

Clinical and genetic heterogeneity of EB
Epidermolysis bullosa (EB) is a heterogeneous group of skin fragility syndromes with the diagnostic hallmark of blistering and erosions of the skin. Proper diagnosis and subclassification of different forms of EB can be challenging to general practitioners due to considerable phenotypic variability, as reflected by the complex classification schemes riddled with eponyms [1]. The most streamlined classification divides EB into three broad categories depending on the precise location of tissue separation within the cutaneous basement membrane zone, as determined by diagnostic transmission electron microscopy and/or by immunoepitope mapping: (a) in the simplex forms (EBS), tissue separation takes place within the basal keratinocytes; (b) the junctional forms (JEB) show blistering within the dermal-epidermal basement membrane, frequently within the lamina lucida; and (c) in the dystrophic forms (DEB), tissue separation is below the lamina densa within the upper papillary dermis at the level of anchoring fibrils. The inheritance of different forms of EB is either autosomal dominant or autosomal recessive [2, 3]. It is now known that mutations in ten different genes expressed within the cutaneous basement membrane zone underlie the classic simplex, junctional, and dystrophic forms of EB [4, 5]. The types and combinations of mutations, their consequences at the mRNA and protein levels, when placed to the context of the individuals’ genetic background, including modifier genes, and the environmental trauma, all contribute to the severity of the disease, explaining the phenotypic variability in this group of disorders.
In addition to skin involvement, different forms of EB can be associated with extracutaneous manifestations; these include hair, nail and tooth abnormalities, ocular findings, and fragility of the epithelia in upper respiratory, urogenital and gastrointestinal tracts [1]. Particularly interesting are two rare forms of EB, one associated with late-onset muscular dystrophy (EB-MD), and another one with congenital pyloric atresia (EB-PA). EB-MD has been shown to result from mutations in the plectin gene (PLEC1) which encodes a large, ~ 500-kD adhesion molecule [6]. In the skin, the binding partners of plectin include basal cell keratins (KRT5 and KRT14), α6β4 integrins, and type XVII collagen/the 180-kD bullous pemphigoid antigen, thus serving as a bridge between the intermediate filament cytoskeleton and hemidesmosomes within the basal keratinocytes (Fig. 1) [7, 8]. In addition to skin, plectin is expressed in a wide variety of tissues, including striated muscle and gastrointestinal epithelia. Specifically, in skeletal muscle, plectin is expressed in the sarcolemma and the Z-lines, thus participating in the formation of the intermyofibrillar-desmin cytoskeleton. Consequently, expression of plectin in the skin and in the skeletal muscle explains the consequences of mutations in two different organ systems in EB-MD, characterized by skin blistering and muscular dystrophy [6].
Figure 1.

Schematic representation of the attachment complexes at the dermal-epidermal basement membrane zone, which form a continuous network of interacting proteins necessary for stable association of epidermis and dermis at the dermal-epidermal junction. Note the presence of hemidesmosomal components, plectin, α6β4 integrin, and the type XVII collagen/the 180 kD bullous pemphigoid antigen 2 (BPAG2). The critical role of the hemidesmosomal proteins in the integrity of the skin is indicated by mutations in the corresponding genes that result in dermal-epidermal separation. (Adapted from Pulkkinen L, Uitto J. Hemidesmosomal variants of epidermolysis bullosa. Mutations in the alpha6beta4 integrin and the 180-kD bullous pemphigoid antigen/type XVII collagen genes. Exp Dermatol 1998;7:46–64., with permission)
Another gene/protein system harboring mutations in patients with EB-PA is the α6β4 integrin; the corresponding subunit polypeptides of this hemidesmosomal protein are encoded by the ITGA6 and ITGB4 genes. This transmembrane protein is an integral part of hemidesmosomes serving both a structural role in the attachment of the basal keratinocytes to the underlying basement membrane, and serving as signaling molecules. Within the intracellular milieu of basal keratinocytes, α6β4 is linked to the cytokeratin network by plectin, and in fact, the α6β4-plectin interaction is crucial for hemidesmosome stability, and it has been proposed that this association acts as an initiation step in the assembly of hemidesmosomes [9]. The α6β4 integrin is characteristically expressed in a variety of epithelial tissues, including human skin and the gastrointestinal tract, where it functions as a receptor for laminin-332 (laminin-5), a major extracellular component of the epidermal basement membrane (Fig. 1). The subunit responsible for intracellular interactions of α6β4, including binding to plectin, occurs through the cytoplasmic moiety of the β4 subunit, which is unusually large (approximately 1000 amino acid residues) and shares little similarity with other integrin β-subunits [7]. The β4 cytodomain has a modular organization with four fibronectin type III (FnIII) domains arranged in two pairs of tandem repeats separated by a connecting segment region (Fig. 2). The N-terminal region of plectin interacts with the β4 subunit at multiple sites. The primary contact is established between the acting binding domain of plectin and the first pair of FnIII domains and a small region of the connecting segment of β4 polypeptide (Figs. 2, 3). In the extracellular domain, β4 integrin has a segment of cysteine rich repeats.
Figure 2.
