


Abstract
The quantitative polymerase chain reaction (qPCR) is widely utilized for gene expression analysis. However, the lack of robust strategies for cross laboratory data comparison hinders the ability to collaborate or perform large multicentre studies conducted at different sites. In this study we introduced and validated a workflow that employs universally applicable, quantifiable external oligonucleotide standards to address this question. Using the proposed standards and data-analysis procedure, we obtained a perfect concordance between expression values from eight different genes in 366 patient samples measured on three different qPCR instruments and matching software, reagents, plates and seals, demonstrating the power of this strategy to detect and correct inter-run variation and to enable exchange of data between different laboratories, even when not using the same qPCR platform.
INTRODUCTION
Gene expression quantification has an important role in many fields of biology, amongst others in the field of clinical diagnostics and fundamental research. From the various methods available, reverse transcription quantitative polymerase chain reaction (RT–qPCR) is the most rapid, sensitive, accurate and precise method that can be used to quantify the expression levels of selected genes and its use in the field of clinical diagnostics is presently growing (1–5). Compared with microarrays, the amount of required RNA as starting material is much lower for RT–qPCR and archival material such as formalin-fixed and paraffin-embedded tissues can be successfully used as template for RT–qPCR. Moreover, the arrival of a new generation of ultra high-throughput microfluidic based RT–qPCR systems opens up the perspective of measuring thousands of genes in parallel. Nevertheless, a major drawback of most gene expression studies is the difficulty or impossibility to compare data generated in different laboratories. Indeed, the use of different instruments, software, reagents, plates or seals can lead to often underestimated run-to-run differences that need to be compensated in order to make data comparable. Currently available strategies to standardize quantitative polymerase chain reaction (qPCR) data, such as Standardized Reverse Transcriptase PCR (StaRT–PCR), are based on internal standards (6,7). This method relies on end-point quantification and is only commercially available through Gene Express. In this paper, we evaluate a strategy that employs quantifiable external oligonucleotide standards to detect and correct inter-experimental variation. Compared to previously described methods our strategy is universally applicable and offers a high level of flexibility. We show that true multicentre collaborations are possible and that data can actually be compared in one study.
MATERIALS AND METHODS
Sample preparation
Total RNA extraction from 423 fresh frozen neuroblastoma tumour samples was done by silica gel-based membrane purification (RNeasy Mini kit or MicroRNeasy kit, Qiagen), or phenol-based (TRIzol reagent, Invitrogen and Tri Reagent product, Sigma) or chaotropic solution-based isolation methods (Perfect Eukaryotic RNA kit, Eppendorf) according to the manufacturer’s instructions and stored at −80°C. All tumour samples were frozen immediately after removal from the patient and stored at −80°C. A validated sample pre-amplification method was applied yielding sufficient cDNA (∼6 µg stored at −80°C) to measure more than 1000 target genes using only 20 ng of total RNA as starting material (WT-Ovation, NuGEN) (8,9). DNAse treatment was not performed as DNA is not co-amplified using the described earlier sample pre-amplification method (Vermeulen et al., in preparation). In order to assess the RNA quality of the 423 collected tumour samples, we used 20 ng of each RNA isolate to perform a PCR-based SPUD assay for the detection of enzymatic inhibitors in nucleic acid preparations (10) and a microfluidic capillary electrophoresis analysis (high sensitivity chips, Experion, software version 3.0, Bio-Rad) to establish an RNA quality index (RQI) based on the ribosomal RNA profile. Based on these tests, we retained the 366 best quality samples (median RQI: 7.6; 90th percentile RQI > 6.1, absence of enzymatic inhibitors).
High-throughput real-time quantitative PCR based gene expression
A qPCR assay was designed for 8 genes by PrimerDesign (Southampton, UK). Amplicon length was comprised between 120 and 150 base pairs. All assays went through an extensive in silico validation analysis using BLAST and BiSearch specificity, amplicon secondary structure, SNP presence and splice variant analysis (11). A standard dilution series was used to test the PCR efficiency of the primers and only primers with an efficiency between 90 and 110% were retained (mean efficiency of the 8 assays: 95.4% (range 91.1–99.6%) (Supplementary Table S1).
