


Abstract
Microfluidic chips can automate biochemical assays on the nanoliter scale, which is of considerable utility for RNA–protein binding reactions that would otherwise require large quantities of proteins. Unfortunately, complex reactions involving multiple reactants cannot be prepared in current microfluidic mixer designs, nor is investigation of long-time scale reactions possible. Here, a microfluidic ‘Riboreactor’ has been designed and constructed to facilitate the study of kinetics of RNA–protein complex formation over long time scales. With computer automation, the reactor can prepare binding reactions from any combination of eight reagents, and is optimized to monitor long reaction times. By integrating a two-photon microscope into the microfluidic platform, 5-nl reactions can be observed for longer than 1000 s with single-molecule sensitivity and negligible photobleaching. Using the Riboreactor, RNA–protein binding reactions with a fragment of the bacterial 30S ribosome were prepared in a fully automated fashion and binding rates were consistent with rates obtained from conventional assays. The microfluidic chip successfully combines automation, low sample consumption, ultra-sensitive fluorescence detection and a high degree of reproducibility. The chip should be able to probe complex reaction networks describing the assembly of large multicomponent RNPs such as the ribosome.
INTRODUCTION
Microfluidic chips can routinely manipulate fluids on the picoliter to nanoliter scale. Low sample consumption, combined with facile automation schemes and the potential to multiplex assays on a large scale, make microfluidic chips an attractive assay format for a wide range of applications. While chip applications in biology range from microbial and mammalian cell culture (1,2) to DNA sequencing (3), the paradigm shift from microliter-scale conventional assays to nanoliter-scale microfluidic assays has been slow and difficult, despite obvious benefits (4).
The advent of inexpensive poly (dimethylsiloxane) (PDMS) soft lithography (5) and construction of dedicated foundry facilities has enabled non-specialist labs to quickly fabricate sophisticated chips, whether of their own design or adapted from others. The time from design to first use is typically under a week. By stacking multiple, independently fabricated layers, multilayer soft lithography (6) enables facile integration of valve and pump elements which control fluid flow. Valves and pumps can be controlled by digital I/O computer hardware, allowing experiments to be automated by computers. Multilayer chips can be rapidly designed by using an appreciable library of fluid control schemes and design templates (7).
Microfluidic chips have been built to measure diverse intermolecular interactions, starting with microfluidic enzyme-linked immunosorbent assay (ELISA) and immunosensing chips (8). Precise quantitation of the binding energy between bovine serum albumin (BSA) and heparin was achieved using on-chip capillary electrophoresis (9), while the IC50 for a small molecule disrupting a transcription factor–DNA complex was determined using an eight-point titration generated with a mixing network (10). By multiplexing (7) thousands of DNA–protein (11) and RNA–protein (12) binding assays on individual chips, it is possible to find consensus binding sites by performing in-chip screens of libraries of nucleic acids.
Microfluidic platforms have been used to probe the kinetics of biomolecular reactions. Extremely fast mixing on the 1–10 μs time scale occurs in sub-millimeter-scale microfluidic manifolds which focus small flows of analyte (nanoliters/s) into large quantities of diluent (13–15). Probing such a chip with confocal optics has resolved sub-milisecond conformational changes of the protein calmodulin upon binding of Ca2+ ions (16). Different mixing designs, which use microfluidic droplets, consume less diluent by mixing equal volumes of two flows on the millisecond timescale and have measured the binding rate constant for the biotin–streptavidin complex (17).
The focus of these previous approaches on fast mixing of two components makes them poorly suited for studying assembly of large RNP complexes such as bacterial 30S ribosome, in which 21 components bind together over a relatively long time scale. Elucidation of a multi-component reaction mechanism requires data sets of many uniquely prepared reactions, essentially one unique reaction per assembly intermediate (18), and the number of intermediates grows factorially with the number of components. To generate unique reactions by systematically varying the relative amounts of starting materials requires a chip which can variably mix more than two components. Current fast mixer designs also do not retain samples long enough to measure slow rates. Assembly of the 30S ribosome is limited by RNA conformational changes and protein binding events with rate constants as slow as 0.001s−1, (19,20) requiring reaction times on the order of thousands of seconds.
The ‘Riboreactor’ microfluidic chip reported here departs from continuous flow mixers by preparing reactions in batch format, using the common-ring metering and mixing scheme developed in the ‘microfluidic formulator’ chip design of Hansen et al. (21). The chip is a slow mixer with the ability to initiate complex reactions from a variety of initial conditions. The chip can mix arbitrary amounts of eight input reagents to create 5-nl scale reactions in a reaction ring (21) which are detected in situ by a two-photon fluorescence microscope. Computer control of the chip and the microscope allows experiments to be developed as software modules. To address unanticipated technical problems and establish proof-of-principle, a well-studied RNP fragment of the bacterial 30S ribosome, the S15 protein–3WJ (Three–Way Junction) RNA complex (22–24), was assembled in the Riboreactor.
MATERIALS AND METHODS
Chip design and fabrication
The microfluidic chip was fabricated using PDMS soft lithography (5) at the Caltech Microfluidic Foundry. Using the multilayer scheme of Unger et al. (6), analytes were plumbed through 100 μm wide channels in the flow layer (blue lines, Figure 1A) and movement of analyte within the flow layer was directed by valve channels etched into the control layer above (red lines, Figure 1A). The flow and control layers were designed using Adobe Illustrator and separately printed on transparent masks. Photolithography used the masks to create molds in Si wafers (photoresists SPR 220–7 for flow layer, SU8–2025 for control layer, MicroChem). Hard-baked flow layer molds had rounded channels 12.5 μm high by 100 μm wide, while control layer molds were 28 μm × 100 μm. The PDMS flow layer (Sylgard 184, A : B = 20 : 1, Dow Corning) was cast against the mold in a thin layer by spin coating at 2500 r.p.m. 1 min and cured at 80°C for 20 min. A 4-mm thick control layer (degassed, A : B = 5 : 1) was polymerized over its mold and cured identically. The control layer was removed from its mold, drilled for control input ports, aligned on the flow layer and baked to bond the layers together. The layers were then removed from the flow-layer mold, drilled for flow input ports and baked overnight onto a No. 1 cover glass to produce the final chip. In operation, analytes were introduced to the flow layer and water was introduced to the control layer by pressurized Tygon tubing [inner diameter (ID) 0.02 in, outer diameter (OD) 0.06 in, VWR] which was mated to the chip by steel pins [0.025 in OD, 0.017 in ID × 0.5 in, New England Small Tube]. Typical flow-layer pressure was 5.5 psi (range 4–6.5 psi), while control-layer pressure was 22 psi (range 18–26 psi).
Figure 1.

