


Abstract
Increased proliferation and elevated levels of protein synthesis are characteristics of transformed and tumor cells. Though components of the translation machinery are often misregulated in cancers, what role tRNA plays in cancer cells has not been explored. We compare genome-wide tRNA expression in cancer-derived versus non-cancer-derived breast cell lines, as well as tRNA expression in breast tumors versus normal breast tissues. In cancer-derived versus non-cancer-derived cell lines, nuclear-encoded tRNAs increase by up to 3-fold and mitochondrial-encoded tRNAs increase by up to 5-fold. In tumors versus normal breast tissues, both nuclear- and mitochondrial-encoded tRNAs increase up to 10-fold. This tRNA over-expression is selective and coordinates with the properties of cognate amino acids. Nuclear- and mitochondrial-encoded tRNAs exhibit distinct expression patterns, indicating that tRNAs can be used as biomarkers for breast cancer. We also performed association analysis for codon usage-tRNA expression for the cell lines. tRNA isoacceptor expression levels are not geared towards optimal translation of house-keeping or cell line specific genes. Instead, tRNA isoacceptor expression levels may favor the translation of cancer-related genes having regulatory roles. Our results suggest a functional consequence of tRNA over-expression in tumor cells. tRNA isoacceptor over-expression may increase the translational efficiency of genes relevant to cancer development and progression.
INTRODUCTION
Misregulation of components of the translation machinery can lead to malignant transformation (1–3). In particular, deregulation of RNA polymerase III (polIII) and its products has been observed in a wide range of transformed cells, including ovarian and breast carcinomas (4–8). Over-expressed polIII RNAs include transfer RNAs (tRNA) which are directly involved in translation. A recent report demonstrates that such deregulation is not only a by-product but can drive transformation (9). In that study, modest over-expression of initiator tRNA drives cell proliferation and results in malignant transformation of immortalized mouse fibroblasts. As for other tRNAs, their expression or activity can control translational elongation because they read mRNA codons embedded in the coding sequence of each gene at various speeds (10). tRNA levels may therefore be relevant for the translation of key genes required for the tumorigenic process, and may play a central role in cancer development and progression.
A major challenge in cancer research is the identification of accurate molecular signatures for diagnosis and treatment (11–13). Though diverse genomic and proteomic approaches have been applied to this problem, no genome-wide studies of tRNA expression have been carried out in the context of breast cancer. Approximately 450 tRNA genes comprised of over 270 different sequences have been annotated in the human genome, with 22 additional genes present in human mitochondrial DNA (14,15). Human tRNAs are grouped into 49 isoacceptor families to decode all codons specified by the genetic code (16).
Here we describe the comparative analysis of tRNA levels for non-cancer-derived breast epithelial cell lines and breast cancer cell lines, as well as breast tumor and normal breast tissue samples. We apply a previously described tRNA microarray used to study tissue-specific expression of human tRNAs (17). We show that tRNA levels are significantly elevated for both tumor cell lines and tumor tissues. We also develop a chemical ligation method and apply it in a microarray format for the detection and comparison of all tRNA isoacceptors in the breast cell lines. We perform association analysis of tRNA isoacceptors to the codon usage of the house-keeping genes, the cell line-specific genes, and cancer-related genes. We find that the cell line-specific genes and cancer-related genes have similar codon usages that are distinct from house-keeping genes. Over-expressed tRNA isoacceptors do not favor the codon usage of the house-keeping genes or the cell line-specific genes. Instead, a subset of the over-expressed tRNAs favors over-represented codons among the cancer-related genes.
MATERIALS AND METHODS
Cell lines and RNA isolation
All cell lines were purchased from American Type Culture Collection (ATCC, Manassas, VA) and maintained according to ATCC recommendations. MCF7 cells were cultured in DMEM medium (Invitrogen, 11965-092) supplemented with 10% FBS and 1% penicillin/streptomycin (P/S). MCF10A cells were cultured in 1:1 (+I factor, Vectro)-DMEM/F12 (Invitrogen, 11330-032) supplemented with 10% FBS, 1% P/S, 5 μg/ml insulin, 10 ng/ml EGF and 0.5 μg/ml hydrocortisone. All other cell lines were cultured in RPMI 1640 1 × medium (Mediatech, 10-040-CV) supplemented with 10% FBS and 1% P/S.
Total RNA for each cell line was obtained at 80–90% confluency using the mirVana™ miRNA Isolation Kit (Ambion, AM1560). This procedure isolates RNA species as short as 15 nucleotides and is therefore not biased against tRNA. Total RNA quality was verified on an agarose gel.
Normal breast and breast tumor total RNA
Two samples of human breast total RNA were purchased from Ambion (FirstChoice® Human Breast Total RNA, AM6952, lot numbers 0808001 and 0812006). An additional sample of human breast total RNA was purchased from Stratagene (MVP Total RNA Human Breast, 540045, lot number 0380023). Both providers certify small RNA content in total RNA samples and provide relevant donor information (Table S1).
Breast tumor samples were obtained from the Human Tissue Resource Center (HTRC) at the University of Chicago (Table S1). Pathology reports were provided for all samples. All breast tumor samples were obtained frozen and embedded in OCT (optimal cutting temperature compound). Following removal of the surrounding OCT and tissue pulverization, total RNA was isolated using TRIzol reagent (Invitrogen, 15596-018). Total RNA quality was checked on agarose gels.
Chemical ligation substrates and templates
All oligodeoxynucleotides were synthesized on an Expedite Nucleic Acid Synthesis System. 5′-Iodo-oligodeoxyribonucleotides were prepared by reacting the 5′-OH of the oligodeoxyribonucleotide which remains on a solid support with methyltriphenoxyphosphonium iodide, followed by standard deprotection. 3′-Phosphorothioate oligodeoxyribonucleotides were prepared using a 3′-phosphate CPG column (Glen Research). After the first coupling with the phosphoramidite, the phosphorus was sulfurized with Sulfurizing Reagent II (Glen research) instead of oxidized with iodine. Synthesis and deprotection proceeded as with standard DNA synthesis.
