Vs PGD1-3: 48%, p=0.6).
Figure 2.


Development of alloantibodies in PGD patients. Serial analysis of A) HLA class I and B) II alloantibodies detected by Flow-PRA in study patients. The development of HLA antibodies was similar in patients with PGD, regardless of grade. Therefore, patients with PGD grades 1-3 were classified together (thick solid line) and compared with patients with no PGD (grade 0, thin short broken line). All patients included in the study were negative for HLA alloantibodies prior to transplant.
Since development of alloantibodies is dependent on CD4+ T-helper cells, we next analyzed the frequency of alloreactive donor HLA class II specific CD4+ T-cells to the mismatched HLA-DR alleles. The peripheral blood mononuclear cells (PBMC) obtained from the patients were tested against mismatched donor HLA class-II peptides (Table 2) using IFN-γ ELISPOT assays. These HLA class-II peptides are capable of stimulating CD4+ T-cells after being presented on autologous antigen presenting cells (APC). Therefore, they elicit an indirect alloreactive CD4+ T-cell response. Samples were selected after 90 days following transplant to avoid any confounding effects of induction immunosuppression as well as perioperative stress. The sampling time was similar between groups (PGD1-3: 135 ± 35.0 days Vs PGD0: 125.4 ± 28.0 days, p=0.6). Patients with PGD were found to have increased donor-specific HLA class-II alloreactive IFN-γ producing CD4+ T-cells compared to those without PGD [(91.4 ± 19.37) × 10−6 PBMC Vs (23.6 ± 15.93) × 10−6 PBMC, p=0.003). No difference was found in the frequency of CD4+ T-cells reactive to a third-party (mumps) antigen (PGD1-3: 35.7 ± 18.3 × 10−6 PBMC Vs PGD0: 24.1 ± 13.8 × 10−6 PBMC, p=0.1). The response of 10 normal subjects (mean age 29.7 ± 11.3 years; male: female 6:4) to mumps antigen was not statistically different (25.5 ± 16.9 × 10−6 PBMC, p=0.08). Taken together, these data indicate that PGD promotes the development of donor-specific HLA class-II alloimmunity.
Table 2.
Expansion of donor HLA class II specific alloreactive T cells in patients with PGD
| Patient | HLA-DR Phenotype | Sampling time (days post- transplant) |
Mismatched HLA class II Peptide |
Alloreactive IFN-γ T cell frequency |
|
|---|---|---|---|---|---|
| Recipient | Donor | ||||
| PGD1-3 | |||||
| 1 | DR15,17 | DR07, - | 107 | DR*0701 | 121 × 10−6 |
| 2 | DR16,17 | DR01,17 | 91 | DR*0101 | 71 × 10−6 |
| 3 | DR11,15 | DR07,15 | 146 | DR*0701 | 88 × 10−6 |
| 4 | DR03,04 | DR01,04 | 160 | DR*0101 | 79 × 10−6 |
| 5 | DR02,98 | DR09,15 | 173 | DR*1501 | 98 × 10−6 |
| Mean | 135 ± 35.0 | (91.4 ± 19.37) × 10−6 | |||
| PGD0 | |||||
| 1 | DR13,16 | DR15,16 | 99 | DR*1501 | 21 × 10−6 |
| 2 | DR04,13 | DR07,- | 102 | DR*0701 | 14 × 10−6 |
| 3 | DR04,12 | DR01,04 | 150 | DR*0101 | 51 × 10−6 |
| 4 | DR07,11 | DR03,11 | 116 | DR*0301 | 21 × 10−6 |
| 5 | DR13,15 | DR03,- | 160 | DR*0301 | 11 × 10−6 |
| Mean | 125.4 ± 28.0 | (23.6 ± 15.93) × 10−6 | |||
Comment
The role of early post-transplant inflammation in increasing allograft immunogenicity and subsequent risk of chronic rejection was initially proposed by seminal work done by Land et al (14) that led to the introduction of “response to injury” hypothesis (15). In this study, we investigated the hypothesis that inflammation associated with PGD promotes the development of alloimmunity following lung transplantation leading to the immunopathogenesis of BOS. PGD represents the end-result of multiple insults that begin with donor brain death, aspiration, ventilator associated lung injury, cold ischemia, and reperfusion at the time of implantation (10). Therefore, PGD can induce inflammatory changes in the allograft. The observation that PGD patients had elevated pro-inflammatory mediators including IP-10, MCP-1, IFN-γ, IL-1β, IL-2, and IL-12, in the early post-transplant period supports this notion. IP-10 and MCP-1 belong to the CXC and CC family of chemokines, respectively, and have been previously implicated in allograft rejection (16, 17). They are also known to upregulate HLA