

Abstract
CaMKII, a major mediator of synaptic plasticity, forms extra-synaptic clusters under ischemic conditions. This study further supports self-aggregation of CaMKII holoenzymes as the underlying mechanism. Aggregation in vitro was promoted by mimicking ischemic conditions: low pH (6.8 or less), Ca2+ (and calmodulin), and low ATP and/or high ADP concentration. Mutational analysis showed that high ATP prevented aggregation by a mechanism involving T286 auto-phosphorylation, and indicated requirement for nucleotide binding but not auto-phosphorylation also for extra-synaptic clustering within neurons. These results clarify a previously apparent paradox in the nucleotide and phosphorylation requirement of aggregation, and support a mechanism that involves inter-holoenzyme T286-region/T-site interaction.
Keywords: phosphorylation, CaMKII, ischemia
1. Introduction
The Ca2+/calmodulin (CaM)-dependent protein kinase II (CaMKII) is an important mediator of physiological neuronal signaling triggered by glutamate, the major excitatory neurotransmitter in mammalian brain (for review see [1–3]). Important physiological neuronal functions of CaMKII depend on T286 auto-phosphorylation [4], which renders the kinase active even in absence of Ca2+/CaM (for review see [1–3]). The role of CaMKII in glutamate-mediated excitotoxic neuronal death under ischemic conditions is less clear, as opposite functions have been reported [5–8]. However, it is well reported that ischemic conditions trigger the conglomeration of many 12meric CaMKII holoenzymes into large clusters [9–11], concomitant with a reduction of CaMKII enzymatic activity in the brain [12,13]. Such extra-synaptic clustering has also been observed in real-time for GFP-CaMKII in cultured hippocampal neurons following excitotoxic glutamate treatment [14]. It has been proposed that these clusters are formed entirely from CaMKII holoenzymes that have aggregated with each other through inter-holoenzyme interactions [9,15,16]. Indeed, sedimentable aggregates can form in vitro using purified CaMKII holoenzymes, and this requires stimulation by Ca2+/CaM and acidic pH (< pH 7)[15,16], conditions that are found during ischemia. However, somewhat paradoxical, this self-aggregation also required at least some ATP, although only at low level (10 μM), while high ATP (>1 mM) prevented self-aggregation [15]. It was assumed that auto-phosphorylation of CaMKII was required for self-association, as the non-hydrolysable analog AMP-PNP did not substitute for the low level ATP [15]. The present study confirmed the pH and CaM requirements of CaMKII self-association, but showed that ATP could be substituted for either AMP-PNP or ADP, although higher concentrations were required (>100 μM). At even higher concentrations (around the cellular level of ~4 mM), ADP and ADP/ATP (4:1) still allowed CaMKII aggregation, while ATP alone protected from aggregation by a mechanism that involved CaMKII auto-phosphorylation at T286. These results support a refined model for CaMKII holoenzyme aggregation as the mechanism for extra-synaptic clustering under ischemic conditions (see Figure 6).
Fig. 6.
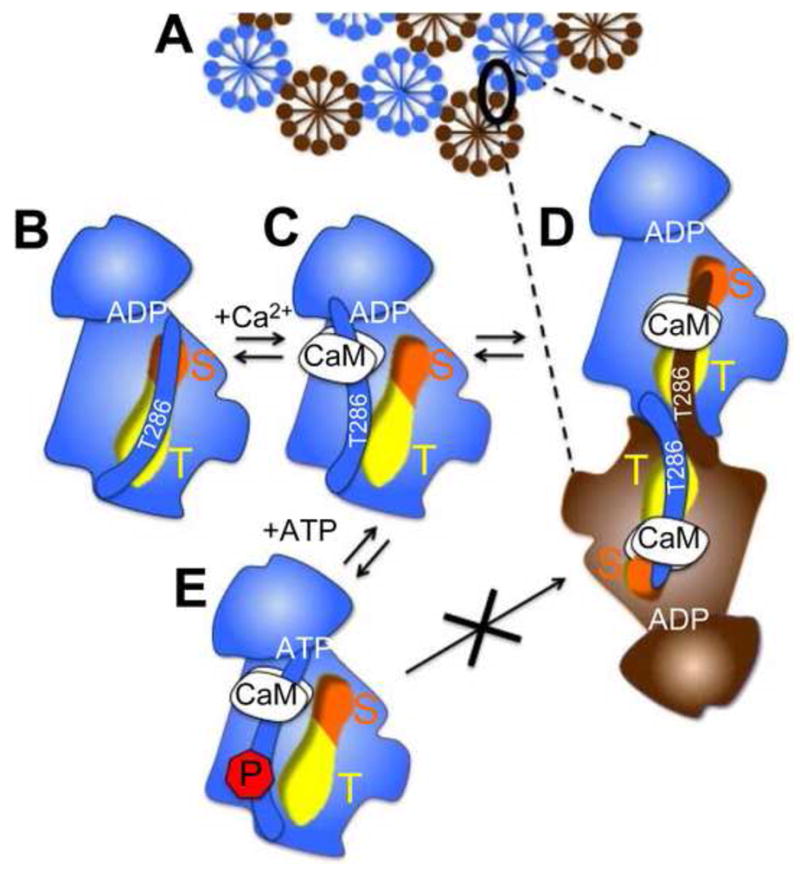
A model for self-aggregation of 12meric CaMKII holoenzymes (A; shown either blue or brown) involving inter-subunit, inter-holoenzyme interaction of the T286-region with the T-site. (B) In the basal state of the kinase, the regulatory region around T286 interacts with the T-site (yellow); the C-terminal part of the regulatory region extends to block the substrate binding S-site (orange). (C) In presence of Ca2+, CaM binds to the regulatory region and makes both S- and T-site accessible. This allows (D) inter-subunit T286-region/T-site interaction and aggregation or (E), in presence of high ATP concentration, T286 auto-phosphorylation which prevents the T-site interaction and aggregation.