30-mer RNA template oligonucleotides were designed to contain minimal secondary structure when the nucleotide at the 15th position is U (18). Four chemically synthesized 30-mer RNAs have the same sequence at all positions except the 16th nucleotide (I, U, G, C). Chemically synthesized 30-mer RNAs were purchased from Dharmacon Research, de-protected, and purified according to manufacturer's; protocols. When needed, RNA oligonucleotides were purified on denaturing polyacrylamide gels containing 7 M urea. Yeast tRNAPhe was purchased from Sigma–Aldrich (R4018) and used without further purification.
Chemical ligation reactions
For 30-mer model RNAs, reactions were carried out with 0.2 μM RNA template, 0.7 μM 5′-iodo probe, and 0.5 μM 5′-32P-labeled, 3′-phosphorothioate probe in 50 mM pH 7.5 Tris–HCl containing 1.5 mM EDTA, 100 mM NaCl, and 1 mM TCEP at 30°C for 1 h. tRNAPhe reactions were carried out with 0.05–0.15 μM RNA template, 0.26 μM 5′-iodo substrate, and 0.065 μM 5′-32P-labeled, 3′-phosphorothioate probe in 20 mM pH 7.5 Tris–HCl containing 1.5 mM EDTA, 200 mM NaCl, and 1 mM TCEP at 60°C for 24 h. Total human RNA reactions were carried out with 1–6 μg total RNA, 0.26 μM each 5′-iodo substrate, and 0.065 μM each Cy3-labeled, 3′-phosphorothioate probe in 20 μl 20 mM pH 7.5 Tris–HCl containing 1.5 mM EDTA, 200 mM NaCl, and 1 mM TCEP. Reactions were incubated at 60°C for 24 h. Following ligation, all reaction mixtures were treated with 0.1 U/μl RNase H (Epicentre Biotechnologies, R0601K). The reactions were quenched with an equal volume of 8 M urea/100 mM EDTA, boiled for 2 min and rapidly cooled on ice prior to loading on 10–15% denaturing polyacrylamide gels containing 7 M urea. Radiolabeled ligation products were visualized on a FUJIX BAS1000 Phosphorimager and quantified using the accompanying software. Fluorescent-labeled ligation products were visualized on a PharosFX Molecular Imager and quantified using QuantityOne software.
Standard tRNA microarrays
The standard tRNA microarray experiment consists of four steps starting from total RNA: (i) deacylation to remove remaining amino acids attached to the tRNA, (ii) selective Cy3/Cy5 labeling of tRNA, (iii) hybridization on commercially printed arrays and (iv) data analysis.
Deacylation: 0.25 μg/μl total RNA premixed with three tRNA standards (Escherichia coli tRNALys, E. coli tRNATyr, and yeast tRNAPhe) at 0.2 μM each was incubated in 100 mM Tris–HCl (pH 9.0) at 37°C for 30 min. The solution was neutralized by the addition of an equal volume of 100 mM Na-acetate/acetic acid (pH 4.8) plus 100 mM NaCl, followed by ethanol precipitation. Deacylated total RNA was dissolved in water, and its integrity verified by agarose gel electrophoresis.
Cy3/Cy5 labeling: tRNA in the total RNA mixture was selectively labeled with either Cy3 or Cy5 fluorophore using an enzymatic ligation method previously described. The ligation reaction relies on an 8-base pair RNA:DNA hybrid helix containing a Cy3 or Cy5 fluorophore pre-attached to the loop and an overhang complementary to the universally conserved 3′CCA nucleotides present in all tRNAs. The ligation reaction was carried out overnight (∼16 h) at 16°C with 1 U/μl T4 DNA ligase (USB Corporation, 70042X) and 9 μM labeling oligonucleotide.
Hybridization: hybridization was performed in a GeneMachines Hyb4 at 60°C overnight (16 h) with 1–2.5 μg each of Cy3- or Cy5-labeled total RNA as previously described. Multiple arrays were run using the MCF10A reference sample labeled with either Cy3 or Cy5.
Data analysis: arrays were scanned using a GenePix 4000b scanner (Axon Instruments). For both Cy3 and Cy5 wavelengths, PMT gain was set at 600 and power at 100%. These settings were chosen to provide optimal signal without saturation. Array images were generated and analyzed using GenePix 6.0 software. GenePix adaptive circle spot segmentation was used for image analysis. To account for differences in labeling and hybridization efficiencies, the following normalization procedure was used: first, the median Cy5/Cy3 ratio at each probe spot was divided by the corresponding MCF10A Cy5/Cy3 reference ratio. Second, the obtained value was divided by the averaged Cy5/Cy3 ratio value for the three tRNA standards added in (i) (E. coli tRNALys, E. coli tRNATyr and yeast tRNAPhe).
The standard microarray method described above uses a very standard, two-color microarray method extensively used for mRNA analysis. Compared to mRNA arrays, the main difference is the previously developed method for the labeling of tRNA in total RNA samples. The reproducibility of the tRNA array method and result validation by Northern blots have been extensively described in previously published papers (17,19–22).
All microarray data has been deposited in GEOarchive (accession numbers GSE17945, GPL9143).
Chemical ligation tRNA Microarrays
These microarray experiments consist of four steps starting from total RNA: (i) chemical ligation to detect target tRNA isoacceptors, (ii) purification of chemical ligation products, (iii) hybridization, and (iv) data analysis.
Chemical ligation: 5 μg total RNA was premixed with 4 pmol each of three tRNA standards (E. coli tRNALys, E. coli tRNATyr, and yeast tRNAPhe). Reactions were carried out with 5 μg total RNA, 0.26 μM of each 5′-iodo probe, and 0.065 μM of each Cy3- or Cy5-labeled, 3′-phosphorothioate probe in 50 μl 15 mM pH 7.5 Tris–HCl containing 1 mM EDTA, 200 mM NaCl, and 1 mM TCEP. Reactions were incubated at 60°C for 24 h, and then treated with 0.1 U/μl RNase H. The reactions were quenched with an equal volume of 8 M urea/100 mM EDTA, then boiled for 2 min and rapidly cooled on ice prior to loading on 10% denaturing polyacrylamide gels containing 7 M urea.
Purification: chemical ligation products were purified on 10% denaturing polyacrylamide gels containing 7 M urea. Ligation products were visualized on a PharosFX Molecular Imager, and the corresponding band cut out for purification.
Hybridization.
Data analysis: these are identical to those for the standard arrays described above.