The Riboreactor microfluidic chip prepares complex reactions using a common-ring approach developed for the microfluidic formulator chip (21). (A) Using PDMS multilayer soft lithography (5,6), the flow layer (blue) and control layer (red) are fabricated independently, and then bonded together. Reagents are shunted, stored and reacted in flow channels that are gated by valves formed by the intersections of flow channels and wide segments of control channels. (B) Up to eight different reagents (α–θ) are introduced to the chip via a manifold gated by six valves. Valve state (open, red; closed, black) is controlled by modulating control line pressure with computer-controlled solenoids. (C, D) Examples show fluids θ and δ entering the chip. (E) The input manifold is connected to a 5 nl reaction ring that prepares mixtures and holds reactions. (F) The ring is flushed with buffer (θ) before use. (G) Staging brings fluid δ to the entrance of the ring. (H) Fluid δ is metered into the ring when a peristaltic pump draws in discrete volumes per pump cycle. (I and J) To mix fluid δ with fluid α, staging and metering are first repeated. (K) Fluids δ and α are mixed by isolating the ring from the chip and circulating them with the peristaltic pump (mixed fluid, brown). (L) A single cycle of the peristaltic pump.
Some chips were passivated by covalent attachment of poly(ethlyene glycol) (PEG) (2-[methoxy(polyethyleneoxy)propyl]trimethoxysilane, SIM6492.7, Gelest) or the teflon-like (tridecafluoro-1,1,2,2-tetrahydrooctyl)trimethoxysilane (SIT8176.0, Gelest) to the PDMS walls (25). The PEG and fluorinated alkane coatings resulted in markedly lower protein adsorption in the absence of detergents. However, both modifications were ultimately unusable in multilayer structures, as the first step of the coupling reaction (oxidation with HCl/H2O2 rendered the valves unable to close fully and the chips inoperative.
Automation
When control valves are pressurized in the Quake multilayer scheme (6), the flow lines beneath collapse and form a fluid-tight seal. Automation is achieved by modulating control line pressure between 0 and ∼22 psi using a bank of solenoid valves (FluidController, Fluidigm) which translate electrical potentials from a digital I/O computer card (PCIe-6251, National Instruments) into pressure differences. The card is driven by custom-written multi-threaded C/NI-DAQmx (National Instruments) programs which coordinate chip operations with microscope functions and data acquisition. As the shortest time delay between valve operations is relatively long (2 ms), timing of valve operations is performed by the computer clock which is found to be accurate to better than one microsecond.
Integration with two-photon optics
Microfluidic reactions were monitored using a custom-built two-photon microscope. Femtosecond pulses from a passively mode-locked Ti–sapphire laser with a center wavelength of ∼860 nm and typical average powers of 5–15 mW (Mira 900-F, Coherent) are focused into the sample using a high-numerical aperture objective (100× 1.45 N.A. α Plan-Fluar Zeiss). Alignment is aided by a secondary microscope built opposite the primary microscope (5× 0.12 N.A. A-Plan, Zeiss). Fluorescence is detected through the primary objective, separated from the laser by a dichroic mirror, and further split into three channels by two dichroic mirrors and three bandpass filters (730DCSPX, 530DCXR, Q625LP, HQ485/70M, HQ580/60M, HQ665/90M, Chroma). Photons are detected by avalanche photodiodes (AQRH-14, PerkinElmer Optoelectronics), counted on a data acquisition card (PCIe-6251, National Instruments) and analyzed using programs written in C/NI-DAQmx (National Instruments).
3WJ and S15 construct
The 3WJ RNA (Figure 4) was prepared by annealing three short RNAs: 20–21: N5.UGG UGC GAG CUG GCG GUC UUC CA, 21–22: N5.UGG AAG ACU UGA GGG CAG GAG AGG ACC, 22–20: N5.UGG UCU GGC CUG CAC CUG ACG CCA GCU CGC ACC A (Dharmacon) where N5 is a primary amine tethered to the RNA by five carbons. Using the three-color FRET scheme (two fluorophores on RNA, one on protein) described by Klostermeier et al. (24) but with fluorophores optimized for two-photon excitation, strand 20–21 was labeled with the fluorophore and FRET donor Atto 425, while strand 22–20 was labeled with FRET acceptor Atto 532 (Atto-Tec GmbH) (Figure 5A).
Figure 4.

The 3WJ RNA construct folds in the presence of Mg2+ and binds protein S15. (A) The minimal binding site for 30S ribosomal protein S15 is a 3WJ located within the central domain of the 16S rRNA (23,29). (B) In the presence of metal ions, the junction folds such that helices 21 and 22 stack coaxially, while helices 20 and 22 dock (22). S15 preferentially binds the folded form (26).
Figure 5.

Mg2+-RNA binding titrations on the 5 nl scale. (A) When Mg2+ ions bind the 3WJ RNA, folding increases the FRET efficiency between fluorophores Atto425 and Atto532 (R0 = 58Å). As both fluorophores are directly excited by two-photon processes (red arrows), FRET changes are measured as a change in the ratio of emissions (blue and orange arrows). (B) The affinity of the RNA–Mg2+ interaction was measured by five independent titrations. Distinct reactions were created by serially diluting a stock solution of Mg2+ in the reaction ring before mixing with ∼700 pl of RNA. The recovered affinities (93 ± 6 μM) are slightly weaker than results of conventional measurements performed on the 200 μl scale (68 ± 5 μM, data not shown) and by others [77 μM (24)].
Both RNA fluorophores were conjugated to primary amines on their respective strands using NHS Ester chemistry on the 30 nmol scale by reacting 125 μM phenol-extracted RNA with 2.25 mM fluorophore in 75 mM sodium tetraborate pH 8.5, 15% DMSO for 6 h in the dark, at room temperature, with constant stirring. RNA was separated from unconjugated fluorophore by three phenol extractions, then ethanol precipitated. Labeled and unlabeled RNA were separated by analytical-scale reverse-phase High Performance Liquid Chromatography (HPLC) (Delta-Pak C18, Waters) using a H2O/acetonitrile, 0.1 M triethylamine acetate buffer system (Glen Research). As judged by 260 nm absorption in the HPLC traces, the reaction proceeded to >60% in each case. Baseline resolution was achieved for Atto 425-labeled RNA only. Within the uncertainty of UV–Vis spectroscopy and the precision by which the extinction coefficients for each fluorophore are known, both purified strands were fully labeled. RNA integrity and the absence of contaminating unbound fluorophores were confirmed by denaturing gel electrophoresis (20% polyacrylamide, 6 M urea) and mass spectrometry.
3WJ RNAs were prepared by annealing strands 20–21, 21–22 and 22–20 in 100 mM NaPO4 pH 7.0, 100 mM KCl by heating to 95°C for 2 min before slow cooling to 35°C at 2°C/min. Labeled strands were present at 5 μM, unlabeled at 10 μM. 3WJ formation was verified by electrophoresis on a native 6% polyacrylamide gel and titrations with Mg2+ and S15 confirmed binding with expected affinities (24).
The Bacillus stearothermophilus protein S15 was recombinantly expressed in Escherichia coli BL21(DE3) with an N-terminal His6 tag and the point mutation R78C, and purified as described by Klostermeir et al. (24). To reduce the cysteine for labeling, S15 was incubated with 50 mM dithiothreitol (DTT), 6 M urea, 50 mM Tris–HCl pH 7.6 for 2 h at 37°C. The protein was separated from DTT by C18 chromatography (218TP, Vydac) using a H2O/acetonitrile 0.1% trifluoroacetic acid buffer system and lyophilized.