2. Materials and methods
2.1. Material
CaMKIIα wild type and mutants were purified after overexpression in a baculovirus/Sf9 cell system using a phospho-cellulose column (P11, Whatman International Ltd) followed by CaM-sepharose (GE Healthcare Biosciences) affinity chromatography, essentially as described [17,18]. CaM was purified after bacterial expression as described [17,18]. Chemicals were obtained from Sigma-Aldrich (St. Louis, MO).
2.2. CaMKII self-association assay
Purified CaMKIIα (0.5 μM) was combined with 25 mM PIPES at pH 6.8, 20 mM KCl, 10 mM MgCl2, 0.1 mg/ml BSA, 0.1% Tween 20, 0.5 mM DTT, 0.5 mM CaCl2, 2 μM CaM and 10 μM ATP, or as indicated. The mixtures were prepared on ice, incubated for 5 min at room temperature, and then centrifuged at 16,000 × g and 4°C for 30 min, following a previously described method [15]. CaMKII in the supernatant and pellet was detected by Western-blot.
2.3. Western-blot analysis
After SDS-PAGE and electro-transfer onto nitrocellulose (Schleicher&Schuell) or PVDF (PerkinElmer) membrane, CaMKIIα was detected using a monoclonal antibody (CBα2; Invitrogen) and the Westen Lightning chemoluminiscence system (PerkinElmer) essentially as described [17,18]
2.4. CaMKII extra-synaptic clustering in hippocampal neurons
Primary rat hippocampal neurons were cultured and transfected with mGFP-CaMKII constructs as previously described [18,19]. Neurons were stimulated with glutamate/glycine as indicated in imaging solution and fixed as described [18]. Images were acquired using SlideBook software (Intelligent Imaging Innovations, Denver, CO), for fixed neurons on an Olympus spinning disk confocal microscope, for live neurons on a Zeiss Axiovert 200M system, as describe [19].
3. Results
3.1. CaMKII self-aggregation requires Ca2+/CaM, low pH, and nucleotide
In order to clarify the requirements for CaMKII self-aggregation in vitro, we utilized a differential centrifugation assay, followed by Western-detection of CaMKII in the pellet and supernatant (Fig. 1). Self-aggregation required Ca2+, CaM, and nucleotide; lack of any of these conditions prevented self-aggregation and all detection of any CaMKII in the pellets (Fig. 1 and Supplementary data). Additionally, low pH (pH 6.8 or less) strongly promoted self-aggregation (Fig. 1). This confirmed the previously described pH and CaM requirement, and formally demonstrated the simultaneous requirement of Ca2+ in addition to CaM (i.e. that CaM without Ca2+ is not sufficient).
Fig. 1.