Cell proliferation
Proliferation rates were measured over four days using the Promega CellTiter Blue metabolic assay. Cells were plated at low (1500 cells/well) and high (5000 cells/well) cell density in 100 μl medium in 96 well-plates. A total of 20 μl CellTiter Blue reagent was added to each well at the indicated timepoints. Fluorescence (560ex/590em) was measured after 2 h incubation at 37°C. Fluorescence is proportional to the number of cells present and is normalized to fluorescence at day 0 to account for differences in metabolic efficiency across cell lines. Doubling times were obtained from the equation N/N0 = e(kt). All measurements were performed in triplicate.
RESULTS
Over-expression of tRNAs in breast cancer cell lines
To our knowledge, no systematic genome-wide studies of tRNA expression have been carried out in cancer. To explore the potential of tRNAs to define breast cancer signatures, we used tRNA microarrays to generate comparative tRNA profiles for three non-cancer-derived breast epithelial cell lines (MCF10A, 184 A1, 184 B5) and six breast cancer cell lines (BT-474, HCC70, MCF7, MDA-MB-231, MDA-MB-436, ZR-75-1). The breast cancer cell lines cover a range of physiological and molecular properties (Table S2). Importantly, genome-wide mRNA expression data are available for these cell lines (23) to allow tRNA expression and codon usage association analysis.
We used a previously described array method in the studies of tissue-specific expression of human tRNAs (17). Arrays used for this study contain 40 probes for human nuclear-encoded tRNAs and 22 probes for human mitochondrial-encoded tRNAs; each probe is repeated 21 times. The array also includes 42 probes for bacterial tRNAs and 275 probes complementary to short regions in yeast and human rRNA as hybridization and specificity controls. After isolation of total RNA from cell culture, tRNAs in the sample are labeled by selective ligation to a fluorophore-containing oligonucleotide. The labeled samples are hybridized directly onto the array (Figure 1A). The MCF10A (a non-cancer-derived breast epithelial cell line) sample was included as a reference in all array hybridizations to correct for variations in fluorescence labeling and array manufacturing. Since tRNA constitutes up to 30% of the total RNA, the method requires no amplification and only 1–2 μg of total RNA per array. The specificity of the array is illustrated by the fluorescence signals derived from human tRNA probes compared to the non-human tRNA probes (Figure 1B and C).
Figure 1.

tRNA overexpression in breast cancer cells. (A) Fluorescence labeling scheme of tRNA in total RNA samples. (B) Fluorescence intensity of a human total RNA sample hybridized to the array. “Human” indicates signals for the designated human tRNA probes, “other” signals for bacterial tRNA probes (no signal expected). (C) One of the 48 blocks from a standard tRNA microarray hybridized with MCF10A (Cy5) and MCF7 (Cy3). The schematic indicates the position of human tRNA probes (black) and other probes (white, include ribosomal RNA and bacterial tRNA probes).
Significant differences are observed in the expression levels of tRNA among non-cancer-derived and cancer-derived breast cell lines (Figure 2A and C). The global level of nuclear- and mitochondrial-encoded tRNAs can be approximated separately by the median and mean sample-to-MCF10A ratio. For nuclear-encoded tRNAs, this ratio is 0.7–0.8 for the other non-cancer-derived cell lines and 2–3 for the cancer-derived cell lines. These differences are even more pronounced for mitochondrial-encoded tRNA levels: the sample-to-MCF10A ratio for non-cancer-derived cell lines is ∼0.75, but as much as 5 for the cancer-derived cell lines.
Figure 2.

Relative abundance of nuclear and mitochondrial encoded tRNAs in breast cancer cells. Data is shown for three breast epithelial cell lines (MCF10A, 184 A1, 184 B5) and six breast cancer cell lines (MDA-MB-231, MCF7, HCC70, ZR-75-1, MDA-MB-436, BT-474), all relative to MCF10A. (A) Mean and median values of the nuclear (left) and mitochondrial (right) encoded tRNAs. (B) Total tRNA quantified by agarose gel electrophoresis for all samples. All RNAs are detected by ethidium bromide staining and quantified using a PharosFX Molecular Imager. Fraction of total tRNA was measured relative to the non-tRNA bands in the same lane, and then normalized to that of MCF10A. (C) Expression of nuclear and mitochondrial encoded tRNAs shown as TreeView image. All values are relative to MCF10A. Green indicates a decreased level of expression; red indicates an increased level of expression relative to MCF10A. Data are grouped according to amino acid type. (D) Expression of nuclear and mitochondrial encoded tRNAs normalized to median shown as TreeView image. All values are relative to MCF10A and normalized to the median value for each sample. Green indicates a decreased level of expression; red indicates an increased level of expression relative to median. Data are grouped according to corresponding amino acid type. (E) Same as (D), data are grouped from high to low expression.
The over-expression of tRNA in cancer-derived relative to non-cancer-derived cell lines is also selective: certain individual tRNAs are more strongly over-expressed than others (Figure 2C). Variations in the relative expression of nuclear-encoded tRNAs carrying certain amino acid types are readily observed across cancer-derived cell lines. For example, nuclear-encoded tRNAs carrying polar amino acids (e.g. Ser, Thr, and Tyr) are up-regulated 3- to 4-fold in breast cancer cell lines relative to MCF10A, while nuclear-encoded tRNAs carrying small amino acids (Ala, Cys, Gly, Pro) are up-regulated only 1.5- to 2-fold. These differences can be observed more clearly when the tRNA expression is normalized to the median value for either nuclear- or mitochondrial-encoded tRNAs within each cell line (Figure 2C and D). Differences in the relative expression of tRNA isoacceptors also become more apparent after normalization. For example, tRNALys(UUU) is expressed strongly above the median, while tRNALys(CUU) is expressed below the median. All breast cancer cell lines exhibit a remarkable similarity in relative tRNA expression patterns: tRNAArg(UCU), tRNAArg(CCU), tRNAThr (CGU), tRNASer(CGA), and tRNATyr(GTA) are among the most over-expressed tRNAs, while tRNAHis(GTG), tRNAPhe (GAA), and tRNAMet(CAT) are among the least over-expressed tRNAs. Selective up-regulation of tRNA levels is also observed for mitochondrial-encoded tRNAs: certain tRNAs are expressed up to 2-fold above median, and others are expressed 4-fold below median. While nuclear-encoded tRNA expression patterns are remarkably similar across cell lines, mitochondrial-encoded tRNA expression patterns exhibit greater variations.