class-I and II, costimulatory molecules such as CD80 and CD86 on immune cells, and cell adhesion molecules such as CD54 and CD58 on allograft airway epithelial cells (18). In addition, they activate APC and Th1-lymphocytes (19) that can augment the inflammatory cascade by producing Th1-cytokines. There are multiple studies published in the past that have demonstrated increase in pro-inflammatory cytokines in patients with PGD immediately post-transplant (20, 21). In this report, we included cytokine analysis done several weeks after PGD in order to demonstrate that these patients remain “immunologically charged” for a significant period of time after the initial injury. However, the duration that this inflammation lasts remain unknown from this present study. It is also unclear if other injury mechanisms like acute rejection, gastro-esophageal reflux disease, and respiratory infections propagate the inflammation. Further, the power of the present study was not sufficient enough to correlate the severity of PGD with cytokine levels. A larger study performed longitudinally would be important to address these questions.
Inflammatory mediators including IFN-γ and IL-2 can induce expression of HLA-class II antigens on donor vascular endothelium (22, 23). We further found that IFN-γ significantly increased the expression of HLA class-II antigens both on the larger (bronchial) as well as the small AEC (data not shown). Human and murine studies have demonstrated that donor airway epithelium may constitute the predominant target of BOS pathogenesis (9). Upregulation of HLA class-II molecules on donor cells in PGD patients can increase the donor antigen presentation to the recipient immune system promoting the expansion of alloreactive CD4+ T-cells. The CD4+ T-cells can further augment anti-donor antibody development that was consistent with the higher incidence of de novo HLA class-II alloantibody production observed in PGD patients (Figure 2). Additional inflammatory risk factors such as acute rejection, gastro-esophageal reflux and respiratory infections, would further propagate donor-specific alloimmunity and promote ligation of HLA class-II alloantibodies to AEC by up-regulating HLA class-II antigens. Binding of the alloantibodies to the AEC can produce deleterious effects like complement-mediated cytotoxicity, apoptosis, and production of stress proteins as well as growth factors that lead to smooth muscle cell proliferation and fibrosis (24). The increased risk of BOS from HLA class-II alloimmunity has been previously reported by our laboratory and others (5, 6, 8).
HLA class-I alloimmunity has also been strongly implicated in the pathogenesis of BOS (9). However, in this study, we found that PGD did not significantly increase the development of HLA class-I antibodies. HLA class-I antigens are constitutively expressed on somatic cells of the donor tissue whereas HLA class-II antigens are upregulated due to inflammatory mediators during PGD. PGD can be hypothesized to promote alloimmunity by increasing the donor antigen load and activating immune cells. While in case of donor HLA class-II alloimmunity both mechanisms are effective, only the latter may play a role in the development of HLA class-I alloimmunity. However, this does not exclude the role of HLA class-I alloimmunity in the development of BOS. We postulate that, within the same PGD grade, patients with HLA class-I antibodies would be more likely to develop BOS compared to those without. It is noteworthy that there was a higher incidence of HLA antibody development in this study compared to the previous reports (3, 25, 26). This is most likely due to the longer and close follow-up of patients combined with using the Flow-PRA technique which is known to have higher sensitivity. We also noted that HLA antibodies developed following transplantation can disappear from the sera, most likely due to immunoadsorption at the allograft site. Therefore, cross-sectional or studies without close follow-up of patients may underestimate the development of HLA antibodies.