Protein labeling was achieved by re-suspending 100 nmol S15 in 940 μl degassed 7 M Guanidinium HCl, 100 mM sodium phosphate pH 7.0, before adding 300 nmol Alexa Fluor 594 C5 Maleimide (‘Alexa594’, Invitrogen) in 60 μl anhydrous DMSO (Sigma). The reaction was shaken for 16 h at room temperature in the dark. Unreacted fluorophore was separated from protein by dialysis against 4L 2% acetic acid (3 × 16 h, 3.5 kDa MWCO, Spectrum Laboratories) and the protein was lyophilized. Unreacted protein was separated from fluorophore-labeled protein by analytical-scale reverse-phase HPLC (Delta-Pac, 300 × 3.9 mm, Waters) using a H2O/acetonitrile, 0.1% trifluoroacetic acid buffer system; pure labeled protein was stored lyophilized. Mass spectra confirmed labeling and S15R78C-Alexa594 binds 3WJ RNA with wild-type affinity (23). A lower bound on the extent of S15 labeling (96%) was set by donor quenching—the product of labeling extent and FRET efficiency.
For binding reactions, S15 was dissolved in 6.8 M guanidinium HCl, 10 mM HEPES pH 7.0 and folded by rapid dilution into 50 mM Tris pH 7.6, 0.5% Tween-20. Residual guanidinium was in the range of 1–40 mM in all assays described. Conventional (non-microfluidic) 3WJ/S15 and 3WJ/Mg2+ affinity assays were performed in a fluorescent plate-reader (α-Fusion, Perkin Elmer), in 200 μl 50 mM Tris pH 7.6, 0.5% Tween-20. Binding was quantified by quenching of the donor (Atto425) and data fit to the Hill equation. Reactions simulating previous buffer conditions (23,26) (10 mM K–Hepes pH 7.5, 50 mM K–acetate, 100 μM K–EDTA, 100 μg/ml tRNA (4 μM), 0.5% Tween-20) and reactions without K–acetate (50 mM Tris–HCl pH 7.6, 0.5% Tween-20) were performed in a fluorimeter (SLM-Aminco 8100).
Microfluidic Mg2+–3WJ folding assay
The input lines of the chip's; manifold were loaded with the following solutions: (α): reference fluorophore (100 nM Alexa 488); (β, δ, ζ, η, θ): Buffer A; (γ): 50 nM 3WJ rRNA in Buffer A; and (ε): 15 mM Mg2+, 50 mM Tris pH 7.6. Buffer A contained 50 mM Tris pH 7.6, 0.5% w/v Tween-20 (Sigma).
The following experimental program was run at 18°C, and consisted of 14 separate trials corresponding to 14 different serial dilutions of Mg2+: (i) flush ring with buffer (θ), 4.5 s; (ii) fill ring with Mg2+ (ε), 50 pump cycles; (iii) wash staging area with buffer (θ); 2 s. (iv) dilute Mg2+ with nine pumps of buffer (θ), and mix to homogeneity (pump speed 33 Hz, 10 s). Repeat the dilution step a total of x times, where x is the number of the trial,1 ≤ x ≤ 14; (v) stage and meter nine pumps 3WJ (γ); and (vi) mix (33 Hz, 10 s) and simultaneously observe (14 s); the last 4 s are used for quantifying FRET signal.
Microfluidic S15–3WJ kinetic assay
The input lines of the chip's; manifold were loaded with the following solutions: (α) reference fluorophore (100 nM Alexa 488); (β): 50 nM 3WJ rRNA in Buffer B with 0.5% w/v Tween-80 (Sigma); (γ, δ, ε, η) Buffer B; (ζ): 3 μM S15 in Buffer B with 0.5% w/v Tween-20; and (θ): Buffer B with 0.5% w/v Tween-80, 50 μg/ml tRNA (Sigma). Buffer B contained 50 mM Tris pH 7.6, 100 μg/ml BSA (Sigma).
The following experimental program was run at 17°C, and consisted of 14 trials corresponding to 14 different dilutions of S15: (i) flush ring with buffer (θ), 10 s. (ii) flush ring with S15 (ζ), 15 s; (iii) wash staging area with buffer (θ), 10 s; (iv) dilute S15 with 14 pumps of buffer (θ), and mix to homogeneity (25 Hz, 13.2 s). Repeat this step a total of x times, where x is the number of the trial; (v) stage and meter eight pumps 3WJ (β); and (vi) immediately mix (25 Hz, 10 s) and simultaneously observe the reaction (250 s).
Tween-20 abrogated RNA–PDMS interactions as judged by scanning microscopy and FCS (as did ‘Nikkol’ octaethylene glycol monododecyl ether, Fluka). Tween-20 prevented preformed S15–3WJ and non-specific S15–tRNA complexes from adsorbing to PDMS, but did not completely prevent adsorption of uncomplexed S15. This in turn was prevented by adding Tween-20 to the S15 stock and Tween-80 in both RNA and buffer stocks. In addition to detergents, BSA increased the apparent activity of S15, and tRNA [frequently used as a weak competitor in gel electrophoretic mobility assays (23)] increased the ratio between the maximum and the minimum obtainable concentrations of S15 from ∼10 to >500, presumably by liberating adsorbed S15. This suggests that even in the presence of detergents the protein existed in a weak equilibrium with the PDMS walls. The apparent off-rate of S15 from the PDMS was indirectly observed as Kapp = 0.2 s−1 (data not shown). As this is similar to the mixing speed of the microfluidic reactor, mixing delays in the experimental program served the dual purpose of mixing fluid within the ring and allowing equilibrium to be re-established between the walls and the fluid within.
Data analysis
Photons were counted in 100 ms bins, and the standard deviation of the photon counts was computed as an estimate of error, which permitted robust fitting of otherwise noisy data using weighted nonlinear least squares. Rare superbursts of intensity—attributed to protein aggregates diffusing through the focal volume—were occasionally observed. Culling of superbursts using a 2.33σ cutoff produced visually cleaner data (Figure 6B,C) but did not improve fitting statistics. Rare time traces which displayed a gradual decay of fluorescence from the ring, likely from leakage of the ring contents, were discarded.
Figure 6.

Microfluidic 3WJ RNA–S15 protein binding kinetics. (A) Binding of the fluorescently labeled protein S15-Alexa594 to the doubly labeled 3WJ RNA results in efficient FRET to S15-Alexa594 (24). (B) Time trace of a 5-nl scale binding reaction prepared by metering 3WJ RNA into a reaction ring filled with serially diluted S15. Mixing creates damped oscillations for the first 10 s, after which the peristaltic pumps are locked closed and fluorescence changes due to FRET. (C) Eleven reactions differing in the initial concentration of S15 were run in quadruplicate and apparent binding constants (Kapp) were recovered by fitting FRET donor intensity to mono-exponential decays. (D) Fitting the averaged Kapp to S15 concentration (Equation 1) recovers the bimolecular on-rate Kon and unimolecular off-rate Koff. The ratio of the rates yields an affinity (KD = 8 ± 2 nm) which agrees with the affinity obtained from 200 μl scale assays with the same materials (10.7 ± 0.8nM, data not shown) and literature values (23,26).