tRNA over-expression determined by the microarray method does not necessarily mean that the amount of total tRNA in the cell is increased by the same extent. The median increase for each tRNA species is weighted equally in the microarray measurements regardless of their absolute abundance, whereas abundant tRNAs are over-represented in the total tRNA. Quantitative comparison of total RNA samples by gel electrophoresis shows that the cancer-derived cells still have globally elevated tRNA levels (1.2- to 1.4-fold, Figure 2B), although this increase in total tRNA is smaller compared to the microarray measurements (2- to 3-fold over the median increase). This result highlights the over-expression of low abundant tRNAs in breast cancer cells. Because each tRNA species functions separately in decoding, changes in low-abundant tRNA levels as measured by microarrays should have greater functional significance.
We also determined that tRNA over-expression in breast cancer cell lines does not simply reflect an increase in proliferation rate (Table S3). Under the culture conditions used in this study, doubling times range from 21 to 47 h for the non-cancer-derived cell lines, versus 15–35 h for the cancer-derived cell lines. Thus, the proliferation of all three non-cancer-derived cell lines with low tRNA content is comparable to that of breast cancer cell lines with much higher tRNA content. Doubling times do not correlate with either nuclear or mitochondrial global tRNA levels (not shown).
tRNA over-expression in patient-derived breast tumor samples
In all breast cancer cell lines examined, we observe global tRNA over-expression and differential tRNA isoacceptor expression. To generalize our results in cell lines to breast tumor tissue, we measured tRNA expression patterns in nine patient-derived breast tumor samples and three normal breast tissue samples (Table S3). The nine breast tumor samples were selected from the three major subtypes of breast cancer: luminal (ER+, HER2−), basal (ER−, HER2−), and HER2+. For consistency of data analysis, all samples were run using MCF10A as a reference in array experiments.
As in the breast cancer cell lines, we observe significant differences in global tRNA expression levels among breast tumor and normal breast samples (Figure 3A and B). For nuclear-encoded tRNAs, the mean sample-to-MCF10A ratio is 0.2 to 0.5 for normal breast tissue samples, versus 2 to 4 for breast tumor samples. This result indicates that global nuclear-encoded tRNA levels are increased up to 20-fold in breast tumors relative to normal breast. For mitochondrial-encoded tRNAs, the mean sample-to-MCF10A ratio is 0.4 to 1 for normal breast tissue samples, versus 1.2 to 5 for breast tumor samples. This result indicates that global mitochondrial-encoded tRNA levels are increased up to 13-fold in breast tumors relative to normal breast.
Figure 3.

tRNA over-expression in breast cancer in vivo. Data is shown for three normal breast tissue samples (A-01, A-03, S-23), four ER−/HER2− tumor samples (59826, 60046, 62706, 62944), 2 ER−/HER2+ tumor samples (46258, 58955), and three ER+/HER2− tumor samples (41299, 57731, 45163). All data are relative to MCF10A. (A) Mean and median values of the nuclear (left) and mitochondrial (right) encoded tRNAs. (B) Expression of nuclear and mitochondrial encoded tRNAs shown as TreeView image. All values are relative to MCF10A. Green indicates a decreased level of expression; red indicates an increased level of expression relative to MCF10A. Data are grouped according to amino acid type. (C) Expression of nuclear and mitochondrial encoded tRNAs normalized to median shown as TreeView image. All values are relative to MCF10A and normalized to the median value for each sample. Green indicates a decreased level of expression; red indicates an increased level of expression relative to median. Data are grouped according to corresponding amino acid type. (D) Same as (C), data are grouped from high to low expression.
Consistent with our results on cell lines, tRNA over-expression in breast tumor samples is also selective: certain individual tRNAs are more strongly over-expressed than others (Figure 3C). Among the top 10 over-expressed, nuclear-encoded tRNAs, six tRNA species overlap between cell lines and tumor tissues: tRNAArg(CCU), tRNAThr(CGU), tRNALeu(UAA), tRNATyr(GUA), tRNASer(GCU) and tRNAArg(UCU). We readily observe variations in the relative expression of both nuclear- and mitochondrial-encoded tRNAs. These variations correlate with both the cognate amino acid type and tRNA isoacceptor identity. For example, tRNAs carrying polar (Ser, Thr, Tyr) and charged (Arg, Glu, Lys) amino acids are upregulated 2- to 8-fold, while tRNAs carrying small amino acids (Ala, Cys, Gly, Pro) are upregulated only 1.5- to 2-fold. We also observe variations in the relative expression of tRNA isoacceptors. For example, tRNAArg(CCU) and tRNALys(UUU) are over-expressed to a greater extent than tRNAArg(ICG) and tRNALys(CUU), respectively. These variations become more clear when tRNA expression levels are normalized to the median value within each sample (Figure 3D). Interestingly, the tRNA expression patterns in the three major sub-types of breast cancer are very similar, suggesting that tRNA over-expression is a general consequence for all breast tumors.
We conclude that elevated tRNA levels are a hallmark of breast cancer. Our results suggest that tRNAs can be used as biomarkers for breast cancer, although it is best suited to determine cancer versus normal breast cells. Furthermore, tRNA expression measured by microarrays more accurately reflects the “functional abundance” of individual tRNA species.
Detecting single-nucleotide differences between tRNA isoacceptors
tRNA over-expression across breast cancer cells suggests that elevated tRNA levels may be of functional significance. For instance, differential expression of tRNA isoacceptors may enhance translation of specific cancer-related genes via their codon usages. In order to perform tRNA expression and codon usage association analysis, more comprehensive profiling of all tRNA isoacceptors is desired. Because the microarray method used above works by hybridization alone, it can only distinguish between tRNAs that differ by at least eight nucleotides (17). However, several human tRNA isoacceptors differ by less than eight nucleotides (Table S4). For example, the proline isoacceptors tRNAPro(IGG), tRNAPro(CGG) and tRNAPro(UGG) differ by only one nucleotide; they all hybridize to the same probe on the array above.