It is intriguing how PGD can impact the development of antibodies and BOS several years later. There are two pathways of alloantigen recognition: direct and indirect. In the direct pathway, the recipient T cells directly react with the donor antigens present on the donor APC (in context of recipient MHC). In contrast, the indirect pathway involves recognition of processed donor antigens on recipient APC (in context of recipient’s MHC). While the direct pathway is more important for acute allograft rejection, the indirect pathway is postulated to play a dominant role in chronic allograft rejection (27). Interestingly, immunosuppressive agents are effective in depleting the direct alloreactive T cells but not the indirect alloreactive T-cells (28). The presence of donor HLA class-II (indirect) alloreactive T-cells in the early post-transplant period (Table 2) further supports this notion. The development of alloantibodies is dependent on CD4+ T-helper cells that recognize donor antigens through the indirect pathway. These indirectly alloreactive “effector” T-cells in lung transplant patients remain suppressed by the naturally occurring CD4+CD25+foxp3+ regulatory T-cells (29). The long-term outcome following lung transplantation may depend on the balance of regulatory T-cells and effector T-cells. There are mechanisms that can lead to impairment of the regulatory T cell function and lead to a favorable effector T-cell response. For example, the widely used calcineurin inhibitors block the production of IL-2 which is also crucial for regulatory T-cell function. Therefore, while effective in preventing acute rejection, the impact of calcineurin inhibitors on BOS has been limited. Recent data from our laboratory also demonstrates that respiratory viral infections induce apoptosis in regulatory T-cells and promote chronic allograft rejection (manuscript under preparation). In addition, there are mechanisms that can augment inflammation, increase donor antigen load and activate effector T-cells. Besides PGD, these include acute rejection, gastro-esophageal reflux, and respiratory viral infections that have been shown to correlate with increased incidence of BOS. Repeated injuries that produce allograft inflammation also lead to tissue remodeling and development of autoimmunity (29). Both allo- as well as auto- antibodies have the ability to stimulate epithelial and endothelial cells and produce growth factors that result in the gradual process of luminal obliteration seen in BOS.
Acknowledgements
This work was supported by a grant from NIH/NHLBI HL56543. We thank Ms Billie Glascock for her secretarial assistance in preparation of this manuscript.
Footnotes
Presented as the J Maxwell Chamberlain Memorial Paper in General Thoracic Surgery at the 44th annual Meeting of the Society of Thoracic Surgeons, Fort Lauderdale, FL, January 28-30, 2008.
References
- 1.Sundaresan S, Trulock EP, Mohanakumar T, Cooper JD, Patterson GA. Prevalence and outcome of bronchiolitis obliterans syndrome after lung transplantation. Washington University Lung Transplant Group. Ann Thorac Surg. 1995;60(5):1341–1346. doi: 10.1016/0003-4975(95)00751-6. discussion 1346-1347. [DOI] [PubMed] [Google Scholar]
- 2.Sundaresan S, Mohanakumar T, Smith MA, et al. HLA-A locus mismatches and development of antibodies to HLA after lung transplantation correlate with the development of bronchiolitis obliterans syndrome. Transplantation. 1998;65(5):648–653. doi: 10.1097/00007890-199803150-00008. [DOI] [PubMed] [Google Scholar]