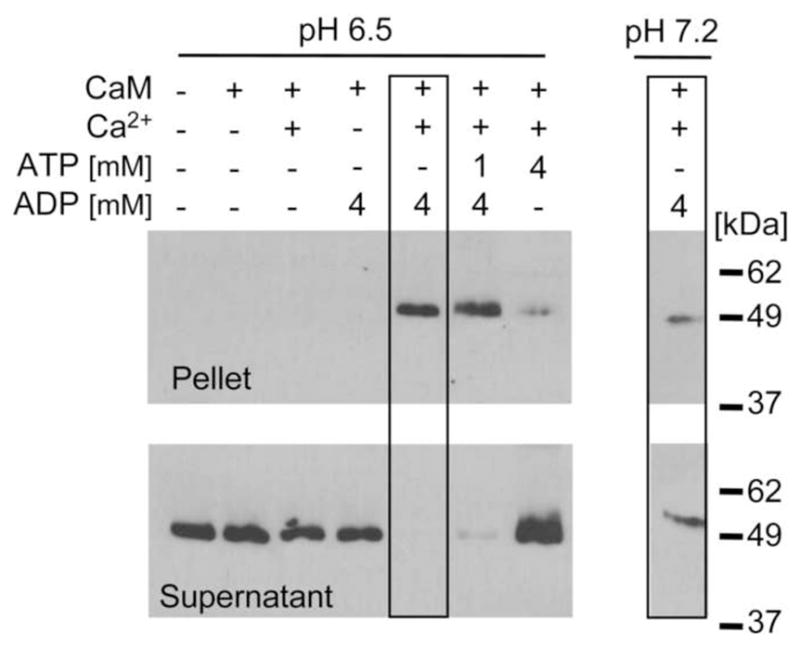
CaMKII self-aggregation requires Ca2+, CaM, low pH and nucleotide (high ADP and/or low ATP concentration). CaMKII aggregates were detected by Western blot in a 16,000 g pellet following incubation with Ca2+, CaM, and 4 mM ADP at low pH (6.5); aggregation was significantly less under the same conditions at pH 7.2 (compare boxed lanes; cropped from different regions of the same gel exposure). Incubation with 1 mM ATP and 4 mM ADP also allowed self-aggregation, while 4 mM ATP reduced it significantly. No aggregates were detected without Ca2+, CaM, or any nucleotide.
3.2. Low ATP can be substituted for higher concentrations of ADP or AMP-PNP
Substitution of 10 μM ATP with 10 μM ADP or the non-hydrolyzable ATP analog AMP-PNP abolished CaMKII self-aggregation (Fig. 2 and Supplementary data), as previously described for AMP-PNP [15]. However, increasing the ADP or AMP-PNP concentration to 0.1 or 1 mM allowed CaMKII self-aggregation (Fig. 2 and Supplementary data). These results are consistent with a lower affinity of both ADP and AMP-PNP compared to ATP. More importantly, the results indicate that nucleotide binding but not auto-phosphorylation is required for CaMKII aggregation.
Fig. 2.
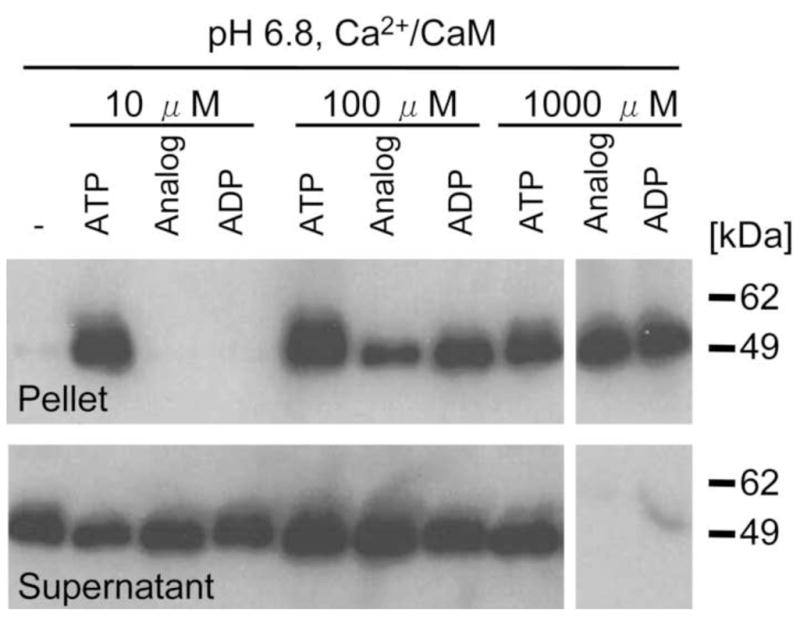
AMP-PNP (Analog) and ADP can substitute for 10 μM ATP and promote self-association, but only at higher concentration. At pH 6.8, CaMKII self-aggregated following incubation with Ca2+/CaM and 10 μM ATP. At concentrations of 100 μM or 1000 μM, Analog or ADP also induced self-association, but 10 μM Analog and ADP was not sufficient. Ca2+/CaM alone could not induce self-association; at least some nucleotide was required.