We therefore modified a previously described non-enzymatic, nucleic acid-templated, phosphorothioate-iodide autoligation chemistry (24,25) to detect single-base differences between tRNA isoacceptors. This method involves the reaction of a phosphorothioate at the 3′-end of one DNA oligo with an iodide leaving group on the 5′-end of the other oligo (Figure 4A).
Figure 4.

The chemical ligation strategy for detection of single base differences between tRNA isoacceptors. (A) Oligonucleotide reactants complementary to target sequence and mechanism of chemical auto-ligation. For tRNA templates, the ligation junction is between a conserved U and the first position of the anticodon. (B) Overview of the strategy. Chemical ligation proceeds efficiently when the nucleotide at the ligation junction is complementary to the template, but poorly when the nucleotide is mismatched. (C) Ligation yield is proportional to the amount of RNA template. Left: ligation reactions using a defined mixture of X = I and C 30-mer RNA templates, and X = C 3′-phosphorothioate substrate. Right: the percent product from gel analysis is plotted against the fraction of X = I 30-mer RNA template. (D) Relative ligation efficiency with matched and mismatched 3′-phosphorothioate substrates on yeast tRNAPhe(GAA) template. (E) Percent ligation product as a function of total RNA. Left: ligation reaction using varying amounts of human total RNA and substrates for tRNAPro(UGG). Right: the percent product from gel analysis is plotted versus the amount of human total RNA.
In order to determine whether the above set-up allows single base discrimination, we performed chemical ligation using a series of model 30-mer RNAs that differ by a single nucleotide at the 16th position (U15I16, U15C16, U15U16, U15G16) (Figure 4B). While the 5′-iodo substrate was always fully complementary to the RNA template, the base at the 3′-end of the 3′-phosphorothioate substrate was varied (X = A, G, C. X = T was not examined because no tRNA isoacceptor contains A34). For example, when ligations are carried out in defined mixtures of U15I16 and U15C16 templates, the amount of product obtained with the X = C 3′-phosphorothioate substrate increases linearly as the amount of U15I16 template increases. Conversely, the amount of product obtained with the X = G 3′-phosphorothioate substrate decreases linearly as the amount of U15C16 template decreases (Figure 4C). Similar results are obtained with other template–substrate combinations.
In order to confirm that the chemical ligation method works with tRNA, we first applied this method to purified yeast tRNAPhe which has G34 (Figure 4D). The ligation product obtained with mismatched 3′-phosphorothioate substrates (X = A, G) is less than 10% the ligation product obtained with the matched 3′-phosphorothioate substrate (X = C). This confirms that phosphorothioate–iodide autoligation chemistry successfully discriminates in favor of complementary nucleotides at the ligation junction on full-length tRNA templates. Finally, we examined whether human tRNAs could be detected and quantified in a total RNA sample using this autoligation chemistry (Figure 4E). The amount of ligation product obtained with 5′-iodo and 3′-phosphorothioate substrates complementary to tRNAPro(UGG), tRNAPro(IGG), and tRNAArg(UCG) increases linearly with the amount of total human RNA, and therefore reflects the amount of tRNA present in the reaction.
We then applied this chemical ligation method to simultaneously detect and compare selected tRNA isoacceptors in total RNA samples on a microarray platform (Figure 5A). 5′-Iodo and 3′-phosphorothioate oligonucleotide substrates were designed against all tRNA isoacceptors in pairs differing by less than eight nucleotides (Table S4). For each pair, two 3′-phosphorothioate substrates were synthesized with the first favoring one and the second favoring the other isoacceptor. The ligation junction was placed between U33 of the tRNA and the first position of the anticodon. Our chemical ligation microarray includes 10 sense tRNA probes complementary to expected ligation products, 6 sense tRNA probes not complementary to expected ligation products, and 79 antisense tRNA probes complementary to human tRNAs; each probe is repeated eight times. We established the specificity of the human tRNA array by examining the cross-hybridization of chemical ligation products to complementary and control probes present on the same array (Figure 5B).
Figure 5.

tRNA microarray for the analysis of all isoacceptors. (A) General strategy for single-nucleotide resolution tRNA microarrays. (B) Fluorescence intensity of a human total RNA sample hybridized to a single-nucleotide resolution tRNA microarray. “Sense” indicates probes complementary to tRNA chemical ligation products; “Other” indicates probes complementary to tRNA sequences, as well as probes with a tRNA sequence but not complementary to chemical ligation products (no signal expected). (C) Overview of target tRNA isoacceptor abundance in three breast epithelial cell lines (MCF10A, 184 A1, 184 B5), and six breast cancer cell lines (MDA-MB-231, MCF7, HCC70, ZR-75-1, MDA-MB-436, BT-474). (D) Expression of target tRNA isoacceptors shown as TreeView image. All values are relative to MCF10A. Green indicates decreased level of expression relative to MCF10A; red indicates increased level of expression relative to MCF10A. Data are grouped according to amino acid type.
Chemical ligation results also show overall elevated tRNA levels in breast cancer cell lines relative to breast epithelial cell lines (Figure 5C and D). Again, the up-regulation of tRNA levels in cancer-derived relative to non-cancer-derived cells is selective. For instance, tRNAVal(CAC) is decreased 2- to 3-fold in the breast cancer cell lines relative to MCF10A while tRNAVal(IAC) remains unchanged. Similar results are observed for tRNASer(UGA)/tRNASer(IGA) and tRNAAla(CGC)/tRNAAla(UGC).
In conclusion, we successfully developed a microarray method that has a single nucleotide resolution for tRNA isoacceptors. Its application provides comprehensive profiles of all tRNA isoacceptors for breast cancer cell lines.
Relative tRNA abundance and codon usage associations
The differential expression of tRNA isoacceptors can be used to regulate translational efficiency via the codon usage of specific genes. In prokaryotes and fungi, differences in the abundance of tRNA isoacceptors are correlated with codon preferences of genes encoding highly expressed proteins and impact the synthesis of these proteins (26–28). In humans, tissue-specific differences in the expression of individual tRNA species can correlate to the codon usage of highly-expressed, tissue-specific genes, although this correlation was seen only for a small number of tissues (17).
tRNA isoacceptor levels in breast cancer cell lines may also correlate or associate with the codon usage of certain genes that are important for cancer. Finding such associations would suggest an additional level of translational regulation for breast cancer cell lines, and by extension for breast cancer in vivo. At least three groups of genes are relevant in seeking codon usage-tRNA association for cancer cell lines: cell-line specific genes, cancer-related genes and house-keeping genes (Figure 6A). Cell-line specific genes can be important for distinct tumorigenic properties across cell lines; cancer-related genes can be important for general tumor initiation and progression; and house-keeping genes are important for cell growth and architecture.