qPCR was done on all three currently available high-throughput 384-well plate instruments (LC480 from Roche (second derivative Cq value determination method), 7900HT from Applied Biosystems (baseline/threshold Cq value determination method), and CFX384 from Bio-Rad (derivative Cq value determination method)) (Supplementary Table S2). PCR plates were prepared using a 96-well head pipetting robot (Tecan Freedom Evo 150). qPCR amplifications were performed in 8 µl containing 4 µl 2× SYBR Green I master mix (LC480 SYBR Green I master (Roche), custom made qPCR SYBR green I Mastermix (Eurogentec) or iQ SYBR Green Supermix (Bio-Rad)), 0.4 µl forward and reverse primer (5 µM each), 0.2 µl nuclease-free water and 3 µl WT-Ovation amplified cDNA (corresponding to 4.5 ng of unamplified cDNA, total RNA equivalents) or 3 µl of standard oligonucleotides (see further). All reactions were performed in 384-well plates (LightCycler 480 Multiwell Plates 384, white and LightCycler 480 Sealing Foils from Roche on the LC480; MicroAmp Optical 384-Well Reaction Plates with Barcode and ABsolute QPCR Seals from Applied Biosystems on the 7900HT; and Hard-Shell 384-well microplates and Microseal ‘B’ clear adhesive seals from Bio-Rad on the CFX384). The cycling conditions were comprised of 3 min (10 min when using Eurogentec mastermix) polymerase activation at 95°C and 45 cycles of 15 s at 95°C and 30 s at 60°C followed by a dissociation curve analysis from 60 to 95°C.
For data analysis, the Cq values of the genes were converted to relative quantities and normalized using the geometric mean of three reference genes (HPRT1, SDHA and UBC) (12), followed by inter-run calibration (IRC) using the standards as inter-run calibrators. Data handling and calculations (normalization, IRC, rescaling and error propagation) were performed in qBasePlus version 1.2 (http://www.qbaseplus.com) (13).
External oligonucleotide standards
A standard was designed for all eight genes. The sequence of each standard consists of the forward primer sequence of that particular gene, a stuffer sequence (sequence consisting of an ACTG repeat) in the middle and the reverse complement sequence of the reverse primer of that gene at the end (total length of 55 nucleotides) (Supplementary Table S1). All standard oligonucleotide sequences were analysed for secondary structure using the DINAMelt Server powered by UNAFold (http://dinamelt.bioinfo.rpi.edu/quikfold.php) and the stuffer sequence was slightly modified in case of formation of a secondary structure. The standard oligonucleotides were PAGE purified and blocked at their 3′-end with a phosphate group to avoid participation in the PCR amplification process (Biolegio, the Netherlands). Manufacturer’s supplied concentration of each oligonucleotide was confirmed using the Nanodrop 1000 Spectrophotometer (Thermo Scientific). All eight standards were pooled together at equimolar concentrations and a dilution series consisting of five 10X serial dilution points, starting from 150 000 molecules down to 15 molecules was created using 10 ng/µl yeast tRNA as carrier. The standards were run in parallel with the samples for each gene using the sample maximisation experiment design (13).
Terminology and data
According to the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) and Real-time PCR Data Markup Language (RDML) guidelines (14,15) we used the proposed terms for the plethora of available descriptions [e.g. quantification cycle value (Cq) as unit of measurements].
RDML is a structured and universal data standard for exchanging qPCR data (http://www.rdml.org). (Supplementary Data, Vermeulen2.rdml).
IRC
IRC can be performed on Cq or normalized relative quantity (NRQ) level (Supplementary Figure S1—‘calculation workflow’).
For IRC on the Cq level, we outline the formulas below. For every replicated PCR reaction r, dilution d, gene g and platform p, we first calculated the mean replicate Cq value (formula 1), followed by the difference in mean Cq between two different platforms j and k (k being a randomly selected reference platform) for a given dilution d and gene g (formula 2). The average difference in Cq value for all dilution points for a given gene g measured on two different platforms j and k (formula 3) is then used as the gene specific Cq IRC factor to obtain calibrated Cq values (CCq) through calibration of gene g Cq values coming from platform j using k as reference platform (formula 4) (Supplementary Table S3—example).
| 1 |
| 2 |
| 3 |
| 4 |
where n is the number of PCR replicates (r); m the number of dilution points (d) used for the serial dilution curve; s the number of platforms (p); t the number of genes (g).
For IRC on the NRQ level, we used the procedure outlined in Hellemans et al. (13) and implemented in the qBasePlus software (http://www.qbaseplus.com). Briefly, using an equimolar mixture of all eight external standards as inter-run calibrators, a gene and run specific calibration factor (CF) was calculated as the geometric mean of the inter-run calibrator NRQ values measured in each run for a given gene. The NRQ values of all samples were subsequently converted to calibrated NRQ (CNRQ) values by division by the cognate CF.