As two-photon excitation excited all three dyes (independent of FRET), it was possible to monitor fluorescence of all species simultaneously. However, there was poor agreement between the concentration of S15 as reported by S15-Alexa594 fluorescence and S15 activity as reported by reactivity with the 3WJ. Unlike S15 activity, which remained constant, S15 fluorescence depended on the history of chip operations, and would typically decrease as experimental replicates were performed. Thus, S15 fluorescence intensity was not used to quantify binding.
Fitting equation for apparent reaction rates
Individual reaction traces were fit to decaying exponentials to obtain apparent reaction rates (Kapp). Errors were estimated as the standard deviation of rates obtained from four replicate mesurements. Apparent rates were equated to on and off rates using the approximation Kapp ≃ Kon[S15]i + Koff, where [S15]i is the concentration of S15 at the beginning of the reaction. This approximation is good for reactions where [S15]i ≫ [3WJ]i (few S15 serial dilutions), but poor for reactions where [S15]i ≈ [3WJ]i (many serial dilutions). Numerical simulations of the discrepancies introduced by the approximation were performed using relaxation methods (27), revealing that the approximation overestimates Kapp by only 6% at the lowest values of [S15]i assayed. Since this is much less than the error in the rate from noise in data at low [S15]i, the approximation for Kapp was used at all [S15]i.
The relationship between [S15]i and the number of serial dilutions, n, is assumed to be [S15]i = (1−χ)[S15]0 (fracdil)n, where the amount of S15 injected into the microfluidic reactor, [S15]0, is known from UV–Vis absorption spectroscopy, fracdil is the dilution factor of the serial dilution, and χ represents the amount of 3WJ RNA injected into the ring, expressed as a fraction of the total reaction ring volume. The final expression to which data were fit was
| 1 |
where three values were fit (kon, koff, fracdil) using weighted nonlinear least squares (Igor Pro, WaveMetrics).
RESULTS
Microfluidic reactor design and implementation
The Riboreactor chip was designed to quantitatively aliquot and mix eight different reagents on the 250 pl–5 nl scale. Computer-driven control valves direct flow of fluid within an architecture which allows for measuring, storing, mixing and observing. The chip was fabricated as a three-layer structure where a cover-glass was bonded to a patterned PDMS flow layer (blue, Figure 1A), which was in turn bonded to a patterned PDMS control layer (red). Valves are formed where wide segments of control lines cross flow lines; narrow control line segments do not appreciably gate flow and allow the creation of topologically complex features (Figure 1B) (6). Reagents flow and mix in channels created by bonding the etched surface of the flow layer with the cover-glass. The cover-glass allows the reagents to be probed by high-numerical aperture microscope objectives. Fluids enter either the upper control layer or the deeper flow layer via narrow-gauge steel pins inserted at the circular ports on the periphery of the chip. To move fluid through the chip, flow-layer pins are connected to a manifold held at constant pressure. To actuate valves, control line pins are connected to a similar manifold and each line is further gated by a computer-controled solenoid valve. Only the pressurized control lines restrict flow. The valves are robust over long periods, and there was no detectable change in either the amount of fluid metered per pump cycle or the rate of mixing after more than 100 000 valve actuations.
In the present implementation of the common-ring metering and mixing scheme (21), eight input lines (α–θ, Figure 1B) deliver fluid either to the entrance of a 5-nl reaction ring (Figure 1E) or to the waste port (O2, Figure 1A). Within the ring, reactions take place and are monitored by the stationary focus of the two-photon optical train. Of the 12 independent control lines in the Riboreactor, six lines (A0–C1) route one of the eight input lines toward the reaction ring or waste port (28), and three lines (Z0–Z2) switch flow between the reaction ring and a waste port and can isolate the reaction ring from the input/waste lines for reactions. The final three control lines (P1–P3) are actuated in a repeating sequence (Figure 1L) to act as a peristaltic pump (6) within the reaction ring. When an input line is coupled into the ring, the peristaltic pump acts as a metering device (Figure 1H), drawing ∼250 pl into the ring per pump cycle. When the ring is isolated from the input and waste ports, the peristaltic pump drives fluid flow in a circle, mixing the contents (Figure 1K).
Together, the 12 control lines allow for both continuous-flow and metered-flow modes of operation. Continuous flow is pressure driven and flow rates are sensitive to minor changes in pressure and viscosity, while metered flow is precisely driven by a peristaltic pump. Continuous flow from a selected line of the input manifold toward the exit port can either flow through the ring or bypass it. Flow through the ring (Figure 1F) flushes the contents of the previous reaction and cleans the ring for next use. Flow bypassing the ring (Figure 1G) delivers fresh reagent to the entrance of the ring in preparation for metered injection. When the pump is driven at low frequencies (<15 complete cycles/second), fluid metering is highly reproducible and suitable for quantitative delivery of analyte. Heterogeneous mixtures of fluids are prepared in the ring by repeating staging and metering with a different input line(s) (Figure 1 F–K). Mixing the contents of the ring to homogeneity is accomplished by isolating the ring from the input/waste lines and running the peristaltic pump at ∼30 Hz for several hundred cycles (Figure 1 K,L). Mixing speed is increased by allowing back-pressure to build in the reaction ring before it is isolated from the binary manifold by inserting a 200 ms delay between closing valves Z0 and Z1 and valve Z2 (for valve notation, see Figure 1A).
Assays were developed as C programs by stringing together a custom-written library of functions corresponding to the main modes of operation (Figure 1 F–K). Each function contained a series of timed valve actuations. In this manner, assays containing hundreds of thousands of valve actuations were routinely designed and executed in minutes. Functions were empirically tuned for speed and reliability using a simple pair of fluorophore and buffer solutions, and did not require experiment-specific tuning. The exception was the pump frequency during mixing, which was tuned on a per-chip basis. The pump was further tuned for maximum fluid speed by varying the flow and control valve pressures, and using air instead of water in the pump control lines. Typical mixing times were slow: 10–12 s.
Integration with a two-photon microscope
The combination of nanoliter samples at nanomolar concentrations results in limited amounts of analyte (∼107molecules), which are difficult to detect and can photobleach over long observation periods. Two-photon fluorescence excitation can solve both problems with selective excitation, which affords both single-molecule sensitivity (29) and has intrinsically low photobleaching effects (30). Two-photon excitation is implemented by mounting the microfluidic chip on the stage of a custom-built microscope, and using the same computer to co-ordinate chip control valves, laser excitation and fluorescence detection.