3.3 Cellular levels of ATP but not ADP inhibit CaMKII aggregation
The ATP concentration within cells is around 4 mM. Such high ATP concentrations inhibited CaMKII aggregation in vitro (Fig. 1). By contrast, high ADP (4 mM) or ADP:ATP (4 mM: 1 mM) still allowed aggregation (Fig. 1). Such high ADP concentrations are found under ischemic conditions, when ATP is not replenished, and further suggests CaMKII self-aggregation as mechanism for extra-synaptic clustering of CaMKII during ischemia.
3.4 T286 auto-phosphorylation prevents CaMKII self-aggregation a high ATP
Prevention of CaMKII aggregation by high levels of ATP but not ADP suggested auto-phosphorylation as possible protective mechanism. Indeed, mutational analysis implicated auto-phosphorylation, specifically at T286, as mechanism for aggregation protection by ATP (Fig. 3): A T286A mutant CaMKII, which can not be auto-phosphorylated at this residue, aggregated even in presence of 4 mM ATP. By contrast, a T286D mutant CaMKII, which mimicks phospho-T286, did not aggregate at all, even with 4 mM ADP without ATP (Fig. 3). These results further support CaMKII self-aggregation as the underlying mechanism for glutamate-induced extrasynaptic clustering in neurons, which is also enhanced by the T286A mutation and reduced by the T286D mutation [14], consistent with our biochemical data using purified enzyme.
Fig. 3.
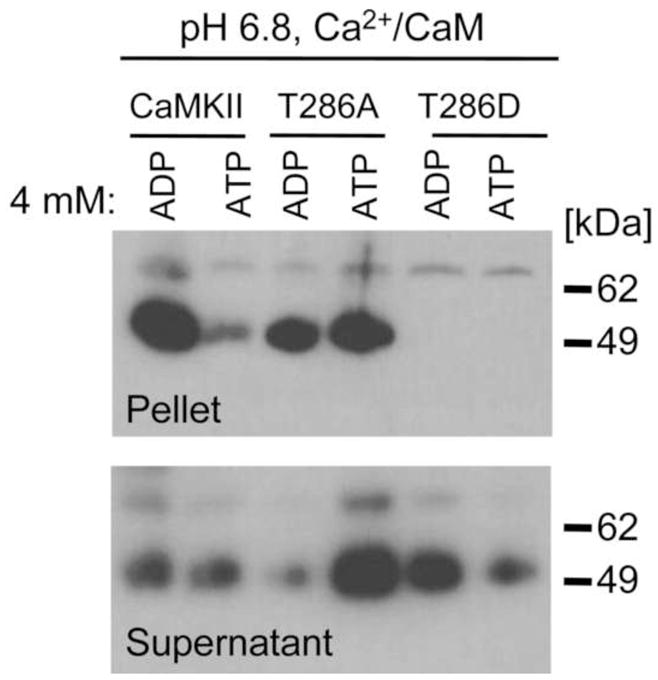
T286A self-aggregated even in presence of 4 mM ATP while T286D did not self-associate at all. CaMKII wild type and T286 mutants were incubated at pH 6.8 with Ca2+/CaM, and 4 mM ADP or 4 mM ATP. After differential centrifugation, pellets were probed for presence of CaMKII aggregates by Western blot.
3.5 Requirement of nucleotide binding for extra-synaptic clustering in neurons
Extra-synaptic clustering of CaMKII within neurons should require nucleotide binding, if it is indeed mediated by self-aggregation of holoenzymes. This prediction was tested using the nucleotide binding-impaired kinase dead mutant K42M. GFP-CaMKII wild type formed extra-synaptic clusters upon prolonged glutamate stimulation of primary hippocampal neurons (Fig. 4). The extra-synaptic localization of clusters was verified by confocal microscopy, which showed that many of the clusters are within the cytoplasm and not associated with the plasma membrane (Fig. 4), and by co-staining with the synaptic marker PSD95 (supplementary data). Live fluorescence microscopy showed that formation of extra-synaptic clusters seen for GFP-CaMKII wild type was indeed impaired by the kinase dead K42M mutation (Fig. 5). The K42M mutation largely abolished cluster formation in the soma but not in the dendrites (Fig. 5), consistent with a specific effect on extra-synaptic but not synaptic clusters. As predicted from our biochemical data, requirement for nucleotide binding was not mediated through a requirement of T286 auto-phosphorylation, as a T286A mutant showed enhanced clustering induced even by spontaneous neuronal activity under basal conditions (Fig. 5). Enhanced cluster formation by T286A is consistent with previous observations [14] and with the phospho-T286-mediated protection from self-aggregation.
Fig. 4.