Gene expression based class prediction
For establishment of a five-gene expression correlation signature (ARHGEF7, HIVEP2, MRPL3, NRCAM and TNFRSF25), the samples were divided into a training and test set. The training set was comprised of 30 randomly selected samples from two patient subgroups with maximally divergent clinical courses: 15 low risk survivors and 15 high risk deceased patients. The expression signature was built using these 30 training samples by calculating the difference between the mean log transformed expression in the low and high risk groups for each of the five target genes. Subsequently, the resulting classifying vector was tested on the remaining test samples by determining the Pearson’s correlation coefficient between the expression signature and the expression profile of a given test sample. A class label was attributed based on positive (bad prognosis) or negative correlation (good prognosis) with the signature (16).
Statistical analysis
Correlation analysis between calibrated normalized relative gene expression levels was performed using Spearman's; rank method.
The R language for statistical computing was used to train and test the correlation signature.
RESULTS
In order to validate the utility of the external oligonucleotide standards, we measured the expression of 8 different genes (five target and three reference genes) in 366 samples using three different commercial PCR reagents, plates and seals and all three 384-well plate real-time PCR instruments and matching software currently available on the market (Supplementary Data, Vermeulen2.rdml). A five-point serial dilution series in triplicate, starting from 150 000 molecules down to 15 molecules, was run in parallel with the samples and used for IRC (Supplementary Table S2 and Supplementary Figure S2).
Comparison of Cq values before and after IRC
Before IRC, the absolute average difference in Cq value of the 366 samples measured on two different platforms 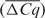 was higher than 1 in 75% of the cases and higher than 2 in 42% of the cases. After Cq level IRC,
was higher than 1 in 75% of the cases and higher than 2 in 42% of the cases. After Cq level IRC, 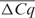 was lower than 0.5 in 75% of the cases and lower than 1 in all cases [mean reduction of 1.4 Cq values (range 0.52–2.86)] (Table 1).
was lower than 0.5 in 75% of the cases and lower than 1 in all cases [mean reduction of 1.4 Cq values (range 0.52–2.86)] (Table 1).
Table 1.
Pairwise IRC on Cq or NRQ level using a five-point serial dilution series of external standards run in parallel with the 366 patient samples on three different qPCR platforms
| Before IRC | After IRC |
|||
|---|---|---|---|---|
| Cqa | CCqb |
CNRQ |
||
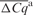 |
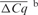 |
r | FC | |
| 7900HT versus LC480 | ||||
| ARHGEF7 | 2.63 (±0.22) | 0.25 (±0.16) | 0.98 | 1.53 (±0.40) |
| HIVEP2 | 2.78 (±0.49) | 0.65 (±0.46) | 0.94 | 1.34 (±0.68) |
| HPRT1 | 2.46 (±0.22) | 0.13 (±0.18) | 0.93 | 1.29 (±0.34) |
| MRPL3 | 3.00 (±0.18) | 0.14 (±0.14) | 0.95 | 1.29 (±0.33) |
| NRCAM | 2.42 (±0.18) | 0.14 (±0.11) | 0.94 | 1.37 (±0.37) |
| SDHA | 2.63 (±0.67) | 0.47 (±0.64) | 0.95 | 1.26 (±0.67) |
| TNFRSF25 | 3.22 (±0.49) | 0.98 (±0.49) | 0.94 | 1.56 (±0.52) |
| UBC | 2.98 (±1.26) | 0.68 (±0.29) | 0.95 | 1.32 (±0.37) |
| Averagec | 2.77 (±0.28) | 0.43 (±0.32) | 1.37 (±0.11) | |
| 7900HT versus CFX384 | ||||
| ARHGEF7 | 1.75 (±0.19) | 0.12 (±0.18) | 0.97 | 1.24 (±0.21) |
| HIVEP2 | 1.86 (±0.44) | 0.51 (±0.39) | 0.91 | 1.23 (±0.29) |
| HPRT1 | 1.54 (±0.19) | 0.26 (±0.18) | 0.91 | 1.12 (±0.18) |
| MRPL3 | 2.02 (±0.16) | 0.12 (±0.11) | 0.93 | 1.32 (±0.26) |
| NRCAM | 1.69 (±0.19) | 0.15 (±0.19) | 0.94 | 1.40 (±0.36) |
