

Abstract
Hinge cleavage of a recombinant human IgG1 antibody, generated during production in a Chinese hamster ovary cell culture, was observed in the purified material. The cleavage products could be reproduced by incubation of the antibody with H2O2 and featured complementary ladders of the C- and N-terminal residues (Asp226–Lys227–Thr228–His229–Thr230) in the heavy chain of the Fab domain and the upper hinge of one of the Fc domains, respectively. Two adducts of +45 and +71 Da were also observed at the N-terminal residues of some Fc fragments and were identified as isocyanate and α-ketoacyl derivatives generated by radical cleavage at the α-carbon position through the diamide and α-amidation pathways. We determined that the hinge cleavage was initiated by radical-induced breakage of the disulfide bond between the two hinge cysteines at position 231 (Cys231-Pro-Pro-Cys-Pro), followed by the formation of a thiyl radical (Cys231-S•) on one cysteine and sulfenic acid (Cys231-SOH) on the other. The location of the initial radical attack and the critical role of Cys231 were demonstrated by the observation that 5,5-dimethyl-1-pyrroline N-oxide only reacted with the Cys231 radical and completely blocked hinge cleavage, suggesting the necessity of an electron/radical transfer from the Cys231 radical to the hinge residues where cleavage was observed. As a precursor of hydroxyl radicals, H2O2 is widely produced in healthy cells and tissues and therefore could be the source for the radical-induced fragmentation of human IgG1 antibodies in vivo.
Introduction
Recombinant human monoclonal antibodies (mAbs)2 have been the main products for the biotechnology industry for more than a decade (1–3). A key strength of antibodies as therapeutics is that their clinical potential can readily be increased by improving their existing properties through a range of antibody engineering technologies (4, 5). As therapeutic agents, mAbs are produced in large scale cell culture, purified, and stored under various conditions and administered to patients (6, 7). Exposure to these production/storage conditions may reduce the stability and efficacy of the mAb by increasing the chance for introducing undesirable modifications such as oxidation, proteolytic cleavage, deamidation, and isomerization. A better understanding of the whole spectrum of possible degradation pathways, particularly new pathways, could facilitate the engineering of mAbs with improvement in the production of stable, efficacious, and safe biotherapeutics.
A recent study indicated that antibodies have the intrinsic capacity to convert molecular oxygen into hydrogen peroxide (H2O2) (8) and in this process to produce some short lived hydroxyl radical species (HO•) at the interface of the light and heavy chains (9–12). These observations were further supported by a more recent observation that the light chains (three κ and three λ types) from the urine of six patients who had multiple myeloma and light chain proteinuria were found capable of generating H2O2 (13). Substantial evidence suggests that the production of H2O2 is an important signaling event triggered by the activation of various cell surface receptors, such as antibody-receptor interaction (14–19). It has been demonstrated that H2O2-mediated redox chemistry can regulate the biological function of proteins through interactions with specific residues such as cysteine (Cys) (20–23); thus, H2O2 may represent a key signaling molecule in mammalian systems. Stamler and Hausladen (20) have proposed a continuum of H2O2-mediated Cys–SH modifications that constitute important biological signaling events on the one hand and irreversible hallmarks of oxidative stress on the other. Quite commonly, Cys–SH reacts with H2O2 and yields oxidized forms of reversible or irreversible modified residues; reversible modified groups can be stabilized within the protein environment and recycled (21). Irreversible oxidation can lead to the degradation of proteins via a hydroxyl radical mediated mechanism to cleave a peptide bond at the α-carbon position through either the diamide or α-amidation pathways (24–27). Although extensive studies have been conducted, because of the transient nature of a radical reaction and unstable intermediate products, the mechanisms underlying the formation of some specific reaction products is still not fully understood. It is well known that Cys residues of an IgG molecule form the intrachain or interchain disulfide bonds (1, 5); thus, the effect of H2O2-mediated Cys redox chemistry on the structure and stability of a human antibody remains unclear.