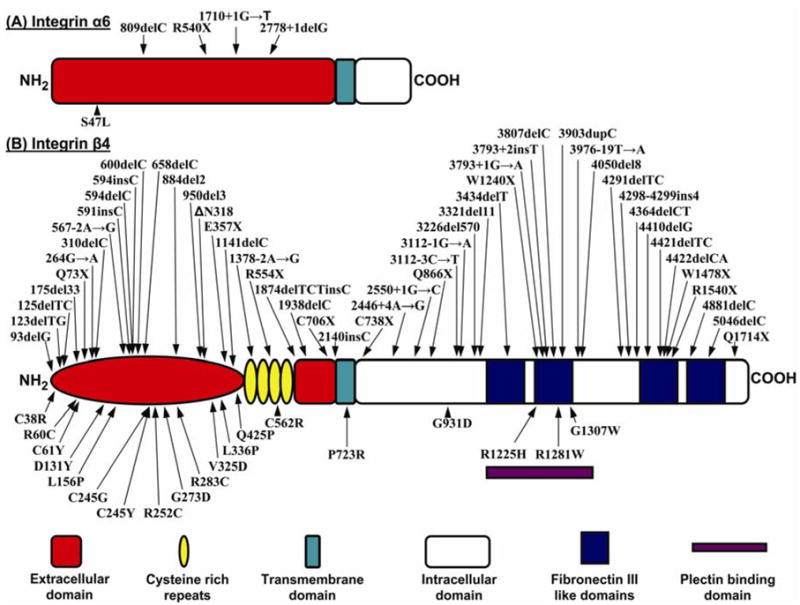
Schematic representation of the domain organizations of the α6 integrin (A) and β4 integrin (B) subunit polypeptides. The color coded explanation of the domains is at the bottom of the figure. The arrows point to the positions of the mutations along the protein structure. Mutations above each of the schematic structure represent premature termination codon-causing mutations, while those below the schematic structures are missense mutations. (Note that the numbering of the mutations may differ from those in the original publications due to the fact that the numbering has been adjusted to conform with the following NCBI database entries: ITGA6, NM-000210; ITGB4, NM-001005731).
Figure 3.
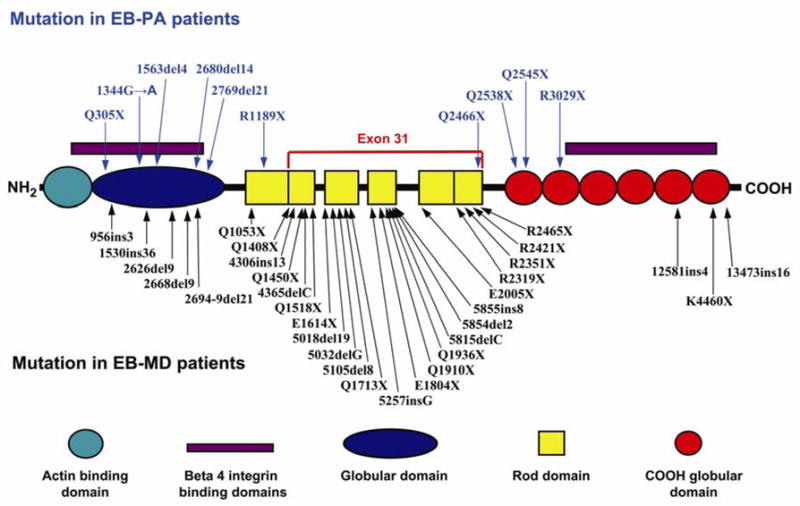
Schematic representation of the domain organization of plectin and mutations in the corresponding gene in patients with EB-PA or with EB-MD. The color code of the domain organization is at the bottom of the figure. The arrows point to the positions of the mutations along the plectin polypeptide. The mutations indicated above the schematic structure are those associated with EB-PA, while those below have been reported to result in EB-MD phenotype. Note the clustering of the mutations causing EB-MD in the rod domain that is encoded by exon 31, while all mutations in EB-PA patients (except one, Q2466X) are outside of exon 31. (Note that the numbering of the mutations may differ from those in original publications due to the fact that the numbering has been adjusted to conform with the plectin sequence in NCBI database entry NM-000445).
The α6β4 integrin stimulates cell migration, invasion and survival of epithelial cells by activating signaling pathways, and these signaling functions are also coupled to disruption of the hemidesmosomal conformation. The extracellular domain of β4 integrin interacts with the α6 integrin which is considerably smaller with a small intracellular domain (Fig. 2). The assembled α6β4 integrin then interacts with laminin 332 which on the other end connects to the lamina densa of the lower portion of the cutaneous basement membrane (Fig. 1). Thus, the α6β4 integrin plays a critical role in the integral stability of the cutaneous basement membrane zone, particularly through its interactions with plectin.
Epidermolysis bullosa with pyloric atresia
EB-PA represents a syndromic association of skin fragility and congenital gastrointestinal atresia, most frequently pyloric, although duodenal atresia with skin fragility has also been reported (Fig. 4) [10, 11]. The inheritance of EB-PA is autosomal recessive (Fig. 5). While association of EB and PA is rare, the overall incidence of PA has been reported to be less than 1% of all gastrointestinal atresias [12] and the incidence of recessively inherited junctional forms of EB has been calculated at 2.04 per 106 [13]. Thus, the segregation of these two disorders must be more than coincidental.
Figure 4.
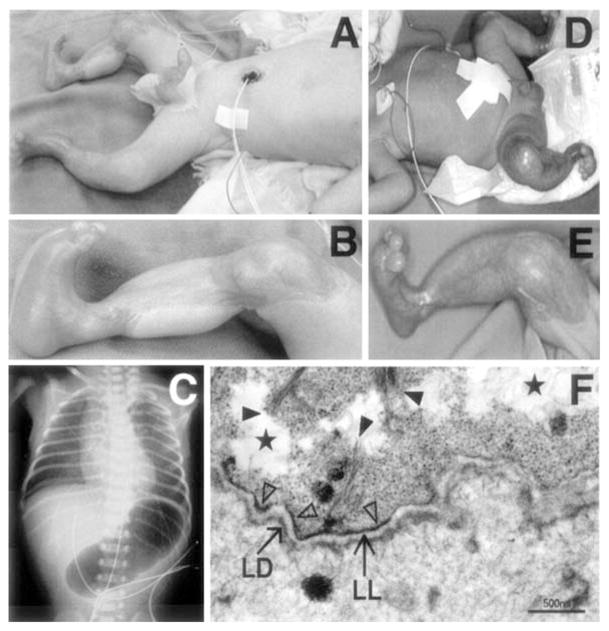
Clinical, roentgenological and ultrastructural features of a patient with EB-PA due to mutations in the PLEC1 gene. (A, B) Note sharply demarcated ulcerations in the skin of the proband. (C) Abdominal bubble of gas in X-ray of the proband. (D, E) The proband’s older brother has similar, but healing cutaneous lesions. (F) Transmission electron microscopy reveals tissue separation within the basal cells (stars). Keratin filaments are sparse (full arrowheads) and not well associated with the hemidesmosomes, which are hypoplastic and reduced in number (open arrowheads). The lamina densa (LD) and lamina lucida (LL) appear intact. Original magnification: bar, 500 nm. (Adapted from Nakamura H, Sawamura D, Goto M, et al. Epidermolysis bullosa simplex associated with pyloric atresia is a novel clinical subtype caused by mutations in the plectin gene (PLEC1). J Mol Diagn 2005;7:28–35, with permission.)