| SDHA | 1.77 (±0.63) | 0.69 (±0.63) | 0.95 | 1.19 (±0.39) |
| TNFRSF25 | 2.11 (±0.31) | 0.33 (±0.30) | 0.95 | 1.18 (±0.28) |
| UBC | 1.68 (±1.22) | 0.40 (±0.20) | 0.97 | 1.14 (±0.21) |
| Averagec | 1.80 (±0.19) | 0.32 (±0.20) | 1.22 (±0.09) | |
| CFX384 versus LC480 | ||||
| ARHGEF7 | 0.88 (±0.15) | 0.29 (±0.13) | 0.97 | 1.25 (±0.20) |
| HIVEP2 | 0.92 (±0.41) | 0.27 (±0.37) | 0.90 | 1.28 (±0.71) |
| HPRT1 | 0.91 (±0.15) | 0.23 (±0.11) | 0.93 | 1.19 (±0.18) |
| MRPL3 | 0.98 (±0.14) | 0.12 (±0.11) | 0.92 | 1.11 (±0.16) |
| NRCAM | 0.73 (±0.21) | 0.12 (±0.18) | 0.93 | 1.11 (±0.20) |
| SDHA | 0.88 (±0.34) | 0.36 (±0.32) | 0.98 | 1.14 (±0.33) |
| TNFRSF25 | 1.11 (±0.37) | 0.67 (±0.35) | 0.92 | 1.58 (±0.42) |
| UBC | 1.30 (±0.25) | 0.30 (±0.22) | 0.94 | 1.23 (±0.21) |
| Averagec | 0.96 (±0.17) | 0.30 (±0.17) | 1.23 (±0.15) | |
IRC: inter-run calibration using a five-point serial dilution series of external standards; CCq: calibrated quantification cycle value; CNRQ: calibrated NRQ value;  absolute average difference in quantification cycle value of 366 samples between both platforms ±SD; r: Spearman's; rank correlation with P-value < 0.0001; FC: mean linear fold change of the CNRQ values of 366 samples between both platforms.
absolute average difference in quantification cycle value of 366 samples between both platforms ±SD; r: Spearman's; rank correlation with P-value < 0.0001; FC: mean linear fold change of the CNRQ values of 366 samples between both platforms.
a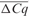 denotes intrinsic and variable inter-run difference which should be removed by a process called IRC.
denotes intrinsic and variable inter-run difference which should be removed by a process called IRC.
b after IRC should be as close to zero as possible, demonstrating removal of inter-run variation using the external standards.
cAverage FC of all genes upon NRQ level IRC are close to one (1.37, 1.22 and 1.23) and almost identical as those upon Cq level IRC on a linear scale (20.43 = 1.35, 20.32 = 1.25 and 20.30 = 1.23, respectively), demonstrating that removal of inter-run variation can be achieved on both levels.
Furthermore, Cq level IRC clearly induced a shift of the correlation plots towards the first bissectrice (45° line through origin) as shown in Figure 1a and a clear shift of the cumulative distribution plots to the left (nearly 100% of the samples with 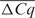 reaching zero) as shown in Figure 1b for one representative target gene. Similar figures were obtained for the other investigated genes (data not shown).
reaching zero) as shown in Figure 1b for one representative target gene. Similar figures were obtained for the other investigated genes (data not shown).
Figure 1.

(a) Correlation plots of the Cq values from 366 samples for a representative target gene (ARHGEF7) measured on two different qPCR platforms before (red) and after (blue) Cq level IRC using a five-point serial dilution series of external standards run in parallel with the patient samples. (b) Cumulative distribution plots of the difference in Cq for a representative target gene (ARHGEF7) and 366 samples measured on two different qPCR platforms before (red) and after (blue) Cq level IRC. Each dot represents a patient sample. The vertical (a) and horizontal (b) distance between two correlation scatterplots (a) or distribution plots (b) designates the platform dependent difference in Cq value between both platforms.
We further investigated the need of using all five different dilution points by measuring the using fewer standard dilution points to correct Cq values (by stepwise leaving out the lowest dilution point). As expected, the more dilution points used for Cq level IRC, the lower the (Table 2).
Table 2.