A lot of the trace reactions involving H2O2 in vivo are also believed to occur in cell-based cell culture systems, and there is convincing evidence linking them to the same pathways of either H2O2 generation or H2O2-mediated cellular signaling (28–30). Most recombinant mAb are produced by large scale bioreactor cell cultures in three principal mammalian cell lines, Chinese hamster ovary (CHO) cells and murine myeloma lines SP2/0 and NS0. The bioreactor cell culture is the industrial standard platform for the production of the mAbs, where the conditions mimic the physiological environment, providing in vivo-like conditions. Here, we describe the degradation of a recombinant human IgG1 antibody that was purified from CHO cells cultured in a bioreactor, and we present evidence for a specific H2O2-mediated radical cleavage in the hinge region of the antibody. The cleavage led to the loss of one Fab domain and a heavily oxidized partial molecule. The mechanism for the formation of this partial molecule could be mimicked by H2O2 treatment of the intact IgG1, which resulted in the breakage of the first interchain disulfide bond in the hinge region and the formation of sulfenic acid (Cys231–SOH) on one cysteine and a thiyl radical (Cys231–S•) on the other. The thiyl radical initializes an electron transfer/radical transfer to the hinge residues, which gives rise to backbone cleavage on a one radical cleavage per molecule basis to generate one free Fab domain fragment with a ladder of the C-terminal heavy chain residues complementary to the N-terminal ladder of one of the heavy chains of the Fc domain in the truncated IgG1 mAb. We propose that the observed sequential hinge cleavage of an IgG1 resulted from a hydroxyl radical reaction mechanism.
EXPERIMENTAL PROCEDURES
Material
The antibody used in this study is a recombinant fully human antibody of the IgG1 subclass. The molecule was expressed in CHO cells and chromatographically purified using conventional techniques, including protein A, cation-exchange chromatography, and hydrophobic interaction chromatography (31).
SEC Analysis and Purification of the Partial Antibody
The partial antibody was separated by SEC on a TSK G3000SWxl dual column (Tosoh Bioscience LLC, Montgomeryville, PA), 7.8 × 300 mm, 5 μm) at a flow rate of 0.5 ml/min. Eluting protein was monitored at 220 nm. The SEC running buffer contained 50 mm sodium phosphate, 300 mm sodium chloride, pH 6.8. The cleavage was measured by the relative percentage of integrated peak area of partial molecules (C1 and C2, see text for details). SEC fractionation was carried out on an Agilent 1200 HPLC system (Agilent Technologies, Santa Clara, CA) equipped with a fraction collector. Purified fractions were pooled and concentrated by centrifugation in Millipore Centriprep YM-30 filter units with a 30-kDa molecular mass cutoff (Millipore, Billerica, MA). The above procedure was repeated as necessary to ensure greater than 95% fraction purity.
Protein Oxidation
A reaction mixture (1.0 ml) containing 10 mg of IgG1 antibody in a buffer containing 10 mm glutamic acid at pH 5.2 was incubated at 25 °C with 20 mm H2O2. To remove H2O2, the samples were buffer-exchanged by centrifugation in filter units as described above.
Reversed-phase Chromatography and Time-of-Flight Mass Analysis
Purified partial molecules were reduced and alkylated as described previously (31). Briefly, antibody was diluted to 1 mg/ml in 50 mm Tris-HCl, pH 8.0 (Sigma), with a final concentration of 4 m guanidine hydrochloride (Mallinckrodt, Hazelwood, MO). A 0.5 m dithiothreitol (DTT, Sigma) stock solution was added to give a final concentration of 5 mm, and the reaction mixture was placed for 30 min at 55 °C. The protein solution was cooled to room temperature, and a 0.5 m iodoacetamide (IAM, Sigma) stock solution was added to reach a final concentration of 15 mm IAM. The alkylation was performed at room temperature for 40 min in the dark. Then, a 0.5 m DTT stock solution was added to obtain a final concentration of 15 mm DTT to quench the alkylation. Reversed-phase high performance liquid chromatography (RP-HPLC) was performed on an Agilent 1200 HPLC system. The mobile phase included water with 0.11% trifluoroacetic acid as solvent A and acetonitrile (Honeywell Burdick & Jackson) with 0.09% trifluoroacetic acid as solvent B. An Agilent Zorbax SB300 C8 (Agilent Technologies, Santa Clara, CA), 2.1 × 150 mm, 5-μm particle size, 300-Å pore size column was used for the RP-HPLC time-of-flight (TOF) mass spectrometric (MS) analysis. The column eluent was analyzed by UV detection at 214 nm and then directed in-line to a TOF mass spectrometer. The initial mobile phase was 25% solvent B for 5 min, and then a two-stage gradient was applied of 1.4% solvent B per min from 25 to 32% solvent B, followed by a second gradient of 0.24% solvent B per min from 32 to 38% solvent B. The separation was performed at 75 °C at a flow rate of 0.3 ml/min. Peak areas were calculated using ChemStation software (Agilent Technologies, Santa Clara, CA) by applying a tangent skimming technique for the integration of incompletely resolved peaks. Electrospray ionization TOF/MS was performed on an Agilent 6210 liquid chromatography-TOF/MS spectrometer equipped with an Agilent 1200 HPLC system. The electrospray ionization-TOF/MS system was set to run in positive ion mode with a capillary voltage of 5,000 V, and an m/z range of 50–3,500. The electrospray ionization mass spectra were analyzed using Agilent BioConfirm protein deconvolution software (Agilent Technologies, Santa Clara, CA).