Figure 5.
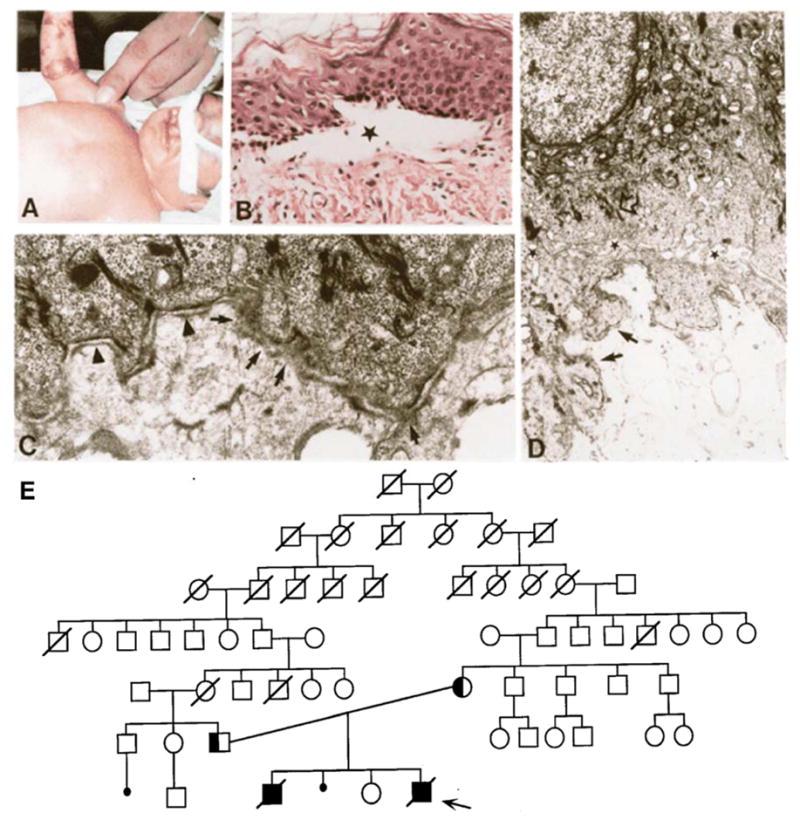
Clinical, genetic, histopathological and ultrastructural features of a patient with EB-PA. The proband (→) is the fourth child of a consanguineous mating (E). The proband demonstrates extensive blistering of the skin (A), which by histopathologic examination shows separation at the dermal-epidermal junction (B, the asterisk indicates the location of a blister). Note extensive blistering on the right arm of the proband who also had bilateral cleft lip and cleft palate (A). Transmission electronmicroscopy of the dermal-epidermal junction reveals the presence of hemidesmosomes lacking the inner plaque (C, arrowheads), and segments of the basement membrane zone were morphologically perturbed (C, arrows). Intermediate filaments were severed from the lower portion of the basal keratinocytes and condensed perinuclearly (D, open arrow). (Adapted from Pulkkinen L, Kimonis VE, Xu Y, et al. Homozygous alpha6 integrin mutation in junctional epidermolysis bullosa with congenital duodenal atresia. Hum Mol Genet 1997;6:669-74, with permission).
In some cases of EB, aplasia cutis congenita (ACC) is also present (Fig. 4) [10]. While congenital absence of skin is clearly a heterogenous group of disorders and can be associated with different subtypes of EB, patients with EB- PA can demonstrate widespread ulcerated lesions, frequently in the extremities, with complete absence of all layers of the skin (Fig. 4). The healed ACC lesions demonstrate a smooth epidermis, proliferation of fibroblasts in loose connective tissue stroma, newly formed capillaries and absence of adnexal structures. While intrauterine mechanical trauma has been suggested as an explanation for the development of ACC in some patients, with or without EB, the more than coincidental association of PA and ACC with EB suggests a common genetic basis for pathogenesis of these complications of EB [10].
The precise subclassification of the type of EB associated with PA has been confusing due to the fact that ultrastructural findings of the location of skin blistering in EB-PA patients has demonstrated two patterns [1]. First, in the majority of the cases, tissue separation occurs within the lamina lucida, associated with small hemidesmosomal plaques and often with attenuated sub-basal dense plate, observations found in classic junctional forms of EB (Fig. 5). Secondly, some patients with EB-PA demonstrate intracellular tissue separation within the lower basal cell layer, just above the level of the hemidesmosomal plaque (Fig. 4). The latter patients also demonstrate reduced integration of keratin filaments with hemidesmosomes. Since the tissue separation in the latter cases is within the basal cells, it has been suggested that these patients belong to the subtype of EB simplex (EBS). Consequently, the latest classification of inherited epidermolysis bullosa, based on report of the Third International Consensus Meeting on Diagnosis and Classification of EB, includes the existence of two different subtypes of EB with pyloric atresia, viz., EBS-PA and JEB-PA [1]. It should be noted that a previously suggested classification attempting to streamline the nomenclature of EB suggested consolidation of these two different forms of EB with pyloric atresia under the newly proposed subgroup of “hemidesmosomal variants of EB” [14]. While this classification highlighted the molecular involvement of hemidesmosomal genes in the etiology of EB-PA, together with plectin abnormalities in EB-MD and type XVII collagen/the 180-kD bullous pemphigoid antigen in generalized atrophic benign EB (GABEB), the International Consensus Meeting suggested that the term “hemidesmosomal EB” not to be used [1]. Thus, EB-PA is separated into two distinct entities based on the ultrastructural findings of the level of tissue separation within the cutaneous basement membrane zone: EBS-PA and JEB-PA.