IRC on Cq level using a five-point serial dilution series of external standards run in parallel with the 366 patient samples on three different qPCR platforms (
n: n highest dilution points used)
| Before IRC | After IRC |
|||||
|---|---|---|---|---|---|---|
|
a
|
5 b
|
4 b
|
3 b
|
2 b
|
1 b
|
|
| 7900HT versus LC480 | ||||||
| ARHGEF7 | 2.63 | 0.25 | 0.19 | 0.14 | 0.14 | 0.16 |
| HIVEP2 | 2.78 | 0.65 | 0.59 | 0.65 | 0.67 | 0.71 |
| HPRT1 | 2.46 | 0.13 | 0.17 | 0.24 | 0.27 | 0.30 |
| MRPL3 | 3.00 | 0.14 | 0.24 | 0.37 | 0.41 | 0.51 |
| NRCAM | 2.42 | 0.14 | 0.15 | 0.21 | 0.26 | 0.31 |
| SDHA | 2.63 | 0.47 | 0.53 | 0.58 | 0.59 | 0.59 |
| TNFRSF25 | 3.22 | 0.98 | 0.97 | 1.12 | 1.17 | 1.30 |
| UBC | 2.98 | 0.68 | 0.73 | 0.77 | 0.85 | 0.93 |
| Averagec | 2.77 | 0.43 | 0.45 | 0.51 | 0.55 | 0.60 |
| 7900HT versus CFX384 | ||||||
| ARHGEF7 | 1.75 | 0.12 | 0.12 | 0.89 | 0.82 | 0.77 |
| HIVEP2 | 1.86 | 0.51 | 0.51 | 0.42 | 0.41 | 0.39 |
| HPRT1 | 1.54 | 0.26 | 0.26 | 0.69 | 0.66 | 0.63 |
| MRPL3 | 2.02 | 0.12 | 0.12 | 0.62 | 0.58 | 0.48 |
| NRCAM | 1.69 | 0.15 | 0.15 | 0.53 | 0.47 | 0.42 |
| SDHA | 1.77 | 0.69 | 0.69 | 0.54 | 0.53 | 0.53 |
| TNFRSF25 | 2.11 | 0.33 | 0.33 | 0.20 | 0.20 | 0.23 |
| UBC | 1.68 | 0.40 | 0.40 | 0.54 | 0.47 | 0.39 |
| Averagec | 1.80 | 0.32 | 0.32 | 0.55 | 0.52 | 0.48 |
| CFX384 versus LC480 | ||||||
| ARHGEF7 | 0.88 | 0.29 | 0.20 | 0.14 | 0.13 | 0.10 |
| HIVEP2 | 0.92 | 0.27 | 0.28 | 0.30 | 0.31 | 0.36 |
| HPRT1 | 0.91 | 0.23 | 0.16 | 0.11 | 0.11 | 0.11 |
| MRPL3 | 0.98 | 0.12 | 0.18 | 0.28 | 0.27 | 0.35 |
| NRCAM | 0.73 | 0.12 | 0.12 | 0.12 | 0.12 | 0.18 |
| SDHA | 0.88 | 0.36 | 0.52 | 0.24 | 0.24 | 0.25 |
| TNFRSF25 | 1.11 | 0.67 | 0.63 | 0.65 | 0.64 | 0.65 |
| UBC | 1.30 | 0.30 | 0.39 | 0.42 | 0.51 | 0.61 |
| Averagec | 0.96 | 0.30 | 0.31 | 0.28 | 0.29 | 0.33 |
IRC: inter-run calibration using a five-point serial dilution series of external standards; : absolute average difference in quantification cycle value of 366 samples between both platforms.
a denotes intrinsic and variable inter-run difference which should be removed by a process called IRC.
b after IRC should be as close to zero as possible, demonstrating removal of inter-run variation using the external standards.
cThe more dilution points used for IRC, the lower the .
In a last step, we compared the technical PCR replicate variability within a run to the inter-run variation before and after calibration. Therefore we calculated the variation in Cq values of the triplicate reactions for each standard dilution point measured on the three qPCR platforms for all eight genes as well as the variation in Cq values for all standards between two platforms before and after calibration. Figure 2 shows that the remaining variation after IRC between two different runs is as small as the PCR replicate variation within a run.
Figure 2.

Cumulative distribution plots depicting the intra-run variation between the PCR replicates for the standard samples as well as the inter-run variation between IRC samples before and after IRC. Results are based on data from all tested genes (ARHGEF7, HIVEP2, HPRT1, MRPL3, NRCAM, SDHA, TNFRSF25 and UBC) on three different qPCR platforms (Cq, quantification cycle value; CCq, calibrated Cq value).
Correlation between calibrated NRQs
Next, we analysed the correlation between the calibrated NRQ (CNRQ) values of the 366 samples measured on the different platforms after NRQ level IRC. The correlation between the CNRQ values calculated for any combination of two different platforms was almost perfect (r > 0.9) for all eight genes as shown in Table 1. Moreover, mean linear fold change (FC) of all genes upon NRQ level IRC were close to one (1.37, 1.22 and 1.23) and almost identical as those upon Cq level IRC on a linear scale (1.35, 1.25 and 1.23, respectively), demonstrating that removal of inter-run variation can be achieved on both levels. Figure 3 shows a good correlation between the calibrated normalized data along the first bissectrice as shown for one representative target gene. In a supplementary analysis, we demonstrated that NRQ level IRC is truly able to detect and correct inter-run variation (Supplementary Figure 3).
Figure 3.

Correlation scatterplots between the CNRQ of 366 samples for a representative target gene (ARHGEF7) measured on two different qPCR platforms indicate almost perfect concordance, validating NRQ level IRC.