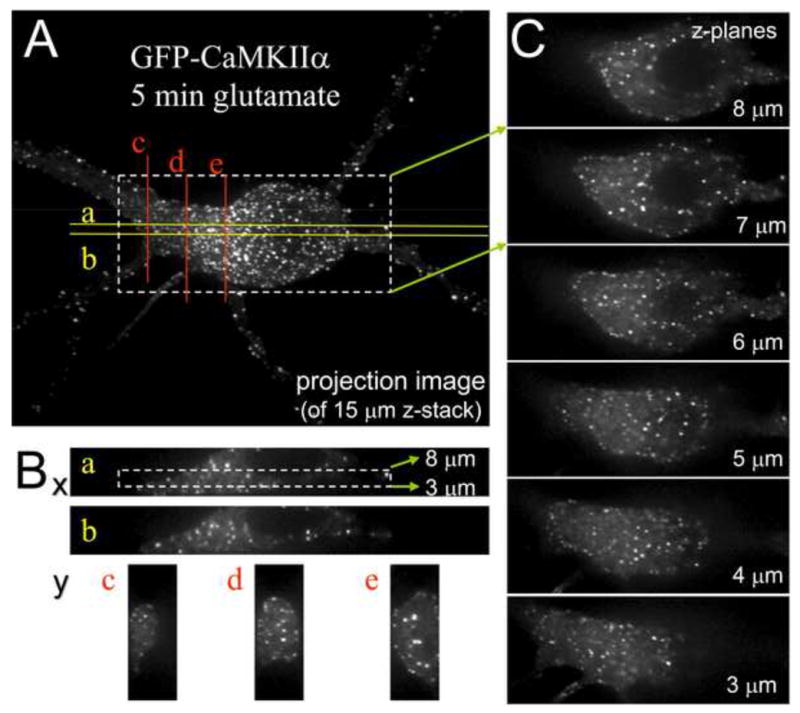
Prolonged glutamate induces formation of extra-synaptic GFP-CaMKII clusters. Primary hippocampal neurons (21 days in vitro) were fixed after 5 min stimulation with glutamate/glycine (100 μM/10 μM) and imaged on an Olympus spinning-disk confocal setup (z-stack of 30 images, 0.5 μm apart). (A) Projection of the z-stack into a single plane. (B) Reconstructed z sections through the confocal image stack. Approximate positions of the sections in the x/y plane are indicated on the projection image shown in A. (C) A series of confocal images in different z-planes, covering 5 μm of the whole stack. Position of the image series shown is indicated in A (x/y-plane) and Ba (z-axis). Panel B and C clearly demonstrate that most of the CaMKII clusters in the soma are not associated with the plasma membrane, thereby ruling out synaptic localization.
Fig. 5.

Nucleotide binding is required for extra-synaptic clustering of GFP-CaMKII in hippocampal neurons, as demonstrated by the nucleotide binding-deficient mutant K42M. Primary hippocampal neurons (14 days in vitro) were stimulated with glutamate/glycine (200 μM/10 μM) two days after transfection and imaged live on a climate controlled Zeiss Axiovert 200M setup at 32°C (z-stack of 24 images, 0.5 μm apart). Shown are projection images after deconvolution using SlideBook software. K42M significantly reduced formation of extra-synaptic clusters in the soma, but not clusters in the dendrites that likely correspond to synapses. T286A showed significant clustering even under basal conditions.
Taken together, these results support a modified model of CaMKII aggregation by inter-holoenzyme T286-region/T-site interaction as mechanistic basis for extra-synaptic clustering during ischemic conditions (Fig. 6).
4. Discussion
The results of this study show that self-aggregation of CaMKII holoenzymes is triggered in vitro by mimicking ischemic conditions. While the requirement for low pH and CaM has been reported before [15,16], this study demonstrated the Ca2+ requirement formally for the first time. More importantly, the results show that the low level of ATP thought to be necessary for aggregation can be substituted by high levels of ADP. Additionally, the results showed that prevention of self-aggregation by high ATP concentrations occurs through a mechanism involving T286 auto-phosphorylation. These findings link CaMKII self-aggregation even further to ischemic conditions, which include low ATP but high ADP concentration.