Clinical and pathologic features of EB-PA
The diagnosis of EB-PA, with or without ACC, is based on clinical observations, imaging studies, histopathology, ultrastructural findings, immunohistochemistry of the skin, and molecular diagnostics (Fig. 4–6) [10]. As indicated above, the cutaneous findings include tissue separation either within the basal cells at the level of hemidesmosomes or within the lamina lucida of the dermal-epidermal basement membrane. The presence of pyloric atresia is often suggested by gestational hydramnion which can be confirmed by ultrasound and X-ray studies (Fig. 4). Histopathology of the gastrointestinal involvement can show obstruction of the esophagus by a fibrotic membrane with disappearance of the mucosal layer. Narrowing of the pylorus and proximal part of duodenum associated with thickening of the gastric submucosal connective tissue can proceed to complete obstruction by a fibrotic membrane rich in blood vessels [10]. These findings are frequently associated with inflammation. It has been postulated that the basic pathology leading to the EB-PA-ACC phenotype involves two elements: (a) the integrity of the basement membrane zone and hemidesmosomes, and (b) the control of processes of fibrosis [10]. This sequence of events may be initiated by the separation of the epidermis or the intestinal mucosal layer as a result of poorly functional or absent hemidesmosomal complexes. Inflammatory responses contribute to the development of secondary fibrosis leading specifically to the obstruction of the intestinal lumina, especially in anatomically narrow spaces, such as pylorus. In support of this cascade of events are ultrastructural demonstrations of hemidesmosomal abnormalities, histologically demonstrated inflammation, and more recently, immunohistochemical demonstration of abnormalities in the expression of α6β4 integrin and/or plectin.
Figure 6.
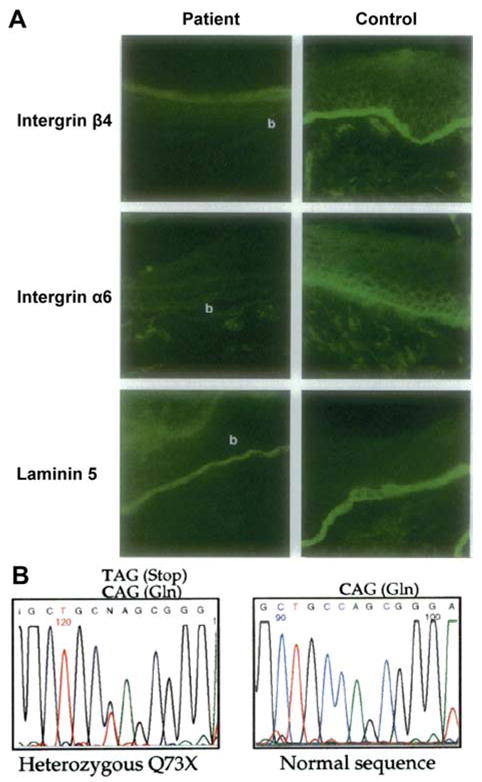
Immunohistochemical analysis and mutation detection in families with EB-PA. Upper panel (A) illustrates immunohistochemical analysis of the skin of an affected infant with EB-PA (left panel), as compared to an unrelated control (right panel). Skin sections were stained with monoclonal antibodies 3E1 recognizing integrin β4, GoH3 recognizing integrin α6, and GB3 recognizing intact laminin-5 (laminin 332), as indicated on the left side. Skin of the affected infant was negative for α6 and β4, which staining for laminin-5 was normal. The blister cavities are indicated by b. (B) Illustration of identification of a mutation in the ITGB4 gene in a patient with EB-PA (lower panel). Note that sequencing of the proband is ITGB4 gene revealed a C→T transition mutation at nucleotide position 217 in one allele, resulting in change of codon for glutamine (CAG) to a stop codon (TAG) at amino acid position 73. In comparison, the control sequences are shown on right. (Adapted from Pulkkinen L, Kim DU, Uitto J. Epidermolysis bullosa with pyloric atresia: novel mutations in the beta4 integrin gene (ITGB4). Am J Pathol 1998;152:157-66., with permission).
The severity of skin involvement in EB-PA can be highly variable [15–17]. In some cases, even upon successful surgical correction of the pyloric or duodenal atresia, skin fragility is so severe that the affected children die from complications, such as infections or electrolyte imbalance, within a few days of weeks post-partum. In other cases, the skin fragility may be relatively mild, and in some cases, age-associated amelioration of the skin fragility allows individuals to conduct normal life activities with relatively minor blistering tendency. In some cases, blisters may develop only upon strenuous mechanical trauma to the skin. The denuded lesions of ACC can heal in surviving individuals, with primarily cosmetic sequela.
Molecular genetics of EB-PA
Early immunohistochemical evidence suggested that expression of the α6β4 integrin is reduced or completely absent in the skin of several patients affected with EB-PA (Fig. 6a) [18–20]. This observation was followed by cloning and sequencing of the genes encoding the two subunit polypeptides of the α6β4 integrin (ITGA6 and ITGB4) (Figs. 2, 3) [21, 22].
The genes encoding α6β4 integrin subunit polypeptides have been shown to harbor a large number of mutations in patients with EB-PA, the majority of them residing in ITGB4 (Fig. 2). Examination of the mutation database in ITGB4 reveals a total of 70 distinct mutations in patients with skin blistering and all but two of them have PA. Among these mutations, 45 were premature termination codon (PTC)-causing mutations (nonsense mutations, small insertions or deletions or putative splice junction mutations resulting in frame-shift), which are predicted to result in the translation of shortened, and presumably non-functional, β4 integrin polypeptides (Fig. 2b). The generation of PTCs can also result in the absence of the corresponding protein due to accelerated mRNA decay or by the truncated polypeptides being sensitive to proteolytic degradation [23, 24]. In addition, a number of missense mutations have been identified, and in many cases the substituted amino acid has been shown to be highly conserved through evolution of the ITGB4 gene in different species or among different members of the human β-integrin proteins (Fig. 7) [25].