- 3.Jaramillo A, Smith MA, Phelan D, et al. Development of ELISA-detected anti-HLA antibodies precedes the development of bronchiolitis obliterans syndrome and correlates with progressive decline in pulmonary function after lung transplantation. Transplantation. 1999;67(8):1155–1161. doi: 10.1097/00007890-199904270-00012. [DOI] [PubMed] [Google Scholar]
- 4.Jaramillo A, Zhang L, Mohanakumar T. Binding of anti-HLA class I antibodies to airway epithelial cells induces activation and growth factor production and indirectly upregulates lung fibroblast proliferation. J Heart Lung Transplant. 2001;20(2):166. doi: 10.1016/s1053-2498(00)00304-1. [DOI] [PubMed] [Google Scholar]
- 5.Bharat A, Narayanan K, Street T, et al. Early posttransplant inflammation promotes the development of alloimmunity and chronic human lung allograft rejection. Transplantation. 2007;83(2):150–158. doi: 10.1097/01.tp.0000250579.08042.b6. [DOI] [PubMed] [Google Scholar]
- 6.Palmer SM, Davis RD, Hadjiliadis D, et al. Development of an antibody specific to major histocompatibility antigens detectable by flow cytometry after lung transplant is associated with bronchiolitis obliterans syndrome. Transplantation. 2002;74(6):799–804. doi: 10.1097/00007890-200209270-00011. [DOI] [PubMed] [Google Scholar]
- 7.SivaSai KS, Smith MA, Poindexter NJ, et al. Indirect recognition of donor HLA class I peptides in lung transplant recipients with bronchiolitis obliterans syndrome. Transplantation. 1999;67(8):1094–1098. doi: 10.1097/00007890-199904270-00002. [DOI] [PubMed] [Google Scholar]
- 8.Reznik SI, Jaramillo A, SivaSai KS, et al. Indirect allorecognition of mismatched donor HLA class II peptides in lung transplant recipients with bronchiolitis obliterans syndrome. Am J Transplant. 2001;1(3):228–235. doi: 10.1034/j.1600-6143.2001.001003228.x. [DOI] [PubMed] [Google Scholar]
- 9.Jaramillo A, Fernandez FG, Kuo EY, Trulock EP, Patterson GA, Mohanakumar T. Immune mechanisms in the pathogenesis of bronchiolitis obliterans syndrome after lung transplantation. Pediatr Transplant. 2005;9(1):84–93. doi: 10.1111/j.1399-3046.2004.00270.x. [DOI] [PubMed] [Google Scholar]
- 10.Daud SA, Yusen RD, Meyers BF, et al. Impact of immediate primary lung allograft dysfunction on bronchiolitis obliterans syndrome. Am J Respir Crit Care Med. 2007;175(5):507–513. doi: 10.1164/rccm.200608-1079OC. [DOI] [PubMed] [Google Scholar]
- 11.Yousem SA, Berry GJ, Cagle PT, et al. Revision of the 1990 working formulation for the classification of pulmonary allograft rejection: Lung Rejection Study Group. J Heart Lung Transplant. 1996;15(1 Pt 1):1–15. [PubMed] [Google Scholar]
- 12.Christie JD, Carby M, Bag R, Corris P, Hertz M, Weill D. Report of the ISHLT Working Group on Primary Lung Graft Dysfunction part II: definition. A consensus statement of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2005;24(10):1454–1459. doi: 10.1016/j.healun.2004.11.049. [DOI] [PubMed] [Google Scholar]
- 13.Jaramillo A, Majumder K, Manna PP, et al. Identification of HLA-A3-restricted CD8+ T cell epitopes derived from mammaglobin-A, a tumor-associated antigen of human breast cancer. Int J Cancer. 2002;102(5):499–506. doi: 10.1002/ijc.10736. [DOI] [PubMed] [Google Scholar]
- 14.Land W, Schneeberger H, Schleibner S, et al. The beneficial effect of human recombinant superoxide dismutase on acute and chronic rejection events in recipients of cadaveric renal transplants. Transplantation. 1994;57(2):211–217. doi: 10.1097/00007890-199401001-00010. [DOI] [PubMed] [Google Scholar]
- 15.Land WG. The role of postischemic reperfusion injury and other nonantigen-dependent inflammatory pathways in transplantation. Transplantation. 2005;79(5):505–514. doi: 10.1097/01.tp.0000153160.82975.86. [DOI] [PubMed] [Google Scholar]