Figure 6.

Analysis of codon usage versus tRNA over-expression. (A) Three gene groups are relevant in this analysis (mRNA expression level is derived from signals on Affymetrix mRNA arrays). tRNA expression or over-expression when comparing cancer and non-cancer cells are unlikely to positively correlate to the codon usage of all three groups. (B) Codon usage comparison between cell-line specific genes, cancer-related genes and house-keeping genes. The degree of association was assessed using Spearman's; rho (rs). Mean rs values are plotted for the following pairs: cell line versus cell line, cell line versus house-keeping, cancer-related versus cancer-related, and cancer-related versus house-keeping. Error bars indicate standard deviation from the mean. (C) Association of relative tRNA levels to ratios of codon usage between cancer-related and house-keeping genes. As discussed in the text, a positive association is only expected for the codons that are over-represented in the cancer-related genes (x > 2). (D) Association of relative tRNA levels to ratios of codon usage between the top-third (nine genes) and bottom-third (nine genes) transcribed cell cycle genes. Again, a positive association is only expected for the codons that are over-represented in the top third genes (x > 1.2). (E) Association of relative arginine tRNA isoacceptor levels to the arginine codon frequency of cancer-related genes.
To identify cell line-specific genes, we used publicly available mRNA expression data ((http://www.ebi.ac.uk/arrayexpress/, accession number E-TABM-157). Cell line-specific genes were selected based on mRNA expression levels (7- to 15-fold above the median expression level determined for all genes) and high cell line/MCF10A expression ratios (top 20–30 genes, Table S5). A cell line-specific gene set was determined for each breast cancer cell line. To identify cancer-related genes, we selected from a comprehensive list of potential breast cancer diagnostic markers (http://www.sabiosciences.com/gene_array_product/HTML/OHS-402.html). Genes in this group are highly associated with breast cancer (29). Functional groupings used in our study include: cell cycle, cell growth and proliferation, ECM molecules, protein kinases, and transcription factors/regulators (Table S6). To identify house-keeping genes, we selected the 30 most highly expressed house-keeping genes as defined in a previous report (30). This list includes ribosomal proteins, actin, ubiquitin, and others (Table S7). In all cases, gene sequences were compiled and analyzed for codon content at http://www.bioinformatics.org/sms2/.
For each gene set, we compiled gene sequences and analyzed them for codon usage (expressed as number per one thousand codons, www.bioinformatics.org/sms2) (Tables S8 and S9). Because certain tRNAs read more than one codon, we converted the obtained codon usage into tRNA-based codon usage. For example, tRNAArg(ICG) reads both CGU and CGC. Its tRNA-based codon usage is therefore equal to the sum of the CGU and CGC codon usages. For simplicity, we refer to tRNA-based codon usage as codon usage throughout our analysis.
We first compared the codon usages of each gene set in the three gene groups (cell line-specific, cancer-related, and house-keeping) against each other (Figure 6B). Though there is limited overlap across cell line-specific gene sets, their codon usage was remarkably similar (average rs = 0.92 ± 0.04). The codon usage of cell-line specific gene sets also correlates with the codon usage of the house-keeping genes (average rs = 0.80 ± 0.08), but to a significantly lesser extent (P < 0.01). Similarly, the codon usages of cancer-related gene sets correlate remarkably well with each other (average rs = 0.92 ± 0.05) but to a lesser extent with the house-keeping genes (average rs = 0.83 ± 0.06, P < 0.01). These results suggest different gene groups have significantly different codon usages.
Is the codon usage of cell-line specific genes related to the over-expression patterns of nuclear-encoded tRNAs in breast cancer cells? We plotted relative isoacceptor levels (derived from comparative tRNA measurements) versus the codon frequency of cell-line specific genes (derived from comparative mRNA analysis) to determine whether the changes in tRNA levels favor the codon usage of these genes. No obvious associations were observed (not shown). The absence of associations may be explained by the low mRNA level of these genes at the global scale (Figure 6A). The cell line-specific genes identified for this study are expressed only 7- to 15-fold above the median expression level of all genes, compared to ∼80-fold above median for the house-keeping genes. Among human tissues examined, a significant tRNA abundance-codon usage association was found only in liver (17). The mRNA levels of liver-specific genes approach those of house-keeping genes (200-fold above median), sufficiently high for tRNAs to adjust to their respective codon usages. Because the codon usage of line-specific genes is different from that of house-keeping genes, adjusting tRNA levels to favor expression of cell line-specific genes would be unfavorable for translation of house-keeping genes.
The codon usage of cancer-related genes, however, appears to have a positive association with the relative tRNA over-expression in breast cancer cell lines (Figure 6C–E). Since the tRNA over-expression pattern is similar for the nuclear-encoded tRNAs across all lines examined, we used the average tRNA over-expression for all six cancer lines for this analysis. Bearing in mind that tRNA over-expression cannot favor all codons of cancer-related genes because such tRNA adjustment would diminish the translational efficiency of house-keeping genes, we reasoned that a positive association should reveal itself only for codons that are strongly over-represented in cancer-related relative to house-keeping genes. A positive association between tRNA over-expression and codon usage is indeed observed for codons over-represented by 2-fold or more for the cell cycle, extracellular matrix, and transcription factor groups (Figure 6C). Among the group of 28 cell cycle genes ranked by their average mRNA expression levels in all lines, a similar relationship can be seen when comparing the codons that are over-represented in the nine genes with the highest mRNA expression versus the nine genes with the lowest mRNA expression (Figure 6D). Finally, tRNAArg isoacceptors seem to be particularly tuned to increase the translational efficiency of Arg-codons of the cell cycle, extracellular matrix and transcription factor genes (Figure 6E). Consistent with this observation, two of the five tRNAArg isoacceptors are among the highest over-expressed tRNAs in cancer cells (Figure 2E).