Figure 7.
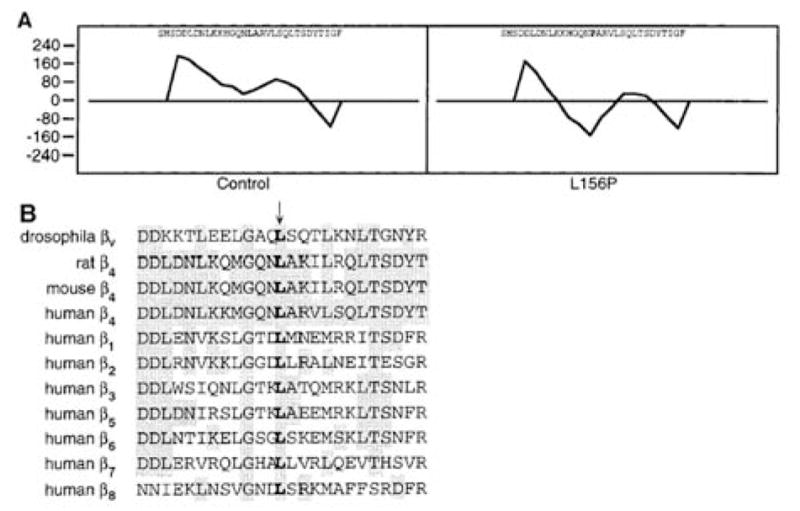
Demonstration of consequences of a leucine-to-proline substitution in the amino acid position 156 of the β4 integrin polypeptide in a patient with EB-PA. (A) Garnier α-helicity plot predicts that introduction of proline into position 156, in place of a leucine, disrupts the α-helix of the mutated polypeptide. (B) Comparison of β4 integrin sequences in different species as well as between different human integrin β-chains reveals conservation of the leucine at position 156. (Adapted from Pulkkinen L, Bruckner-Tuderman L, August C, et al. Compound heterozygosity for missense (L156P) and nonsense (R554X) mutations in the beta4 integrin gene (ITGB4) underlies mild, nonlethal phenotype of epidermolysis bullosa with pyloric atresia. Am J Pathol 1998;152:935-41., with permission.)
Among the missense mutations in ITGB4, there are five cysteine substitutions which affect the extracellular domain of the β4 subunit (Fig. 2b). While some of the cysteine substitution mutations (such as homozygous p.C61Y) have been associated with lethal outcome [14], non-lethal cases from consangineous union have also been described. For example, homozygous, p.C562R mutation in the cysteine-rich region of the β4 integrin resulted in pyloric atresia noted at birth but developed only localized, relatively mild skin blistering with subsequent tooth and nail dystrophy [14]. In addition to homozygous mutations, a number of compound heterozygous mutations in ITGB4 have been described. For example, two non-lethal cases of EB-PA, one of them compound heterozygous for two distinct arginine substitution mutations (p.R252C/p.R1281W) and another one for a leucine-to-proline substitution mutation, in combination with a nonsense mutation (p.L156P/p.R554X), have been reported [14, 25]. The first one of these two cases was moderately affected at birth, but the condition considerably improved with time. The second case had very mild blistering tendency and dystrophic nails. One of the mutations (p.R1281W) affects the intracellular domain of β4 integrin polypeptide within the putative region interacting with plectin. On the other hand, the other mutation in this case, p.R252C, creates a new cysteine residue in the extracellular domain of β4 integrin and may participate in the formation of new intra- or intermolecular disulfide bonds, possibly disrupting ligand binding or affecting non-covalent associations between the α6 and β4 integrin subunits. The missense mutations, particularly those affecting highly conserved amino acid residues within the extracellular domain of β4 integrin can result in conformational changes of the β4 integrin polypeptide. For example, in a patient with p.L156P mutation, the Garnier α-helicity plot analysis has suggested that the region spanning Leu-156 has propensity for α-helix formation, and introduction of a proline residue to this position disrupts the helical conformation (Fig. 7) [25]. These and related observations suggest that the invariably conserved Leu-156 residue plays a critical role in the structure/function characteristics of β4 integrin.
In addition to mutations in the ITGB4 gene, a total of five mutations in the α6 integrin subunit gene (ITGA6) have been reported in patients with EB-PA (Fig. 2a). Examination of the ITGA6 mutation database reveals that three of the total of five mutations are PTC-causing ones, while one is a nonsense mutation. All of them reside within the extracellular domain of the deduced protein (Fig. 2a). The clinical features of the cases with ITGA6 mutations are largely indistinguishable from those caused by mutations in ITGB4, indicating that both subunit polypeptides of the α6β4 integrin complex play a critical role in the function of this integrin. Why the mutations in the ITGA6 gene are less frequent than those in ITGB4 is currently unknown.
As indicated above, ultrastructural observations on the skin lesions in EB-PA have suggested that tissue separation can occur at the level of lamina lucida or within the basal cells, thus leading to suggestion that there are two subtypes, JEB-PA and EBS-PA. While mutations in the α6β4 integrin genes, which encode a transmembrane protein both with intracellular and extracellular domains, the majority of the ITGB4 and ITGA6 mutations appear to be associated with lamina lucida split, suggesting that they are the primary cause of the JEB-PA subtype. In addition, deletion of a cytoplasmic domain of integrin β4 polypeptide has been shown to cause a relatively mild EBS without association of PA [26]. Specifically, a 2-bp deletion, c.4364delCT, in ITGB4 resulted in in-frame skipping of 50 amino acids (p.del1450-1499) within the third fibronectin Type III repeat in the cytoplasmic domain of the integrin β4 polypeptide. This domain of integrin β4 is thought to interact with type XVII collagen/the180-kD bullous pemphigoid antigen, attesting to the importance of the α6β4 integrin in epidermal integrity.