Two-photon excitation selectively excites a ∼0.3 fl focal volume, which is held stationary within the reaction ring (Figure 2A and B). Alignment of the focal volume within the chip was accomplished with the aid of a second microscope, which images the same focal plane from the top of the chip (micrograph, Figure 2A). The selective focus (lateral diameter ≃0.5 μm, vertical diameter;3 μm) probes the interior of the flow lines and is insensitive to fluorescent biomolecules adsorbed to the walls (Figure 2B). Scans of a chip which had fluorophores adsorbed to the walls showed a clear distinction between the interior of the chip and the walls (intensity profiles, Figure 2B). Light scattering from the PDMS substrate was not detectable (800 nm ≤ λex ≤ 900 nm). Single molecule sensitivity is evident in traces of dilute RNA diffusing through the flow channels (Figure 2C). Analysis of dilute fluorescently labeled RNA using fluorescence correlation spectroscopy (FCS) confirms that the optical setup is stable, and that RNA diffusing in the center of the ring is unperturbed by the channel walls as evidenced by the FCS diffusion time, τD = 108 ± 2 μs (Figure 2D) compared to the time measured on a coverslip, τD = 96 ± 2 μs (data not shown).
Figure 2.

Sensitive and low-photobleaching two-photon excited fluorescence monitors reactions. (A) The reaction ring (filled with concentrated green fluorescent dye for visualization purposes) is probed by a stationary ∼0.3 femtoliter excitation volume (orange spot) created by focusing a femtosecond-pulsed laser beam with an 100× objective. The chip is aligned over the focal volume with the aid of an opposing low power microscope. (B) Two-photon excitation selectively excites the interior of the flow channel. Selectivity is demonstrated by intensity scans through a channel which has fluorophore deliberately adsorbed to the walls. (C) Spikes in the time trace are caused by single molecules of fluorescent RNA diffusing through the focal volume, and demonstrate single-molecule sensitivity. (D) FCS of an RNA solution confirms that the RNA in the channel diffuses at the expected rate and is free to react with protein. (E) The minimal photobleaching properties of two-photon excitation permit continuous observation of 5 nl samples with a bleaching half-life on the order of days (40 h).
At the laser powers employed, photobleaching was slow with a half-life on the order of days for a 5 nl sample (Figure 2E), enabling long reaction read times. Higher laser powers up to 25 mW caused no detectable damage to the PDMS substrate when the focus was held in the flow channel or even in the PDMS material itself for longer than 100 min. Constant excitation of labeled RNA at the nanomolar concentrations used in biochemical assays displayed a stable average with large fluctuations (Figure 2E). Large fluctuations are expected from the convolution of photon shot noise (∼10 ± 3 photons detected per molecule) with occupancy shot noise (∼10 ± 3 molecules observed at any given time) (31).
Chip calibration and reproducibility
The metering performance of each chip was calibrated prior to use and calibration parameters were used to calculate the final concentrations of reactants in assays. The amount of fluid metered per pump cycle was determined by diluting the reference fluorophore Alexa 488 with Buffer A with varying numbers of pump cycles, and measuring the fluorescence (Figure 3A). The amount metered is not a linear function of pump cycles. Deviations are presumably due to the same fluid-wall interactions which slow the fluid near the walls and make rotary mixing possible. The repeatability of metering was assayed by performing n serial dilutions of the fluorophore Alexa 488 and measuring the fluorescence (Figure 3B). Data were weighted by the standard deviation of the photon counts and fit to y(n) = y0(fracdil)n + y∞. The major variability was typically from the optical baseline, y∞, while the fraction diluted, fracdil, was identical within error. Thus, even though calibration was required on a per-chip basis, each chip is capable of highly reproducible fluid manipulations.
Figure 3.

Metering requires calibration but is highly reproducible. The peristaltic pump (6) meters discrete amounts of fluid into the ring every time it is cycled (21). (A) To calibrate metering volumes against the number of pump cycles, fluorophore in the reaction ring (cartoon, blue) was displaced by metering a variable number of pump cycles of buffer (cartoon, grey) into the ring. The remaining fluorophore was mixed with buffer, and quantified by measuring fluorescence. Eight pump cycles (orange circle) displace (1–0.58)(5 nl) = 2.1 nl, where 5 nl is the calculated ring volume. (B) Repeatedly metering and mixing the same amount of buffer creates a serial dilution. Here, eight pump cycles serially dilute the sample by a factor of 1/0.58 = 1.7 per cycle. As serial dilutions would amplify metering error, the similar curves demonstrate a high degree of reproducibility.
Monitoring Mg2+-induced RNA folding
The 3WJ RNA construct is a synthetic fragment of the bacterial 30S ribosomal subunit, an 85 nt RNA corresponding to the junction of 16S rRNA helices 20, 21 and 22 (Figure 4A), that is the minimal binding site for the 12 kDa protein S15 (23,22,26). Binding of Mg2+ ions or S15 induces a conformational change in the 3WJ which can be monitored by Förster resonance energy transfer (FRET, Figure 4B). The affinity of the 3WJ RNA for Mg2+ was measured by mixing 3WJ with serial dilutions of Mg2+ ions and observing the change in FRET between the two fluorophores on the RNA (Figure 5A). Upon folding with Mg2+, FRET between Atto425 and Atto532 (Figure 5A) quenches Atto425 emission by ∼55%. To quantify folding, relative FRET change was measured as the ratio of acceptor photons over the sum of both donor and acceptor photons. As substantial direct excitation of the acceptor occurs due to the broad two-photon excitation spectra, this measurement of relative FRET is not equivalent to the FRET efficiency, E, but over the small range of relative FRET efficiencies reported (0.4–0.5), the ratio is expected to be proportional to the fraction of RNA folded. The high rate of direct excitation of acceptor does not appreciably change the behavior of the donor fluorophore. Errors were generated by propagating the measured standard deviations of the donor and acceptor photon counts.
Binding reactions were initiated in the chip's; reaction ring by washing with buffer before staging and metering a stock solution of 15 mM MgCl2. The Mg2+ in the reaction ring was then diluted with 0.42 ± 0.01 reaction ring volumes (eight pump cycles) of buffer before the ring was isolated from the supply manifold and the contents were mixed using rotary mixing. For a dilution series, this step was repeated incrementally in each subsequent experiment to generate the logarithmic series of Mg2+ -ion concentrations such that in the n-th trial, [Mg2+] = (15 mM)(1–0.42)n. The dilution factor (1–0.42 ± 0.01) was empirically determined by repeating an identical series of serial dilutions with the fluorophore Alexa488 in place of Mg2+ and fitting the fluorescence intensity as described in ‘Materials and Methods’. Finally, the 3WJ RNA was staged and 0.14 ± 0.02 ring volumes were metered and mixed in the reaction ring while fluorescence was monitored.
Five independent titration series were recorded (Figure 5B) and independently fit to the Hill equation to obtain the apparent Mg2+-3WJ affinity, Kapp. The average apparent affinity, Kapp = 93 ± 6 μM, was comparable with the value obtained in a conventional fluorometric plate reader, Kapp = 68 ± 5 μM (data not shown) and by others, Kapp = 77 μM (24).