- 16.Belperio JA, Keane MP, Burdick MD, et al. Critical role for the chemokine MCP-1/CCR2 in the pathogenesis of bronchiolitis obliterans syndrome. J Clin Invest. 2001;108(4):547–556. doi: 10.1172/JCI12214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Melter M, Exeni A, Reinders ME, et al. Expression of the chemokine receptor CXCR3 and its ligand IP-10 during human cardiac allograft rejection. Circulation. 2001;104(21):2558–2564. doi: 10.1161/hc4601.098010. [DOI] [PubMed] [Google Scholar]
- 18.Nakajima J, Ono M, Takeda M, Kawauchi M, Furuse A, Takizawa H. Role of costimulatory molecules on airway epithelial cells acting as alloantigen-presenting cells. Transplant Proc. 1997;29(4):2297–2300. doi: 10.1016/s0041-1345(97)00334-5. [DOI] [PubMed] [Google Scholar]
- 19.Baggiolini M, Dewald B, Moser B. Human chemokines: an update. Annu Rev Immunol. 1997;15:675–705. doi: 10.1146/annurev.immunol.15.1.675. [DOI] [PubMed] [Google Scholar]
- 20.Mathur RS, Jenkins JM, Bansal AS. The possible role of the immune system in the aetiopathogenesis of ovarian hyperstimulation syndrome. Hum Reprod. 1997;12(12):2629–2634. doi: 10.1093/humrep/12.12.2629. [DOI] [PubMed] [Google Scholar]
- 21.Fisher AJ, Donnelly SC, Hirani N, et al. Elevated levels of interleukin-8 in donor lungs is associated with early graft failure after lung transplantation. Am J Respir Crit Care Med. 2001;163(1):259–265. doi: 10.1164/ajrccm.163.1.2005093. [DOI] [PubMed] [Google Scholar]
- 22.Cunningham AC, Zhang JG, Moy JV, Ali S, Kirby JA. A comparison of the antigen-presenting capabilities of class II MHC-expressing human lung epithelial and endothelial cells. Immunology. 1997;91(3):458–463. doi: 10.1046/j.1365-2567.1997.d01-2249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zissel G, Ernst M, Rabe K, et al. Human alveolar epithelial cells type II are capable of regulating T-cell activity. J Investig Med. 2000;48(1):66–75. [PubMed] [Google Scholar]
- 24.Jaramillo A, Smith CR, Maruyama T, Zhang L, Patterson GA, Mohanakumar T. Anti-HLA class I antibody binding to airway epithelial cells induces production of fibrogenic growth factors and apoptotic cell death: a possible mechanism for bronchiolitis obliterans syndrome. Hum Immunol. 2003;64(5):521–529. doi: 10.1016/s0198-8859(03)00038-7. [DOI] [PubMed] [Google Scholar]
- 25.Girnita AL, Duquesnoy R, Yousem SA, et al. HLA-specific antibodies are risk factors for lymphocytic bronchiolitis and chronic lung allograft dysfunction. Am J Transplant. 2005;5(1):131–138. doi: 10.1111/j.1600-6143.2004.00650.x. [DOI] [PubMed] [Google Scholar]
- 26.Smith MA, Sundaresan S, Mohanakumar T, et al. Effect of development of antibodies to HLA and cytomegalovirus mismatch on lung transplantation survival and development of bronchiolitis obliterans syndrome. J Thorac Cardiovasc Surg. 1998;116(5):812–820. doi: 10.1016/S0022-5223(98)00444-9. [DOI] [PubMed] [Google Scholar]
- 27.Benichou G, Valujskikh A, Heeger PS. Contributions of direct and indirect T cell alloreactivity during allograft rejection in mice. J Immunol. 1999;162(1):352–358. [PubMed] [Google Scholar]
- 28.Sawyer GJ, Dalchau R, Fabre JW. Indirect T cell allorecognition: a cyclosporin A resistant pathway for T cell help for antibody production to donor MHC antigens. Transpl Immunol. 1993;1(1):77–81. doi: 10.1016/0966-3274(93)90063-e. [DOI] [PubMed] [Google Scholar]
- 29.Bharat A, Fields RC, Steward N, Trulock EP, Patterson GA, Mohanakumar T. CD4+CD25+ regulatory T cells limit Th1-autoimmunity by inducing IL-10 producing T cells following human transplantation. American Journal of Transplantation. 2006;6(8):1799–1808. doi: 10.1111/j.1600-6143.2006.01383.x. [DOI] [PubMed] [Google Scholar]