A recent report has suggested that ITGB4 can harbor mutations which do not lead into significant skin blistering but are associated with pyloric atresia [27]. Specifically, two Kuwaiti siblings had PA and life-threatening intestinal desquamation, without significant skin abnormalities. Analysis of α6β4 integrin genes identified a novel mutation in ITGB4, a homozygous deletion of a single isoleucine 1314 residue within the intracellular plectin-binding domain. Interestingly, expression of α6 and β4 integrin within skin, duodenal and colonic epithelium was normal or slightly reduced as detected by immunohistochemical techniques. This demonstration of a mutation within the ITGB4 gene, together with the existence of desquamated enteropathy associated with congenital PA, suggests existence of an overlap condition with JEB-PA without cutaneous signs. While the deletion of this single amino acid (Ile1314), which resides within a fibronectin III-like domain, potentially interacting with plectin, it is unclear as to why the phenotypic expression in these patients is limited to intestinal epithelium which does not contain hemidesmosomes. In this case, there was evidence of mucosal inflammatory response, possibly secondary to loss of epithelial barrier function, and co-localization of IgG and C1q within the intestinal basement membrane and low-titer circulating IgG autoantibody was noted. Significant improvement in the desquamative enteropathy was noted with immune modulatory therapy, leading to suggestion that such treatment of these patients may give significant clinical benefits in other patients as well. Finally, a case with classic features of JEB, without PA and no history of gastrointestinal diseases, has been reported to harbor an ITGB4 mutation, of critical amino acid residues leading to selected, tissue-specific manifestations in some patients [28].
Mutations in the plectin gene (PLEC1) can cause EB-PA
Plectin, a 500-kD intermediate filament-binding protein and intracellular component of hemidesmosomes interacts with β4 integrin (Fig. 1). As indicated above, mutations in the plectin gene were originally shown to result in an autosomal recessive variant of EB with late-onset muscular dystrophy [6]; up to now, 31 mutations in the plectin gene associated with EB-MD have been reported (Fig. 3). Considering the interactions of plectin and α6β4 integrin, it was not entirely surprising that a subset of patients with EB-PA was shown by immunohistochemical staining to have reduced or absent expression of plectin in the skin, and subsequently, 10 distinct mutations in the PLEC1 gene were demonstrated in 11 patients with EB-PA (Fig. 3) [29, 30]. Among these, there are a total of 9 PTC-causing mutations, one mutation causes an in-frame deletion of 7 amino acids, and none of the mutations are missense ones. All these patients with EB have definite pyloric atresia and none of them have been reported to develop muscular dystrophy. Most of these patients also have extensive ACC, and at least 9 of them died at early post-natal period.
A remarkable observation relates to the distribution of the mutations in PLEC1 gene in patients with EB-PA. While the mutations in EB-MD patients are distributed along the entire length of the plectin gene, many of them cluster in exon 31 which encodes the rod domain in the middle of the protein (Fig. 3). In contrast, the PLEC1 mutations in EB-PA patients, with one exception, are outside of the exon 31. This observation would provide an explanation for the phenotypic differences as a result of plectin mutations leading to EB-PA or EB-MD. Specifically, cells such as skin fibroblasts express two different plectin isoforms consisting of either full-length polypeptides or a rodless form generated by alternative splicing of exon 31 [31]. In case of mutations within exon 31, the rodless isoform of plectin may be expressed leading to milder skin manifestations, yet associated with late-onset muscular dystrophy. In case of PTC-causing, loss-of-function mutations outside the exon 31, both the full-length and rodless isoforms of plectin are lost, thus leading to more severe cutaneous blistering phenotype associated with PA [31]. This pathomechanistic difference might provide an explanation for the genotype/phenotype correlation resulting in two different phenotypes, EB-PA and EB-MD, as a result of mutations in the same gene.
Clinical implications of molecular genetics on EB-PA
Analogous to other major forms of EB, identification of precise mutations in EB-PA has profound implications for the management of patients and counseling of families affected with these diseases. Specifically, identification of mutations in families has provided molecular confirmation of the diagnosis coupled with prognostication, and will also provide a means for prenatal testing in families at risk for recurrence of the disease [32, 33]. In fact, prenatal testing in families at risk for EB-PA has been performed from chorionic villus sampling, which can be performed as early as the tenth week of gestation [32]. As an extension of DNA-based prenatal diagnosis, it has also been suggested that immunofluorescence analysis of villus trophoblasts may provide additional prenatal information [34]. Specifically, first-trimester chorionic villi in normal pregnancies were shown to strongly express both integrin α6β4 and plectin by immunofluorescence analysis. Subsequently, in a cohort of 25 pregnancies at risk for EB-PA, 3 fetuses were predicted to be affected, a finding that was confirmed by DNA-based tests and clinical observations. As an extension of the DNA-based prenatal testing, preimplantation genetic diagnosis has been implemented for EB and related skin fragility syndromes [35, 36]. Although preimplantation genetic diagnosis avoids the potential ethical issues related to termination of pregnancy, its wider adoption has been hampered by the relatively high cost and extensive clinical involvement of the mother in preparation for the test as part of in vitro fertilization procedure. Finally, identification of the genes and precise mutations involved in the pathomechanisms of EB-PA provides the basis for molecularly based therapies, either gene therapy, protein replacement, or stem-cell based therapies [37, 38]. These approaches are supported by development of animal models for different forms of EB [39], including those generated through targeted ablation of the genes, ITGB4, ITGA6, and PLEC1, which harbor mutations in EB-PA.