Comparison of class prediction
In a third step of the validation procedure of the proposed strategy, we evaluated the impact of calibration on gene expression based class prediction. Therefore we built a gene expression signature (composed of five target genes, randomly selected from a prognostic multigene expression signature) (17) using 30 training samples measured on one of the three different qPCR platforms and tested the signature on the 336 test samples measured on all three platforms before and after NRQ level IRC. Subsequently we evaluated how similar class prediction was on samples run on two different platforms by calculating the accuracy as the proportion of true results (both true positives and true negatives) in the population. Table 3 shows a very high concordance in class prediction between the different platforms. This concordance is significantly higher after than before NRQ level IRC (P = 0.003, paired t-test).
Table 3.
Impact of IRC on class prediction
| Class prediction accuracy on test samples |
|||
|---|---|---|---|
| 7900HT versus LC480 | 7900HT versus CFX384 | CFX384 versus LC480 | |
| Training on LC480 | |||
| Before IRC | 92.7% | 93.9% | |
| After IRC | 98.5% | 97.6% | |
| Training on 7900HT | |||
| Before IRC | 90.0% | 84.5% | |
| After IRC | 98.5% | 96.4% | |
| Training on CFX384 | |||
| Before IRC | 85.8% | 93.3% | |
| After IRC | 98.2% | 98.8% | |
DISCUSSION
The quantitative polymerase chain reaction (qPCR) has become the method of choice for fast and accurate gene transcript measurements. As gene expression quantification is currently performed using different qPCR instruments, software, reagents, plates and seals, a robust method is required in order to compare data generated in different laboratories. In this study we assess the value of long oligonucleotides as universally applicable, quantifiable external standards in cross laboratory data comparison. This study demonstrates for the first time the power of this strategy to detect and correct inter-run variation and to enable exchange of data between different laboratories, even when not using the same PCR platform.
The basic principle of IRC is based on the use of identical samples—called inter-run calibrators—in different runs to correct for often underestimated technical inter-run variation. The qBase framework and accompanying qBasePlus software perfected the IRC procedure by allowing more than one inter-run calibrator to be used and by doing the calibration after normalization of the gene expression levels, resulting in more accurate IRC, fewer calculations (and hence smaller error bars due to less error propagation) and higher flexibility (allowing re-synthesis of cDNA of the same IRC RNA sample) (13). In this study, we relied on the same mathematical framework using a five-point serial dilution series of external standards to correct for experimentally induced variation, not only from run to run, but also related to the use of different qPCR instruments, Cq value determination methods, mastermixes and plastics.
While external standards based on serial dilutions of e.g. plasmids or cDNA are often being used to calculate PCR efficiency, in this study we used them to ensure reproducibility and validation of the results across laboratories and experiments. The applied standards consist of synthetic oligonucleotide controls—one for each gene—that need to be run in parallel with the samples. The proposed strategy is universally applicable and offers a high level of flexibility as everyone can design, order and use this kind of standards.
As the principle of this strategy is based on the fact that Cq or NRQ values are corrected with a gene and run specific IRC factor reflecting the mean Cq or NRQ value obtained from IRC samples (here, a series of standards) with known copy number run in parallel with the samples, it is crucial to ensure that the IRC samples input is exactly the same for both runs. This can be achieved by actually using the same synthesized lot of external standard as usually more than 1014 molecules are supplied, providing enough material to create standards for multiple thousands of IRC experiments. However, if standards from different synthesis rounds or suppliers are used, an accurate copy number measurement of the yield is needed. Indeed, standards synthesized by different companies or in successive rounds might lead to differences in supplied concentration compromising the results if used for IRC. To overcome this problem, a digital PCR pilot experiment could be performed to quantify the number of molecules in the supplied standards before using them in actual experiments (18,19). Alternatively, manufacturers could provide kits for a particular assay with inclusion of a standardized standard. Of note, when using the preferred way of IRC (i.e. on NRQ values instead of Cq values) which is ideally suited for gene expression studies, it is sufficient to have the same target ratios (instead of actual identical copy numbers) for the matching IRC sample pair measured on both runs; a simple concentration measurement of the standardized external oligonucleotides would be adequate in this case.
In order to avoid an additional potential source of inter-run variation, ideally all RNA samples should be extracted using the same method and standard operating procedures. This was not the case in this study, where RNA samples were coming from different international laboratories. However, this type of variation is possibly effectively removed by the normalization step as recently demonstrated in a large gene expression study on the same series of neuroblastoma samples in which a prognostic multigene expression signature was successfully tested on a large cohort of samples irrespective of possible confounding factors related to different RNA extraction procedures (17).