Initially, CaMKII self-aggregation was thought to require auto-phosphorylation, based on the observation that 10 μM ATP could not be substituted for 10 μM of the non-hydrolysable analog AMP-PNP [15]. While our results confirmed this finding, they also showed that higher concentrations (>100 μM) of AMP-PNP or of ADP could effectively substitute for low ATP levels. Thus, nucleotide binding but not auto-phosphorylation is required for CaMKII self-aggregation. In fact, auto-phosphorylation at T286 prevented self-aggregation and nucleotide binding was required for also extra-synaptic clustering in neurons, as shown by mutational analysis. This protective mechanism further supports a previously proposed mechanism for self-aggregation [16]: Binding of the regulatory region around T286 of one CaMKII subunit to a T-site (T286 binding site [17]) on the kinase domain of another CaMKII subunit on a different holoenzyme (see Figure 6). Each 12meric CaMKII holoenzyme could engage in multiple of such interactions, thereby aiding the formation of large aggregated clusters. In the basal state of CaMKII, the regulatory region around T286 interacts with the T-site of the same kinase subunit, and the C-terminal part of the regulatory region extends to block the nearby substrate binding S-site [20,21]. Thus, the T286 region and the T-site are indeed able to bind to each other in principle. Furthermore, peptides that occupy the T-site inhibit CaMKII self-aggregation [16], indicating that a T-site interaction is necessary. Consistent with the regulatory region around T286 providing the binding partner for the T-site, we found that phosphorylation of T286 prevented self-aggregation. This finding would be predicted from the model, as T286 phosphorylation does interfere with the intra-subunit T286/T-site interaction to generate Ca2+-independent autonomous activity (for review see [1–3]), and thus should also interfere with a similar but inter-subunit interaction.
In addition to mediating extra-synaptic clustering in ischemic conditions, self-aggregation has been proposed to contribute also to the CaMKII translocation to synaptic sites that is observed after physiological glutamate stimuli [14,22]. Interestingly, the other main protein interaction of CaMKII that is thought to contribute to this synaptic targeting is with the NMDA-type glutamate receptor subunit NR2B [17,18,23–25], an interaction also mediated by the CaMKII T-site [17,18], in this case with a region on NR2B homologous to the CaMKII T286 region [17,25]. Like self-aggregation, the interaction of CaMKII with NR2B requires an initial Ca2+/CaM stimulus in order to make the T-site accessible [17,18,23]. However, the CaMKII/NR2B interaction is not inhibited by T286 auto-phosphorylation, but, if any, further enhanced by it [17,18,23,25]. Such regulation would actually be predicted by the T-site model, as the T286 region is the interacting partner only in self-aggregation, but not for NR2B-binding. In fact, the binding site on NR2B is around S1303, and its phosphorylation also prevents CaMKII binding [25]. The major differences in the conditions required for self-aggregation compared to NR2B-binding are presence of low pH and low ATP, which are associated with ischemic conditions but not physiological glutamate signaling. However, glutamateric synapses are formed onto dendritic spines, small ~1 μm3 protrusions that can form separate compartments by narrow, diffusion limiting spine necks connecting to the dendrite (for review see [1,3]). This could conceivably allow a temporary ATP depletion and lowering of pH in individual spines even during physiological glutamate signaling. However, while self-aggregation is sufficient to explain extra-synaptic clustering, on its own it does not appear sufficient to explain synaptic targeting, which consists of a transient translocation peak followed by a plateau of persistent localization (which depends on the length of the initial stimulus)[18]. In our model, CaMKII binding to NR2B would be the initial event during translocation, as it does require Ca2+/CaM but no drop in pH or ATP concentration. Then, additional CaMKII holoenzymes would aggregate onto the initial NR2B-bound holoenzymes when the drop in pH and ATP occurs; this amplification process would contribute substantially to the amount of synaptic CaMKII during the peak translocation phase. When the physiological glutamate signal subsides, pH and ATP concentration would rise again sharply and the CaMKII aggregates would be released. NR2B-binding would then support the remaining persistent synaptic CaMKII. Indeed, while binding to NR2B requires an initial Ca2+/CaM stimulus, this stimulus is no longer required once the interaction is fully formed [17,18].
The role of self-aggregation in synaptic CaMKII targeting in response to physiological glutamate signals is still unclear and further studies are required. However, this study provides significant additional support for self-aggregation as the underlying mechanism for extra-synaptic CaMKII clustering in response to the pathological glutamate signals under ischemic conditions.
Supplementary Material
Supplementary Fig. 1. Additional experiments showing that AMP-PNP (Analog) and ADP can substitute for ATP and promote self-aggregation, but only at high concentrations. Self-associated CaMKII aggregates were identified by Western blot in 16,000 g pellets (P or Pellet) following incubation at pH 6.8 with Ca2+/CaM and the indicated concentration of ATP, Analog, or ADP, at pH 6.8. Soluble, non-aggregated CaMKII was detected in the supernatant (S or supernatant).