Acknowledgments
GianPaolo Guercio assisted in preparation of this manuscript. The authors’ original studies were supported by DHHS, NIH/NIAMS grants P01 AR38923 and by Dermatology Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fine JD, Eady RA, Bauer EA, et al. The classification of inherited epidermolysis bullosa (EB): Report of the Third international Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008;58:931–50. doi: 10.1016/j.jaad.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 2.Uitto J, Richard G. Progress in epidermolysis bullosa: genetic classification and clinical implications. Am J Med Genet C Semin Med Genet. 2004;131C:61–74. doi: 10.1002/ajmg.c.30035. [DOI] [PubMed] [Google Scholar]
- 3.Uitto J, Richard G. Progress in epidermolysis bullosa: from eponyms to molecular genetic classification. Clin Dermatol. 2005;23:33–40. doi: 10.1016/j.clindermatol.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 4.Varki R, Sadowski S, Pfendner E, et al. Epidermolysis bullosa. I. Molecular genetics of the junctional and hemidesmosomal variants. J Med Genet. 2006;43:641–52. doi: 10.1136/jmg.2005.039685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Varki R, Sadowski S, Uitto J, et al. Epidermolysis bullosa. II. Type VII collagen mutations and phenotype-genotype correlations in the dystrophic subtypes. J Med Genet. 2007;44:181–92. doi: 10.1136/jmg.2006.045302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pfendner E, Rouan F, Uitto J. Progress in epidermolysis bullosa: the phenotypic spectrum of plectin mutations. Exp Dermatol. 2005;14:241–9. doi: 10.1111/j.0906-6705.2005.00324.x. [DOI] [PubMed] [Google Scholar]
- 7.de Pereda JM, Lillo MP, Sonnenberg A. Structural basis of the interaction between integrin alpha6beta4 and plectin at the hemidesmosomes. EMBO J. 2009;28:1180–90. doi: 10.1038/emboj.2009.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andrä K, Kornacker I, Jörgl A, et al. Plectin-isoform-specific rescue of hemidesmosomal defects in plectin (−/−) keratinocytes. J Invest Dermatol. 2003;120:189–97. doi: 10.1046/j.1523-1747.2003.12027.x. [DOI] [PubMed] [Google Scholar]
- 9.Schaapveld RQ, Borradori L, Geerts D, et al. Hemidesmosome formation is initiated by the beta4 integrin subunit, requires complex formation of beta4 and HD1/plectin, and involves a direct interaction between beta4 and the bullous pemphigoid antigen 180. J Cell Biol. 1998;142:271–84. doi: 10.1083/jcb.142.1.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maman E, Maor E, Kachko L, et al. Epidermolysis bullosa, pyloric atresia, aplasia cutis congenita: histopathological delineation of an autosomal recessive disease. Am J Med Genet. 1998;78:127–33. [PubMed] [Google Scholar]
- 11.Dang N, Klingberg S, Rubin AI, et al. Differential expression of pyloric atresia in junctional epidermolysis bullosa with ITGB4 mutations suggests that pyloric atresia is due to factors other than the mutations and not predictive of a poor outcome: three novel mutations and a review of the literature. Acta Derm Venereol. 2008;88:438–48. doi: 10.2340/00015555-0484. [DOI] [PubMed] [Google Scholar]
- 12.Thompson NW, Parker W, Schwartz S, et al. Congenital pyloric atresia. Arch Surg. 1968;97:792–6. doi: 10.1001/archsurg.1968.01340050132020. [DOI] [PubMed] [Google Scholar]
- 13.Pfendner E, Uitto J, Fine JD. Epidermolysis bullosa carrier frequencies in the US population. J Invest Dermatol. 2001;116:483–4. doi: 10.1046/j.1523-1747.2001.01279-11.x. [DOI] [PubMed] [Google Scholar]
- 14.Pulkkinen L, Uitto J. Hemidesmosomal variants of epidermolysis bullosa. Mutations in the alpha6beta4 integrin and the 180-kD bullous pemphigoid antigen/type XVII collagen genes. Exp Dermatol. 1998;7:46–64. doi: 10.1111/j.1600-0625.1998.tb00304.x. [DOI] [PubMed] [Google Scholar]
- 15.Nakano A, Pulkkinen L, Murrell D, et al. Epidermolysis bullosa with congenital pyloric atresia: novel mutations in the beta 4 integrin gene (ITGB4) and genotype/phenotype correlations. Pediatr Res. 2001;49:618–26. doi: 10.1203/00006450-200105000-00003. [DOI] [PubMed] [Google Scholar]
- 16.Pulkkinen L, Kim DU, Uitto J. Epidermolysis bullosa with pyloric atresia: novel mutations in the beta4 integrin gene (ITGB4) Am J Pathol. 1998;152:157–66. [PMC free article] [PubMed] [Google Scholar]
- 17.Mellerio JE, Pulkkinen L, McMillan JR, et al. Pyloric atresia-junctional epidermolysis bullosa syndrome: mutations in the integrin beta4 gene (ITGB4) in two unrelated patients with mild disease. Br J Dermatol. 1998;139:862–71. doi: 10.1046/j.1365-2133.1998.02515.x. [DOI] [PubMed] [Google Scholar]
- 18.Jonkman MF, de Jong MC, Heeres K, et al. Expression of integrin alpha 6 beta 4 in junctional epidermolysis bullosa. J Invest Dermatol. 1992;99:489–96. doi: 10.1111/1523-1747.ep12616168. [DOI] [PubMed] [Google Scholar]
- 19.Phillips RJ, Aplin JD, Lake BD. Antigenic expression of integrin alpha 6 beta 4 in junctional epidermolysis bullosa. Histopathology. 1994;24:571–6. doi: 10.1111/j.1365-2559.1994.tb00578.x. [DOI] [PubMed] [Google Scholar]