Protease Digestion and Peptide Maps
Prior to digestion, samples were buffer-exchanged into 50 mm Tris-HCl, pH 7.5, using Bio-Spin 6 columns (Bio-Rad) according to the manufacturer's instructions. Recombinant proteomics grade trypsin (Roche Applied Science), or sequencing grade Lys-C or Asp-N (Roche Applied Science) was added to samples at an enzyme to protein ratio of 1:10 (w/w). Digestion occurred for 4 h at 37 °C for which aliquots at different time points were frozen away at −20 °C until analysis. In some cases, prior to reduction and alkylation, any unpaired cysteine residues were blocked by N-ethylmaleimide (NEM, Thermo Fisher Scientific Inc., Waltham, MA) according to Martinez et al. (33). Analytical peptide maps consisted of loading 50 μg of the digest onto a Phenomenex Jupiter Proteo C12 column (Phenomenex, Torrance, CA), 2.0 × 250 mm, 4-μm particle size, 90-Å pore size column, heated to a temperature of 60 °C. The separation was performed by gradient elution on an Agilent HP 1200 HPLC system. The column was held at the initial condition of 0.5% solvent A (0.11% trifluoroacetic acid in water) at a flow rate of 0.2 ml/min for 5 min, and the digest was then eluted with a linear gradient to 60% solvent B (0.09% trifluoroacetic acid in acetonitrile) over 160 min. Peptides were identified by data-dependent MS2 and MS3 fragmentation using a Thermo LTQ mass spectrometer (Thermo Fisher Scientific Inc., Waltham, MA).
RESULTS
Discovery of a Partial Human IgG1 Antibody
We evaluated the heterogeneity of a recombinant human IgG1 mAb by measuring its apparent size using SEC. The SEC profile was characterized by a homogeneous major peak eluting between 32 and 33 min and two minor peaks eluting at 27 and 34.5 min, respectively (Fig. 1A). The minor peak with the retention time of 34.5 min represented a smaller size, partial molecule (Fig. 1A, P1). This minor heterogeneity could not be explained by variation in glycosylation, as deglycosylation of the IgG1 did not affect the SEC profile. The heterogeneous SEC profile of the IgG1 was consistent between lots and found in other IgG1 molecules (data not shown), suggesting that the formation of this partial form is inherent in the nature of the IgG1 mAb or the cell culture production process or both.
FIGURE 1.

SECs of the partial molecule (P1) and the H2O2-induced fragments C1 and C2. A, IgG1-untreated control sample included a partial molecule (P1) eluting after 34.5 min. B, time-dependent formation of the C1 and C2 fragments upon incubation in the presence of H2O2. The direction of the chromatographic changes with increasing incubation time is indicated by the arrows. The reaction was carried out at 2 °C in 10 mm glutamic acid buffer, pH 5.2. The fragmentation was initialized by the addition of H2O2 to a final concentration of 20 mm and monitored for 21 days. The H2O2 treatment was stopped at the noted time points by adding 200 units/ml catalase. AU, absorbance unit.