As shown previously, the more inter-run calibrators used, the more accurate and precise the results are (13). In this study we used a five-point serial dilution series of external standards. While we could confirm that more dilution points used for IRC result in better calibration, the difference is marginal here, presumably because carefully diluted synthetic oligonucleotides were used within the limits of accurate quantification. The use of complex cDNA samples (with variable and potentially unknown variation in gene expression levels) as inter-run calibrators [as done in Hellemans et al. (13)] will most likely contribute to higher inter-run variation, necessitating more than one IRC sample. In general, the use of more than one IRC sample enables quality control by inspecting results when calibrating with one or the other. Furthermore, using five IRC points like in this study also enables accurate and precise estimation of the PCR efficiency in each run.
Concordance in class prediction between the different platforms after calibration was nearly perfect and significantly higher after than before NRQ level IRC. While the results without IRC at first sight might seem satisfactory, it is important to consider the following. In this study, we observed similar shifts in Cq value between different genes when comparing two platforms. As this difference is depending on various parameters and in principle unpredictable, this information cannot be used a priori without proper control, this is the use of an IRC sample to measure and correct for the run-to-run differences. A simple change in e.g. baseline/threshold settings for Cq value determination or the use of a new primer pair or PCR reagent batch could completely abrogate the observed so-called systematic difference in Cq value. Another explanation for the unexpected relatively good correlation in class prediction without IRC is the use of the same patient cohort on all platforms. On the one hand, this was required to demonstrate occurrence of inter-run variation and effective removal. On the other hand, this caused each platform to be calibrated to some extent by itself. For classification purposes whereby multiple genes are incorporated in a score or classifier, this appears to work to some extent; for accurate and precise analysis of the expression levels of a single gene, clearly a universal and robust IRC procedure is needed, as outlined in this article.
The proposed strategy employs external standards and qPCR, both of which have been extensively evaluated and are widely used. Other strategies to standardize qPCR data, such as StaRT–PCR, are based on internal standards (6,7). Based on competitive PCR, StaRT–PCR is a patented technique for measuring multigene expression in samples and relies on end-point quantification. The advantage of the method is the incorporation of competitive templates into standardized mixtures of internal standards (SMIS) which allows comparison of generated data since the values are determined relative to the same standardized mixtures. Compared to our strategy, StaRT–PCR, is characterized by a more limited dynamic range of linear quantification, is more labour intensive, and is only commercially available through Gene Express. Our strategy is directly accessible to anyone by the simple ordering of the oligonucleotide sequence of interest and thus offers a high flexibility.
In conclusion, our study clearly demonstrates that the use of external oligonucleotide standards is a powerful method for accurate cross laboratory data comparison. Amongst others, it enables to test a gene signature on a single patient sample in any lab in the world and compare the results with a reference set established in another lab. The proposed strategy truly enables multicentre studies conducted at different sites, greatly advancing this field of application.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Belgian Foundation Against Cancer [grant number SCIE2006-25]; the Children Cancer Fund Ghent; the Fondation Fournier Majoie pour l’Innovation; the Belgian Society of Paediatric Haematology and Oncology, the Belgian Kid’s Fund [to J.VERM.]; the Fondation pour la recherche Nuovo-Soldati [to J.V.]; the Fund for Scientific Research Flanders [to K.D.P. and J.H.]; the Fund for Scientific Research Flanders [grant number G.0198.08]; the Institute for the Promotion of Innovation by Science and Technology in Flanders [to S.D.]; the Ghent University Research Fund [BOF; to P.M., S.L. and F.P.]; and the European Community under the FP6 [project: STREP: EET-pipeline; number: 037260]. Funding for open access charge: Biolegio, the Netherlands.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank Els De Smet, Nurten Yigit and Justine Nuytens for their excellent technical assistance and Ellen Lefebvre for review of the mathematical formulas. They also would like to acknowledge Rob Powel (PrimerDesign, UK) for support with primer design and Biolegio (the Netherlands) for their support with the implementation of the external standards. They are indebted to all members of the International Society of Paediatric Oncology, European Neuroblastoma Group (SIOPEN) and the Gesellschaft fuer Paediatrische Onkologie und Haematologie (GPOH) for providing tumour samples.