3WJ–S15 binding kinetics
Microfluidic assembly assays require that all components are active inside the chip, and present at the assumed concentration. This is not the case in the absence of detergents, as S15 adsorbs to the PDMS walls of the microfluidic reactor and is unable to react. These interactions were disrupted with a specific cocktail of detergents and blocking agents (Tween-20, Tween-80, BSA and tRNA). The detergents and blocking agents were chosen from a small screen which analyzed their ability to eliminate fluorescent S15 adhesion to PDMS walls, both during initial injection and after prolonged use, while still preserving activity in single-concentration binding assays (data not shown). Activity was later verified by quantitative binding assays (below). Reliability was further increased by flushing the chip with a continuous flow of S15 prior to metering the protein. This increased S15 consumption to roughly 200 nl/reaction. Detergents which failed in the present work include Pluronic F68 (inhibited S15·3WJ binding), Pluronic F127 (not resistant to continuous flow of protein, promoted aggregation), and Dodecyl–β–D–maltoside (32) (not resistant to continuous flow of protein).
Binding between the labeled 3WJ RNA and protein S15 was detected by introducing a third dye to the FRET system. Fluorophore Alexa 594 was conjugated to S15 (Figure 6A). When labeled 3WJ and S15 bind, the third dye acts as an efficient terminal FRET acceptor and quenches both dyes on the RNA (E > 96%). Reactions were again run on the 5 nl scale. Binding was initiated by metering 3WJ RNA into the reaction ring pre-filled with varying concentrations of S15 and mixing immediately (Figure 6B), counting photons with the microscope during the entire process. The dead-time of the mixer is apparent as decaying oscillations in the log plot in Figure 6B; mixing occurs at a rate of 0.4 s−1. Fluorescence of S15-Alexa594 decreases ∼12% when mixing is stopped, irrespective of the presence of RNA (data not shown). This is attributed to a buildup of photo-bleached fluorophores which would otherwise have washed out of the focal volume during mixing.
To measure the bimolecular association rate (Kon) and the unimolecular disassociation rate (Koff) of the S15–3WJ complex, a series of reactions were run at different initial protein concentrations. Similar to the microfluidic manipulations used to generate different concentrations of Mg2+, on-chip serial dilutions created different starting concentrations of S15-A594 in the range of [3 nM–1.4 μM], while the RNA concentration was held constant at 17 nM. Four replicate data sets were collected, one such set is shown in Figure 6C.
Reactions were quantitated by monitoring quenching of the primary FRET donor, Atto425. The intensity of the terminal FRET acceptor Alexa594 was unreliable due to the intensity fluctuations described above. In addition, the donor quenching method provides a linear response over the large FRET change exhibited by the RNA–protein FRET construct. Reaction curves were fit to a single exponential decay, 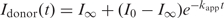 , with a globally fixed initial amplitude, I0, and a varying reaction extent, I∞. Reduced χ2 values for the fits fell within the range (0.2–0.3). The fraction of 3WJ which is bound to S15 is apparent in the reaction traces as the ratio I∞/I0, and increases as expected when serial dilutions decrease and S15 concentrations increases]. The fraction does not level off cleanly at low serial dilutions, and this likely indicates non specific binding of S15 to the 3WJ rRNA, a phenomenon previously observed in electrophoretic gel mobility assays with S15 (23). The fastest rate measured was 0.2 s−1, and in this case observation of binding kinetics is limited by the dead time of the reactor.
, with a globally fixed initial amplitude, I0, and a varying reaction extent, I∞. Reduced χ2 values for the fits fell within the range (0.2–0.3). The fraction of 3WJ which is bound to S15 is apparent in the reaction traces as the ratio I∞/I0, and increases as expected when serial dilutions decrease and S15 concentrations increases]. The fraction does not level off cleanly at low serial dilutions, and this likely indicates non specific binding of S15 to the 3WJ rRNA, a phenomenon previously observed in electrophoretic gel mobility assays with S15 (23). The fastest rate measured was 0.2 s−1, and in this case observation of binding kinetics is limited by the dead time of the reactor.
Apparent rates were averaged from four replicate data sets, and the error was estimated as the standard deviation. The average is plotted against the number of serial dilutions in Figure 6D. As expected, increasing numbers of serial dilutions lower the concentration of S15 at the beginning of the reaction and reactions are slower. Data were fit according to Equation 1, with a reduced χ2 of 0.67. The fit value of fracdil is 16% lower than the value obtained by a fluorophore calibration scheme similar to the one used for the Mg2+ experiments. This difference is likely attributable to the observed weak protein–PDMS interactions. From the fits, the bimolecular association rate is kon=6 × 105 ± 0.4 × 105 M−1 s−1, and the unimolecular disassociation rate is koff=5 × 10−3 ± 0.9 × 10−3 s−1. The equilibrium disassociation constant was calculated as the ratio of koff/kon, KD = 8 ± 2 nM.
The association rate is ∼30× faster than previously reported rates (23,26). The difference in rates is due to different salt concentrations in buffer, notably 50 mM K–acetate in previous reactions. Microliter-scale reactions with the same materials but with buffer conditions similar to previous works were 30× slower than microfluidics, as expected (kon = 2 × 104 M−1 s−1, data not shown). The association rate measured in the microfluidic reactor is 2× slower than the rate measured with 150 μl-scale reactions with the same materials and no salt (kon = 1.4 × 106 ± 0.5 × 106 M−1 s−1, data not shown). The binding constant measured in the microfluidic reactor agrees with microliter-scale direct titration, KD = 10.7 ± 0.8 nM (data not shown) and with a variety of similar constructs and buffer conditions [0.24–16 nM (23,24,26,33)].
DISCUSSION
The ‘Riboreactor’ is a microfluidic mixer designed to measure macromolecular assembly kinetics by preparing a wide variety of mixtures of two to eight components and monitoring reactions over long time scales. Ultimately, the chip and optical platform are intended to dissect the kinetics and mechanism of complex reactions that have been inaccessible to date due to the difficulty of preparing and analyzing a large number of systematically differing reactions in a consistent and reproducible manner.
Previous microfluidic mixer chips (13–15,34) function by mixing two continuous streams of reagents. While this allows mixing on the order of microseconds, input is limited to two solutions which are mixed at a ratio fixed by chip geometry and input pressure. Retaining a sample on a continuous flow chip for more than a few seconds requires a tradeoff between impractically low flow rates and high sample consumption. The Riboreactor does not use continuous flow, but prepares and analyzes reactions in batch using quantized fluid metering and a storage ring (21) at the expense of slow mixing times that are on the order of 10 s. 5-nl scale reactions are prepared and initiated in a reaction ring, and held in place for an arbitrary length of time. Evolution of the reaction can be directly observed in situ with fluorescence detection, and the microscope focus does not need to move through the chip to capture different time points. Reaction compositions are not determined by chip architecture, but by software, as computer-controlled valve actuations meter and mix arbitrary amounts of the eight input solutions within the reaction ring. Repeated metering and mixing with buffer affords serial dilutions (35) with variable carry-over fractions.