DISCUSSION
tRNA over-expression in breast cancer cells
Consistent with previous reports on individual tRNAs, our genome-wide results show that elevated tRNA levels are characteristic of all breast cancer cells analyzed. To our knowledge, however, our study is the first to analyze expression levels for all tRNAs in cancerous versus non-cancerous cells. Our results reveal an unexpected selectivity in the over-expression of individual tRNA species, based on cognate amino acid properties and isoacceptor identities. Each breast cancer cell line generates tRNA profiles that are markedly different from that of non-cancer-derived breast epithelial cell lines. These observations hold true for breast tumor samples compared to normal breast tissue. However, we find no significant differences in tRNA expression patterns across the three major subtypes of breast cancer we examined. The small sample size used in this study is likely insufficient to reveal subtle but significant differences between breast cancer subtypes. Also, tRNA expression at different tumor stages remains to be examined. Overall, our results highlight the potential of using both nuclear- and mitochondrial-encoded tRNAs as biomarkers for malignancy, tumor type, or tumor progression.
Remarkably, tRNAs carrying the amino acids serine, threonine, and tyrosine are among the most over-expressed across all breast cancer cell lines and breast tumors analyzed. Since these amino acid residues are targets for protein kinases and phosphatases, this result suggests these tRNAs might be part of a potential mechanism for potentiating post-translational regulation of proteins involved in signal transduction. How such a mechanism may work remains obscure at this time.
Furthermore, significant differences are observed in the relative expression levels of tRNA isoacceptors. Differential expression of tRNA isoacceptors may provide an additional level of translational regulation for key genes involved in tumorigenesis. Tumors display both quantitative and qualitative changes in protein expression, with preferential translation of growth factors, cell-cycle promoters, and proto-oncogenes, such as VEGF, cyclin D1, and c-Myc (2). It is possible that selective over-expression of tRNA isoacceptors enhances translational efficiency of some of these genes.
Another mechanism linking translational control by tRNA and oncogenic transformation has recently been reported (9). In that work, the translation initiator 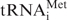 was shown to have oncogenic capacity. A modest over-expression of initiator
was shown to have oncogenic capacity. A modest over-expression of initiator 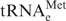 (<2-fold) results in malignant transformation of immortalized mouse fibroblasts, whereas over-expression of the elongator to a similar level does not. Consistent with these observations, our results indicate that is also over-expressed by 2- to 3-fold in all cancer-derived versus non-cancer-derived breast cell lines. However, over-expression of is below that of other tRNAs such as the Ser/Thr/Tyr-tRNAs in the breast cancer cells. Over-expression of these tRNAs may simply be the consequence of over-expression of , although a more exciting possibility is that some of these tRNAs also have oncogenic potential and may be regulated independently. Further studies are needed to elucidate the regulatory relationships between tRNA expression and cancer.
(<2-fold) results in malignant transformation of immortalized mouse fibroblasts, whereas over-expression of the elongator to a similar level does not. Consistent with these observations, our results indicate that is also over-expressed by 2- to 3-fold in all cancer-derived versus non-cancer-derived breast cell lines. However, over-expression of is below that of other tRNAs such as the Ser/Thr/Tyr-tRNAs in the breast cancer cells. Over-expression of these tRNAs may simply be the consequence of over-expression of , although a more exciting possibility is that some of these tRNAs also have oncogenic potential and may be regulated independently. Further studies are needed to elucidate the regulatory relationships between tRNA expression and cancer.
Variations of tRNA–codon usage associations
A main regulatory function of tRNA is to tune the translation efficiency of individual mRNAs via their codon usages (10). A common hypothesis is that during active cell growth, the amount of tRNA isoacceptors correlates with the codon usage of genes translated at high levels. This hypothesis has been validated for bacteria and yeast (26–28). In mammalian cells, this hypothesis also holds for isolated instances where tRNA amounts have been measured in specialized cells, e.g. red blood cells (31).
The common analysis of tRNA-codon usage correlation/association relies on knowing the amount of tRNA isoacceptors in each sample. Information on absolute tRNA abundance levels, however, is difficult to obtain in microarray measurements due to variations in labeling and hybridization efficiencies. Therefore, a direct assessment of tRNA-codon usage association in breast cancer cells cannot be carried out in this study.
Instead, the array results compare the relative abundance of each isoacceptor between two samples. Among the three groups of genes relevant for codon usage-tRNA over-expression association analysis, positive associations are found only for certain codons among cancer-related genes. Our results suggest that during active cell growth (as examined in this work), a complete adjustment of tRNA abundance to fit the codon usage of regulatory genes would be very difficult. tRNAs in the same cell must balance promoting translation of regulatory genes versus demoting translation of house-keeping genes with different codon usage. From the codon usage perspective, tRNA over-expression can only benefit the translation of a subset of regulatory genes or a subset of codons in the regulatory genes. Thus, over-expression of tRNA in breast cancer cells likely affects other regulatory processes that are yet to be explored or understood.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Congressionally Directed Medical Research Programs grant [W81XWH-07-1-0595] to M.R. and T.P.; National Institutes of Health training grant [T32GM007183-33] to M.P.-E. Funding for open access charge: W81XWH-07-1-0595.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr. Lucy Godley for help with the procurement of tumor samples.