- 20.Vidal F, Aberdam D, Miquel C, et al. Integrin beta 4 mutations associated with junctional epidermolysis bullosa with pyloric atresia. Nat Genet. 1995;10:229–34. doi: 10.1038/ng0695-229. [DOI] [PubMed] [Google Scholar]
- 21.Pulkkinen L, Kimonis VE, Xu Y, et al. Homozygous alpha6 integrin mutation in junctional epidermolysis bullosa with congenital duodenal atresia. Hum Mol Genet. 1997;6:669–74. doi: 10.1093/hmg/6.5.669. [DOI] [PubMed] [Google Scholar]
- 22.Pulkkinen L, Kurtz K, Xu Y, et al. Genomic organization of the integrin beta 4 gene (ITGB4): a homozygous splice-site mutation in a patient with junctional epidermolysis bullosa associated with pyloric atresia. Lab Invest. 1997;76:823–33. [PubMed] [Google Scholar]
- 23.Iacovacci S, Cicuzza S, Odorisio T, et al. Novel and recurrent mutations in the integrin beta 4 subunit gene causing lethal junctional epidermolysis bullosa with pyloric atresia. Exp Dermatol. 2003;12:716–20. doi: 10.1034/j.1600-0625.2003.00052.x. [DOI] [PubMed] [Google Scholar]
- 24.Micheloni A, De Luca N, Tadini G, et al. Intracellular degradation of beta4 integrin in lethal junctional epidermolysis bullosa with pyloric atresia. Br J Dermatol. 2004;151:796–802. doi: 10.1111/j.1365-2133.2004.06206.x. [DOI] [PubMed] [Google Scholar]
- 25.Pulkkinen L, Bruckner-Tuderman L, August C, et al. Compound heterozygosity for missense (L156P) and nonsense (R554X) mutations in the beta4 integrin gene (ITGB4) underlies mild, nonlethal phenotype of epidermolysis bullosa with pyloric atresia. Am J Pathol. 1998;152:935–41. [PMC free article] [PubMed] [Google Scholar]
- 26.Jonkman MF, Pas HH, Nijenhuis M, et al. Deletion of a cytoplasmic domain of integrin beta4 causes epidermolysis bullosa simplex. J Invest Dermatol. 2002;119:1275–81. doi: 10.1046/j.1523-1747.2002.19609.x. [DOI] [PubMed] [Google Scholar]
- 27.Salvestrini C, McGrath JA, Ozoemena L, et al. Desquamative enteropathy and pyloric atresia without skin disease caused by a novel intracellular beta4 integrin mutation. J Pediatr Gastroenterol Nutr. 2008;47:585–91. doi: 10.1097/MPG.0b013e31817af98d. [DOI] [PubMed] [Google Scholar]
- 28.Inoue M, Tamai K, Shimizu H, et al. A homozygous missense mutation in the cytoplasmic tail of beta4 integrin, G931D, that disrupts hemidesmosome assembly and underlies Non-Herlitz junctional epidermolysis bullosa without pyloric atresia? J Invest Dermatol. 2000;114:1061–4. doi: 10.1046/j.1523-1747.2000.00960-3.x. [DOI] [PubMed] [Google Scholar]
- 29.Pfendner E, Uitto J. Plectin gene mutations can cause epidermolysis bullosa with pyloric atresia. J Invest Dermatol. 2005;124:111–5. doi: 10.1111/j.0022-202X.2004.23564.x. [DOI] [PubMed] [Google Scholar]
- 30.Nakamura H, Sawamura D, Goto M, et al. Epidermolysis bullosa simplex associated with pyloric atresia is a novel clinical subtype caused by mutations in the plectin gene (PLEC1) J Mol Diagn. 2005;7:28–35. doi: 10.1016/S1525-1578(10)60005-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sawamura D, Goto M, Sakai K, et al. Possible involvement of exon 31 alternative splicing in phenotype and severity of epidermolysis bullosa caused by mutations in PLEC1. J Invest Dermatol. 2007;127:1537–40. doi: 10.1038/sj.jid.5700707. [DOI] [PubMed] [Google Scholar]
- 32.Pfendner EG, Nakano A, Pulkkinen L, et al. Prenatal diagnosis for epidermolysis bullosa: a study of 144 consecutive pregnancies at risk. Prenat Diagn. 2003;23:447–56. doi: 10.1002/pd.619. [DOI] [PubMed] [Google Scholar]
- 33.Fassihi H, Eady RA, Mellerio JE, et al. Prenatal diagnosis for severe inherited skin disorders: 25 years’ experience. Br J Dermatol. 2006;154:106–13. doi: 10.1111/j.1365-2133.2005.07012.x. [DOI] [PubMed] [Google Scholar]
- 34.D’Alessio M, Zambruno G, Charlesworth A, et al. Immunofluorescence analysis of villous trophoblasts: a tool for prenatal diagnosis of inherited epidermolysis bullosa with pyloric atresia. J Invest Dermatol. 2008;128:2815–9. doi: 10.1038/jid.2008.143. [DOI] [PubMed] [Google Scholar]
- 35.Cserhalmi-Friedman PB, Tang Y, Adler A, et al. Preimplantation genetic diagnosis in two families at risk for recurrence of Herlitz junctional epidermolysis bullosa. Exp Dermatol. 2000;9:290–7. doi: 10.1034/j.1600-0625.2000.009004290.x. [DOI] [PubMed] [Google Scholar]
- 36.Fassihi H, Grace J, Lashwood A, et al. Preimplantation genetic diagnosis of skin fragility-ectodermal dysplasia syndrome. Br J Dermatol. 2006;154:546–50. doi: 10.1111/j.1365-2133.2005.07102.x. [DOI] [PubMed] [Google Scholar]
- 37.Uitto J. Progress in heritable skin diseases: translational implications of mutation analysis and prospects of molecular therapies*. Acta Derm Venereol. 2009;89:228–35. doi: 10.2340/00015555-0648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tamai K, Kaneda Y, Uitto J. Molecular therapies for heritable blistering diseases. Trends Mol Med. 2009;15:285–92. doi: 10.1016/j.molmed.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 39.Jiang QJ, Uitto J. Animal models of epidermolysis bullosa--targets for gene therapy. J Invest Dermatol. 2005;124:xi–xiii. doi: 10.1111/j.0022-202X.2005.23652.x. [DOI] [PubMed] [Google Scholar]