Data from samples in microfluidic chip channels can be recorded using a variety of fluorescence microscopy techniques, including widefield imaging and the more technically demanding two-photon-based microscopy. Two-photon excitation was particularly useful to reduce photobleaching of the 5 nl sample, though it was by no means essential for monitoring samples in the chip. Two-photon excitation reduced typical photo-bleaching half-lives to approximately two days, while enabling ultra-sensitive, localized probing of the microfluidic chip, and facile implementation of single-molecule and small-ensemble experiments such as fluorescence correlation spectroscopy (FCS) (36) or fluorescence intensity distribution analysis (FIDA) (31).
As previous work with S15 required detergents and blocking agents to prevent nonspecific absorption to quartz cuvettes and polystyrene plates (24,23), and as nonspecific protein–PDMS interactions had been reported (37,25), care was taken to optimize buffer conditions. With detergents (Tween-20, Tween-80), a blocking agent (BSA), and a low-affinity interaction partner (tRNA), S15–PDMS interactions were weak enough and exchanged fast enough that they did not interfere with measurements. Efforts are ongoing to make multilayer soft-lithography microfluidic chips more biocompatible by developing PDMS derivatives (38) or channel coating chemistries (25), that should only increase the range of applicability. Nevertheless, PDMS chips are already extremely versatile and useful platforms for quantitative biochemistry.
The Riboreactor was used to directly measure RNA–protein binding kinetics and RNA-ion binding, using a well-studied 3WJ RNA which constitutes the minimal binding site for ribosomal protein S15 (22–24,26). The bimolecular association constant and the unimolecular disassociation constant were obtained (kon=6 × 105 ± 0.4 × 105 M−1 s−1, koff = 5 × 10−3 ± 0.9 × 10−3s−1) in good agreement with conventional microliter-scale assays using the same materials and with previous studies (23,26,24). Mg2+-dependent RNA folding was observed inside the chip, with FRET changes of the expected direction and magnitude and modest differences between the chip-based affinity (Kapp = 93 ± 6 μM) and the conventionally obtained affinity (Kapp = 68 ± 5 μM).
RNA–protein and RNA–Mg2+ binding reactions were highly reproducible, which is likely the result of robust automation and mechanical stability of the chip. Due to long staging times before sample injection, sample consumption was greater than expected for S15 (200 nl per binding reaction) but was still small by conventional standards. Together, these measurements establish the validity of the Riboreactor setup for the study of RNP assembly kinetics.
The Riboreactor platform is generally applicable to any complex reaction with slow kinetics and combinatorial complexity, although it was specifically developed to analyze assembly of the bacterial 30S ribosomal subunit, in which 20 proteins bind one large rRNA in a hierarchical manner. Complex reaction mechanisms can only be unraveled by systematically varying the starting concentration of each of the reactants and globally analyzing the entire data set (18). The ability to automate sample dilution and sample mixing and low sample consumption are the key advantages of the microfluidic platform for this class of kinetic problems. The Riboreactor is a powerful new tool for quantitative investigation of highly complex reactions that can be readily adapted to many other applications involving multicomponent mixing.
FUNDING
National Institutes of Health (R37-GM-53757 to J.R.W., DP-OD000217 to R.P.); National Science Foundation Graduate Research Fellowship (to W.K.R.). Funding for open access charge: National Institutes of Health (R37-GM-53757 to J.R.W.).
Conflict of Interest. None declared.
ACKNOWLEDGEMENTS
The authors would like to thank E. Kompfner for expression of the His-tagged S15 construct, Y. Gambin, R. Gómez-Sjöberg, C. Hansen, A. Karnaukhov, H.-J. Lee and S. Quake for helpful discussions, and D. Millar and J. Tainer for access to laboratory equipment.
REFERENCES
- 1.Clausell-Tormos J, Lieber D, Baret J, El-Harrak A, Miller OJ, Frenz L, Blouwolff J, Humphry KJ, Köster S, Duan H, et al. Droplet-based microfluidic platforms for the encapsulation and screening of mammalian cells and multicellular organisms. Chem. Biol. 2008;15:427–437. doi: 10.1016/j.chembiol.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 2.Weibel D, DiLuzio W, Whitesides G. Microfabrication meets microbiology. Nat. Rev. Microbiol. 2007;5:209–218. doi: 10.1038/nrmicro1616. [DOI] [PubMed] [Google Scholar]
- 3.Kartalov EP, Quake SR. Microfluidic device reads up to four consecutive base pairs in DNA sequencing-by-synthesis. Nucleic Acids Res. 2004;32:2873–2879. doi: 10.1093/nar/gkh613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whitesides G. The origins and the future of microfluidics. Nature. 2006;442:368–373. doi: 10.1038/nature05058. [DOI] [PubMed] [Google Scholar]
- 5.Duffy D, McDonald J, Schueller O, Whitesides G. Rapid prototyping of microfluidic systems in poly(dimethylsiloxane) Anal. Chem. 1998;70:4974–4984. doi: 10.1021/ac980656z. [DOI] [PubMed] [Google Scholar]
- 6.Unger M, Chou H, Thorsen T, Scherer A, Quake S. Monolithic microfabricated valves and pumps by multilayer soft lithography. Science. 2000;288:113–116. doi: 10.1126/science.288.5463.113. [DOI] [PubMed] [Google Scholar]
- 7.Melin J, Quake SR. Microfluidic large-scale integration: the evolution of design rules for biological automation. Ann. Rev. Biophys. Biomol. Struct. 2007;36:213–231. doi: 10.1146/annurev.biophys.36.040306.132646. [DOI] [PubMed] [Google Scholar]
- 8.Henares T, Mizutani F, Hisamoto H. Current development in microfluidic immunosensing chip. Anal. Chim. Acta. 2008;611:17–30. doi: 10.1016/j.aca.2008.01.064. [DOI] [PubMed] [Google Scholar]
- 9.Liu X, Liu X, Liang A, Shen Z, Zhang Y, Dai Z, Xiong B, Lin B. Studying protein-drug interaction by microfluidic chip affinity capillary electrophoresis with indirect laser-induced fluorescence detection. Electrophoresis. 2006;27:3125–3128. doi: 10.1002/elps.200500890. [DOI] [PubMed] [Google Scholar]
- 10.Yeh H, Puleo C, Lim T, Ho Y, Giza P, Huang R, Wang T. A microfluidic-FCS platform for investigation on the dissociation of Sp1-DNA complex by doxorubicin. Nucleic Acids Res. 2006;34:e144. doi: 10.1093/nar/gkl787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maerkl S, Quake S. A systems approach to measuring the binding energy landscapes of transcription factors. Science. 2007;315:233–237. doi: 10.1126/science.1131007. [DOI] [PubMed] [Google Scholar]