REFERENCES
- 1.Bjornsti MA, Houghton PJ. Lost in translation: dysregulation of cap-dependent translation and cancer. Cancer Cell. 2004;5:519–523. doi: 10.1016/j.ccr.2004.05.027. [DOI] [PubMed] [Google Scholar]
- 2.Pandolfi PP. Aberrant mRNA translation in cancer pathogenesis: an old concept revisited comes finally of age. Oncogene. 2004;23:3134–3137. doi: 10.1038/sj.onc.1207618. [DOI] [PubMed] [Google Scholar]
- 3.Ruggero D, Pandolfi PP. Does the ribosome translate cancer? Nat Rev Cancer. 2003;3:179–192. doi: 10.1038/nrc1015. [DOI] [PubMed] [Google Scholar]
- 4.White RJ. RNA polymerase III transcription and cancer. Oncogene. 2004;23:3208–3216. doi: 10.1038/sj.onc.1207547. [DOI] [PubMed] [Google Scholar]
- 5.Winter AG, Sourvinos G, Allison SJ, Tosh K, Scott PH, Spandidos DA, White RJ. RNA polymerase III transcription factor TFIIIC2 is overexpressed in ovarian tumors. Proc. Natl Acad. Sci. USA. 2000;97:12619–12624. doi: 10.1073/pnas.230224097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marshall L, White RJ. Non-coding RNA production by RNA polymerase III is implicated in cancer. Nat. Rev. Cancer. 2008;8:911–914. doi: 10.1038/nrc2539. [DOI] [PubMed] [Google Scholar]
- 7.Chen W, Bocker W, Brosius J, Tiedge H. Expression of neural BC200 RNA in human tumours. J. Pathol. 1997;183:345–351. doi: 10.1002/(SICI)1096-9896(199711)183:3<345::AID-PATH930>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 8.Chen W, Heierhorst J, Brosius J, Tiedge H. Expression of neural BC1 RNA: induction in murine tumours. Eur. J. Cancer. 1997;33:288–292. doi: 10.1016/s0959-8049(96)00453-4. [DOI] [PubMed] [Google Scholar]
- 9.Marshall L, Kenneth NS, White RJ. Elevated tRNA(iMet) synthesis can drive cell proliferation and oncogenic transformation. Cell. 2008;133:78–89. doi: 10.1016/j.cell.2008.02.035. [DOI] [PubMed] [Google Scholar]
- 10.Zouridis H, Hatzimanikatis V. Effects of codon distributions and tRNA competition on protein translation. Biophys. J. 2008;95:1018–1033. doi: 10.1529/biophysj.107.126128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bertucci F, Goncalves A. Clinical proteomics and breast cancer: strategies for diagnostic and therapeutic biomarker discovery. Future Oncol. 2008;4:271–287. doi: 10.2217/14796694.4.2.271. [DOI] [PubMed] [Google Scholar]
- 12.Payne SJ, Bowen RL, Jones JL, Wells CA. Predictive markers in breast cancer – the present. Histopathology. 2008;52:82–90. doi: 10.1111/j.1365-2559.2007.02897.x. [DOI] [PubMed] [Google Scholar]
- 13.Rennstam K, Hedenfalk I. High-throughput genomic technology in research and clinical management of breast cancer. Molecular signatures of progression from benign epithelium to metastatic breast cancer. Breast Cancer Res. 2006;8:213. doi: 10.1186/bcr1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 15.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soll D. The RNA World. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1993. Transfer RNA: an RNA for all seasons; pp. 157–184. [Google Scholar]
- 17.Dittmar KA, Goodenbour JM, Pan T. Tissue-specific differences in human transfer RNA expression. PLoS Genet. 2006;2:e221. doi: 10.1371/journal.pgen.0020221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saikia M, Dai Q, Decatur WA, Fournier MJ, Piccirilli JA, Pan TA. Systematic, ligation-based approach to study RNA modifications. RNA. 2006;12:2025–2033. doi: 10.1261/rna.208906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dittmar KA, Sorensen MJ, Elf J, Ehrenberg M, Pan T. Selective charging of tRNA isoacceptors induced by amino-acid starvation. EMBO Rep. 2005;6:151–157. doi: 10.1038/sj.embor.7400341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dittmar KA, Mobley EM, Radek AJ, Pan T. Exploring the regulation of tRNA distribution on the genomic scale. J. Mol. Biol. 2004;337:31–47. doi: 10.1016/j.jmb.2004.01.024. [DOI] [PubMed] [Google Scholar]
- 21.Zaborske JM, Narasimhan J, Jiang L, Wek SA, Dittmar KA, Freimoser F, Pan T, Wek RC. Genome-wide analysis of tRNA charging and activation of the eIF2 kinase Gcn2p. J. Biol. Chem. 2009;284:25254–25267. doi: 10.1074/jbc.M109.000877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou Y, Goodenbour JM, Godley LA, Wickrema A, Pan T. High levels of tRNA abundance and alteration of tRNA charging by bortezomib in multiple myeloma. Biochem. Biophys. Res. Commun. 2009;385:160–164. doi: 10.1016/j.bbrc.2009.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu Y, Kool ET. High sequence fidelity in a non-enzymatic DNA autoligation reaction. Nucleic Acids Res. 1999;27:875–881. doi: 10.1093/nar/27.3.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu Y, Karalkar NB, Kool ET. Nonenzymatic autoligation in direct three-color detection of RNA and DNA point mutations. Nat. Biotechnol. 2001;19:148–152. doi: 10.1038/84414. [DOI] [PubMed] [Google Scholar]
- 26.Ikemura T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: a proposal for a synonymous codon choice that is optimal for the E. coli translational system. J. Mol. Biol. 1981;151:389–409. doi: 10.1016/0022-2836(81)90003-6. [DOI] [PubMed] [Google Scholar]
- 27.Ikemura T. Correlation between the abundance of yeast transfer RNAs and the occurrence of the respective codons in protein genes. Differences in synonymous codon choice patterns of yeast and Escherichia coli with reference to the abundance of isoaccepting transfer RNAs. J. Mol. Biol. 1982;158:573–597. doi: 10.1016/0022-2836(82)90250-9. [DOI] [PubMed] [Google Scholar]
- 28.Kanaya S, Yamada Y, Kudo Y, Ikemura T. Studies of codon usage and tRNA genes of 18 unicellular organisms and quantification of Bacillus subtilis tRNAs: gene expression level and species-specific diversity of codon usage based on multivariate analysis. Gene. 1999;238:143–155. doi: 10.1016/s0378-1119(99)00225-5. [DOI] [PubMed] [Google Scholar]
- 29.Hu Y, et al. Analysis of genomic and proteomic data using advanced literature mining. J. Proteome Res. 2003;2:405–412. doi: 10.1021/pr0340227. [DOI] [PubMed] [Google Scholar]
- 30.Eisenberg E, Levanon EY. Human housekeeping genes are compact. Trends Genet. 2003;19:362–365. doi: 10.1016/S0168-9525(03)00140-9. [DOI] [PubMed] [Google Scholar]
- 31.Hatfield D, Rice M. Aminoacyl-tRNA(anticodon): codon adaptation in human and rabbit reticulocytes. Biochem. Int. 1986;13:835–842. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.