Supplementary Fig. 2. Synaptic and extrasynaptic translocation of GFP-CaMKII in hippocampal neurons (21 days in vitro) during 5 min stimulation with 100 μM glutamate/10 μM glycine). (A) Live imaging shows transition from uniform cytosolic distribution to formation of puncta (single z-plane image acquisition). (B) The same neuron as in A was imaged after fixation and immunostaining of PSD95. A z-stack of 18 images, 0.5 μm apart, was acquired and deconvoluted against the nearest neighbor. The upper panel and the cropped panel show images after maximum projection of the stack into one plane. The lower panel shows a reconstructed x-axis side view of the z-stack; the position of the crossection is indicated in the upper panel. Many CaMKII puncta (green), especially in dendrites, are synaptic, as judged by co-localization with PSD95 (red). However, there are also many extrasynaptic CaMKII puncta, as indicated both by lack of PSD95 co-staining (examples marked by arrows) and by their position in the x/z-plane, showing that they are not membrane-associated.
Acknowledgments
R.S.V was supported by NIH training grants F31NS061584 and T32GM007635. The research was supported by NIH grants P30NS048154 (UCD center grant) and R01NS052644 (to K.U.B.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–90. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- 2.Hudmon A, Schulman H. Neuronal CA2+/calmodulin-dependent protein kinase II: the role of structure and autoregulation in cellular function. Annu Rev Biochem. 2002;71:473–510. doi: 10.1146/annurev.biochem.71.110601.135410. [DOI] [PubMed] [Google Scholar]
- 3.Colbran RJ, Brown AM. Calcium/calmodulin-dependent protein kinase II and synaptic plasticity. Curr Opin Neurobiol. 2004;14:318–27. doi: 10.1016/j.conb.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 4.Giese KP, Fedorov NB, Filipkowski RK, Silva AJ. Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science. 1998;279:870–3. doi: 10.1126/science.279.5352.870. [DOI] [PubMed] [Google Scholar]
- 5.Hajimohammadreza I, Probert AW, Coughenour LL, Borosky SA, Marcoux FW, Boxer PA, Wang KK. A specific inhibitor of calcium/calmodulin-dependent protein kinase-II provides neuroprotection against NMDA- and hypoxia/hypoglycemia-induced cell death. J Neurosci. 1995;15:4093–101. doi: 10.1523/JNEUROSCI.15-05-04093.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laabich A, Cooper NG. Neuroprotective effect of AIP on N-methyl-D-aspartate-induced cell death in retinal neurons. Brain Res Mol Brain Res. 2000;85:32–40. doi: 10.1016/s0169-328x(00)00226-6. [DOI] [PubMed] [Google Scholar]
- 7.Mabuchi T, et al. Phosphorylation of cAMP response element-binding protein in hippocampal neurons as a protective response after exposure to glutamate in vitro and ischemia in vivo. J Neurosci. 2001;21:9204–13. doi: 10.1523/JNEUROSCI.21-23-09204.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Waxham MN, Grotta JC, Silva AJ, Strong R, Aronowski J. Ischemia-induced neuronal damage: a role for calcium/calmodulin-dependent protein kinase II. J Cereb Blood Flow Metab. 1996;16:1–6. doi: 10.1097/00004647-199601000-00001. [DOI] [PubMed] [Google Scholar]
- 9.Dosemeci A, Reese TS, Petersen J, Tao-Cheng JH. A novel particulate form of Ca(2+)/calmodulin-dependent [correction of Ca(2+)/CaMKII-dependent] protein kinase II in neurons. J Neurosci. 2000;20:3076–84. doi: 10.1523/JNEUROSCI.20-09-03076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tao-Cheng JH, Vinade L, Smith C, Winters CA, Ward R, Brightman MW, Reese TS, Dosemeci A. Sustained elevation of calcium induces Ca(2+)/calmodulin-dependent protein kinase II clusters in hippocampal neurons. Neuroscience. 2001;106:69–78. doi: 10.1016/s0306-4522(01)00262-7. [DOI] [PubMed] [Google Scholar]
- 11.Tao-Cheng JH, Vinade L, Winters CA, Reese TS, Dosemeci A. Inhibition of phosphatase activity facilitates the formation and maintenance of NMDA-induced calcium/calmodulin-dependent protein kinase II clusters in hippocampal neurons. Neuroscience. 2005;130:651–6. doi: 10.1016/j.neuroscience.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 12.Aronowski J, Grotta JC, Waxham MN. Ischemia-induced translocation of Ca2+/calmodulin-dependent protein kinase II: potential role in neuronal damage. J Neurochem. 1992;58:1743–53. doi: 10.1111/j.1471-4159.1992.tb10049.x. [DOI] [PubMed] [Google Scholar]
- 13.Hanson SK, Grotta JC, Waxham MN, Aronowski J, Ostrow P. Calcium/calmodulin-dependent protein kinase II activity in focal ischemia with reperfusion in rats. Stroke. 1994;25:466–73. doi: 10.1161/01.str.25.2.466. [DOI] [PubMed] [Google Scholar]