REFERENCES
- 1.Weis JH, Tan SS, Martin BK, Wittwer CT. Detection of rare mRNAs via quantitative RT-PCR. Trends Genet. 1992;8:263–264. doi: 10.1016/0168-9525(92)90242-v. [DOI] [PubMed] [Google Scholar]
- 2.Bustin SA. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol. 2000;25:169–193. doi: 10.1677/jme.0.0250169. [DOI] [PubMed] [Google Scholar]
- 3.Ginzinger DG. Gene quantification using real-time quantitative PCR: an emerging technology hits the mainstream. Exp. Hematol. 2002;30:503–512. doi: 10.1016/s0301-472x(02)00806-8. [DOI] [PubMed] [Google Scholar]
- 4.Bustin SA. Real-time quantitative PCR – opportunities and pitfalls. Eur Pharm Rev. 2008;4:18–23. [Google Scholar]
- 5.Murphy J, Bustin SA. Reliability of real-time reverse-transcription PCR in clinical diagnostics: gold standard or substandard? Expert. Rev. Mol. Diagn. 2009;9:187–197. doi: 10.1586/14737159.9.2.187. [DOI] [PubMed] [Google Scholar]
- 6.Willey JC, Crawford EL, Jackson CM, Weaver DA, Hoban JC, Khuder SA, DeMuth JP. Expression measurement of many genes simultaneously by quantitative RT-PCR using standardized mixtures of competitive templates. Am. J. Respir. Cell Mol. Biol. 1998;19:6–17. doi: 10.1165/ajrcmb.19.1.3076. [DOI] [PubMed] [Google Scholar]
- 7.Crawford EL, Peters GJ, Noordhuis P, Rots MG, Vondracek M, Grafstrom RC, Lieuallen K, Lennon G, Zahorchak RJ, Georgeson MJ, et al. Reproducible gene expression measurement among multiple laboratories obtained in a blinded study using standardized RT (StaRT)-PCR. Mol. Diagn. 2001;6:217–225. doi: 10.1007/BF03262057. [DOI] [PubMed] [Google Scholar]
- 8.Dafforn A, Chen P, Deng G, Herrler M, Iglehart D, Koritala S, Lato S, Pillarisetty S, Purohit R, Wang M, et al. Linear mRNA amplification from as little as 5 ng total RNA for global gene expression analysis. Biotechniques. 2004;37:854–857. doi: 10.2144/04375PF01. [DOI] [PubMed] [Google Scholar]
- 9.Kurn N, Chen P, Heath JD, Kopf-Sill A, Stephens KM, Wang S. Novel isothermal, linear nucleic acid amplification systems for highly multiplexed applications. Clin. Chem. 2005;51:1973–1981. doi: 10.1373/clinchem.2005.053694. [DOI] [PubMed] [Google Scholar]
- 10.Nolan T, Hands RE, Ogunkolade W, Bustin SA. SPUD: a quantitative PCR assay for the detection of inhibitors in nucleic acid preparations. Anal. Biochem. 2006;351:308–310. doi: 10.1016/j.ab.2006.01.051. [DOI] [PubMed] [Google Scholar]
- 11.Lefever S, Vandesompele J, Speleman F, Pattyn F. RTPrimerDB: the portal for real-time PCR primers and probes. Nucleic Acids Res. 2009;37:D942–D945. doi: 10.1093/nar/gkn777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-7-research0034. RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007;8:R19. doi: 10.1186/gb-2007-8-2-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bustin SA, Benes, Garson JA, Hellemans J, Huggett J, Kubista M, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009;55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 15.Lefever S, Hellemans J, Pattyn F, Przybylski DR, Taylor C, Geurts R, Untergasser A, Vandesompele J. RDML: structured language and reporting guidelines for real-time quantitative PCR data. Nucleic Acids Res. 2009;37:2065–2069. doi: 10.1093/nar/gkp056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu R, Wang X, Chen GY, Dalerba P, Gurney A, Hoey T, Sherlock G, Lewicki J, Shedden K, Clarke MF. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N. Engl. J. Med. 2007;356:217–226. doi: 10.1056/NEJMoa063994. [DOI] [PubMed] [Google Scholar]
- 17.Vermeulen J, De Preter K, Naranjo A, Vercruysse L, Van Roy N, Hellemans J, Swerts K, Bravo S, Scaruffi P, Tonini GP, et al. Predicting outcomes for children with neuroblastoma using a multigene-expression signature: a retrospective SIOPEN/COG/GPOH study. Lancet Oncol. 2009;10:663–71. doi: 10.1016/S1470-2045(09)70154-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kalinina O, Lebedeva I, Brown J, Silver J. Nanoliter scale PCR with TaqMan detection. Nucleic Acids Res. 1997;25:1999–2004. doi: 10.1093/nar/25.10.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vogelstein B, Kinzler KW. Digital PCR. Proc. Natl Acad. Sci. USA. 1999;96:9236–9241. doi: 10.1073/pnas.96.16.9236. [DOI] [PMC free article] [PubMed] [Google Scholar]