- 12.Einav S, Gerber D, Bryson PD, Sklan EH, Elazar M, Maerkl SJ, Glenn JS, Quake SR. Discovery of a hepatitis C target and its pharmacological inhibitors by microfluidic affinity analysis. Nat. Biotechnol. 2008;26:1019–1027. doi: 10.1038/nbt.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brody JP, Yager P, Goldstein RE, Austin RH. Biotechnology at low Reynolds numbers. Biophys. J. 1996;71:3430–3441. doi: 10.1016/S0006-3495(96)79538-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knight JB, Vishwanath A, Brody JP, Austin RH. Hydrodynamic focusing on a silicon chip: mixing nanoliters in microseconds. Phys. Rev. Lett. 1998;80:3863–3866. [Google Scholar]
- 15.Yao S, Bakajin O. Improvements in mixing time and mixing uniformity in devices designed for studies of protein folding kinetics. Anal. Chem. 2007;79:5753–5759. doi: 10.1021/ac070528n. [DOI] [PubMed] [Google Scholar]
- 16.Park HY, Kim SA, Korlach J, Rhoades E, Kwok LW, Zipfel WR, Waxham MN, Webb WW, Pollack L. Conformational changes of calmodulin upon Ca2+ binding studied with a microfluidic mixer. Proc. Natl Acad. Sci. USA. 2008;105:542–547. doi: 10.1073/pnas.0710810105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Srisa-Art M, Dyson EC, deMello AJ, Edel JB. Monitoring of real-time streptavidin and biotin binding kinetics using droplet microfluidics. Anal. Chem. 2008;80:7063–7067. doi: 10.1021/ac801199k. [DOI] [PubMed] [Google Scholar]
- 18.Karnaukhov AV, Karnaukhova EV, Williamson JR. Numerical matrices method for nonlinear system identification and description of dynamics of biochemical reaction networks. Biophys. J. 2007;92:3459–3473. doi: 10.1529/biophysj.106.093344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Talkington M, Siuzdak G, Williamson J. An assembly landscape for the 30S ribosomal subunit. Nature. 2005;438:628–632. doi: 10.1038/nature04261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adilakshmi T, Bellur D, Woodson S. Concurrent nucleation of 16S folding and induced fit in 30S ribosome assembly. Nature. 2008;455:1268–1272. doi: 10.1038/nature07298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hansen CL, Sommer MOA, Quake SR. Systematic investigation of protein phase behavior with a microfluidic formulator. Proc. Natl Acad. Sci. USA. 2004;101:14431–14436. doi: 10.1073/pnas.0405847101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Orr J, Hagerman P, Williamson J. Protein and Mg2+-induced conformational changes in the S15 binding site of 16 s ribosomal RNA. J. Mol. Biol. 1998;275:453–464. doi: 10.1006/jmbi.1997.1489. [DOI] [PubMed] [Google Scholar]
- 23.Batey RT, Williamson JR. Interaction of the Bacillus stearothermophilus ribosomal protein S15 with 16 S rRNA: I. Defining the minimal RNA site. J. Mol. Biol. 1996;261:536–549. doi: 10.1006/jmbi.1996.0481. [DOI] [PubMed] [Google Scholar]
- 24.Klostermeier D, Sears P, Wong C, Millar D, Williamson J. A three-fluorophore FRET assay for high-throughput screening of small-molecule inhibitors of ribosome assembly. Nucleic Acids Res. 2004;32:2707–2715. doi: 10.1093/nar/gkh588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sui G, Wang J, Lee C, Lu W, Lee SP, Leyton J, Wu A, Tseng H. Solution-phase surface modification in intact poly (dimethylsiloxane) microfluidic channels. Anal. Chem. 2006;78:5543–5551. doi: 10.1021/ac060605z. [DOI] [PubMed] [Google Scholar]
- 26.Batey RT, Williamson JR. Effects of polyvalent cations on the folding of an rRNA three-way junction and binding of ribosomal protein S15. RNA. 1998;4:984–997. doi: 10.1017/s1355838298980426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eigen M, DeMaeyer L. Relaxation methods. In: Friess SL, Lewis S, Weissberger A, editors. Technique of Organic Chemistry. VIII, Part II. New York: Wiley; 1963. pp. 895–1054. [Google Scholar]
- 28.Thorsen T, Maerkl S, Quake S. Microfluidic large-scale integration. Science. 2002;298:580–584. doi: 10.1126/science.1076996. [DOI] [PubMed] [Google Scholar]
- 29.Mertz J, Xu C, Webb W. Single-molecule detection by two-photon-excited fluorescence. Opt. Lett. 1995;20:2532–2524. doi: 10.1364/ol.20.002532. [DOI] [PubMed] [Google Scholar]
- 30.Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- 31.Kask P, Palo K, Ullmann D, Gall K. Fluorescence-intensity distribution analysis and its application in biomolecular detection technology. Proc. Natl Acad. Sci. USA. 1999;96:13756–13761. doi: 10.1073/pnas.96.24.13756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang B, Wu H, Kim S, Zare R. Coating of poly (dimethylsiloxane) with n-dodecyl-β-d-maltoside to minimize nonspecific protein adsorption. Lab Chip. 2005;5:1005–1007. doi: 10.1039/b509251e. [DOI] [PubMed] [Google Scholar]
- 33.Ha T, Zhuang X, Kim HD, Orr JW, Williamson JR, Chu S. Ligand-induced conformational changes observed in single RNA molecules. Proc. Natl Acad. Sci. USA. 1999;96:9077–9082. doi: 10.1073/pnas.96.16.9077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lipman E, Schuler B, Bakajin O, Eaton W. Single-molecule measurement of protein folding kinetics. Science. 2003;301:1233–1235. doi: 10.1126/science.1085399. [DOI] [PubMed] [Google Scholar]
- 35.Paegel BM, Grover WH, Skelley AM, Mathies RA, Joyce GF. Microfluidic serial dilution circuit. Anal. Chem. 2006;78:7522–7527. doi: 10.1021/ac0608265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dittrich PS, Schwille P. Spatial two-photon fluorescence cross-correlation spectroscopy for controlling molecular transport in microfluidic structures. Anal. Chem. 2002;74:4472–4479. doi: 10.1021/ac025625p. [DOI] [PubMed] [Google Scholar]
- 37.Linder V, Verpoorte E, Thormann W, de Rooij N, Sigrist H. Surface biopassivation of replicated poly(dimethylsiloxane) microfluidic channels and application to heterogeneous immunoreaction with on-chip fluorescence detection. Anal. Chem. 2001;73:4181–4189. doi: 10.1021/ac010421e. [DOI] [PubMed] [Google Scholar]
- 38.Rolland J, VanDam R, Schorzman D, Quake S, DeSimone J. Solvent-resistant photocurable “liquid teflon” for microfluidic device fabrication. J. Am. Chem. Soc. 2004;126:2322–2323. doi: 10.1021/ja031657y. [DOI] [PubMed] [Google Scholar]
- 39.Cannone JJ, Subramanian S, Schnare MN, Collett JR, D'S;ouza LM, Du Y, Feng B, Lin N, Madabusi LV, Muller KM, et al. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics. 2002;3:1471–2105. doi: 10.1186/1471-2105-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]