- 14.Hudmon A, Lebel E, Roy H, Sik A, Schulman H, Waxham MN, De Koninck P. A mechanism for Ca2+/calmodulin-dependent protein kinase II clustering at synaptic and nonsynaptic sites based on self-association. J Neurosci. 2005;25:6971–83. doi: 10.1523/JNEUROSCI.4698-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hudmon A, Aronowski J, Kolb SJ, Waxham MN. Inactivation and self-association of Ca2+/calmodulin-dependent protein kinase II during autophosphorylation. J Biol Chem. 1996;271:8800–8. doi: 10.1074/jbc.271.15.8800. [DOI] [PubMed] [Google Scholar]
- 16.Hudmon A, Kim SA, Kolb SJ, Stoops JK, Waxham MN. Light scattering and transmission electron microscopy studies reveal a mechanism for calcium/calmodulin-dependent protein kinase II self-association. J Neurochem. 2001;76:1364–75. doi: 10.1046/j.1471-4159.2001.00119.x. [DOI] [PubMed] [Google Scholar]
- 17.Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature. 2001;411:801–5. doi: 10.1038/35081080. [DOI] [PubMed] [Google Scholar]
- 18.Bayer KU, LeBel E, McDonald GL, O’Leary H, Schulman H, De Koninck P. Transition from reversible to persistent binding of CaMKII to postsynaptic sites and NR2B. J Neurosci. 2006;26:1164–74. doi: 10.1523/JNEUROSCI.3116-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vest RS, Davies KD, O’Leary H, Port JD, Bayer KU. Dual Mechanism of a Natural CaMKII Inhibitor. Mol Biol Cell. 2007;18:5024–33. doi: 10.1091/mbc.E07-02-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang E, Schulman H. Structural examination of autoregulation of multifunctional calcium/calmodulin-dependent protein kinase II. J Biol Chem. 1999;274:26199–208. doi: 10.1074/jbc.274.37.26199. [DOI] [PubMed] [Google Scholar]
- 21.Rosenberg OS, Deindl S, Sung RJ, Nairn AC, Kuriyan J. Structure of the autoinhibited kinase domain of CaMKII and SAXS analysis of the holoenzyme. Cell. 2005;123:849–60. doi: 10.1016/j.cell.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 22.Shen K, Meyer T. Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science. 1999;284:162–6. doi: 10.1126/science.284.5411.162. [DOI] [PubMed] [Google Scholar]
- 23.Strack S, Colbran RJ. Autophosphorylation-dependent targeting of calcium/calmodulin-dependent protein kinase II by the NR2B subunit of the N-methyl- D-aspartate receptor. J Biol Chem. 1998;273:20689–92. doi: 10.1074/jbc.273.33.20689. [DOI] [PubMed] [Google Scholar]
- 24.Gardoni F, Caputi A, Cimino M, Pastorino L, Cattabeni F, Di Luca M. Calcium/calmodulin-dependent protein kinase II is associated with NR2A/B subunits of NMDA receptor in postsynaptic densities. J Neurochem. 1998;71:1733–41. doi: 10.1046/j.1471-4159.1998.71041733.x. [DOI] [PubMed] [Google Scholar]
- 25.Strack S, McNeill RB, Colbran RJ. Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 2000;275:23798–806. doi: 10.1074/jbc.M001471200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1. Additional experiments showing that AMP-PNP (Analog) and ADP can substitute for ATP and promote self-aggregation, but only at high concentrations. Self-associated CaMKII aggregates were identified by Western blot in 16,000 g pellets (P or Pellet) following incubation at pH 6.8 with Ca2+/CaM and the indicated concentration of ATP, Analog, or ADP, at pH 6.8. Soluble, non-aggregated CaMKII was detected in the supernatant (S or supernatant).
Supplementary Fig. 2. Synaptic and extrasynaptic translocation of GFP-CaMKII in hippocampal neurons (21 days in vitro) during 5 min stimulation with 100 μM glutamate/10 μM glycine). (A) Live imaging shows transition from uniform cytosolic distribution to formation of puncta (single z-plane image acquisition). (B) The same neuron as in A was imaged after fixation and immunostaining of PSD95. A z-stack of 18 images, 0.5 μm apart, was acquired and deconvoluted against the nearest neighbor. The upper panel and the cropped panel show images after maximum projection of the stack into one plane. The lower panel shows a reconstructed x-axis side view of the z-stack; the position of the crossection is indicated in the upper panel. Many CaMKII puncta (green), especially in dendrites, are synaptic, as judged by co-localization with PSD95 (red). However, there are also many extrasynaptic CaMKII puncta, as indicated both by lack of PSD95 co-staining (examples marked by arrows) and by their position in the x/z-plane, showing that they are not membrane-associated.