To further study this partial molecule (P1), SEC was used to separate and enrich P1 to a purity of greater than 95% according to the procedure described under “Experimental Procedures.” The purified P1 was reduced with 5 mm DTT, alkylated with 15 mm IAM, and then subjected to RP-HPLC-TOF/MS analysis. The results showed that P1 was a heavily oxidized partial molecule, which was lacking one of the Fab domains and contained a truncated Fc domain, in which one of the Fc domain heavy chains (Fc-HC) were characterized by a unique N-terminal ladder consisting of the hinge residues DKTHT (Table 1 and Fig. 2).
TABLE 1.
The average masses of the P1 and C1 fragments generated by DTT reduction and IAM alkylation, followed by RP-HPLC-TOF/MS analysis
The data for the C1 fragment were from the 2-week samples incubated with H2O2. The HC contains four methionine residues, three of which are located in the truncated Fc-HC. Most of the fragments contain oxidations indicated by “O” (+16, +32, and +48 Da), as indicated by the differences in mass compared with the theoretical mass. The two glycosylation chains are composed of biantennary glycans with a variable number of galactose residues (G). The most common isoforms are G0, G1, and G2. N-terminal sequencing indicated that the N terminus of the HC was completely cyclized to pyroglutamic acid (data not shown). Conversion of N-terminal heavy chain (HC) glutamine (Q) to pyroglutamic acid (pE) is shown. Some fragments contained adducts that were proposed to conform to carboxylic acid (–COOH, +45 Da) and α-ketoacyl groups (CH3COCO, +71 Da), which were consistent with the peptide mapping results. Therefore, the additional masses can be explained by the number of oxidation products plus one of these adducts (for example, 118 Da = 3 oxidations + 71-Da adduct, and 93 Da = 3 oxidations + 45-Da adduct). Some fragments (Thr230–Gly451) were identified in both peak 2 and peak 3 because of incomplete separation (Fig. 2).
| Peak | Residues (start to end) | Theoretical mass | Observed mass | Mass difference | Assignment |
|---|---|---|---|---|---|
| Da | Da | Da | |||
| IgG1 | |||||
| LC | |||||
| 1 | Glu1–Cys215 | 23,679.3 | 23,678.5 | −0.8 | |
| HC | |||||
| 2 | pE1–Gly451 | 51,373.9 (G0) | 51,373.9 (G0) | 0.0 | |
| P1 | |||||
| Fc | |||||
| 1 | Thr228–Gly451 | 26,997.6 (G0) | 27,029.3 (G0) | 31.7 | 2 O |
| Lys227–Gly451 | 27,125.5 (G0) | 27,156.6 (G0) | 31.1 | 2 O | |
| Asp226–Gly451 | 27,240.6 (G0) | 27,270.6 (G0) | 30.0 | 2 O | |
| Asp226–Gly451 | 27,240.6 (G0) | 27,315.9 (G0) | 75.3 | 45 Da adduct +2 O | |
| Asp226–Gly451 | 27,402.7 (G1) | 27,433.4 (G1) | 30.7 | 2 O | |
| 2 | Thr230–Gly451 | 26,759.4 (G0) | 26,774.5 (G0) | 15.1 | 1 O |
| Thr230–Gly451 | 26,921.6 (G1) | 26,937.1 (G1) | 15.5 | 1 O | |
| Thr228–Gly451 | 26,997.6 (G0) | 26,996.2 (G0) | −1.4 | ||
| Thr228–Gly451 | 27,159.8 (G1) | 27,158.8 (G1) | −1.1 | ||
| 3 | Thr230–Gly451 | 26,759.4 (G0) | 26,758.4 (G0) | −1.0 | |
| Thr230–Gly451 | 26,921.6 (G1) | 26,920.0 (G1) | −1.6 | ||
| Thr230–Gly451 | 26,921.6 (G1) | 26,993.7 (G1) | 72.1 | 71-Da adduct | |
| Thr230–Gly451 | 27,083.8 (G2) | 27,082.1 (G2) | −1.7 | ||
| Thr230–Gly451 | 26,759.4 (G0) | 26,774.9 (G0) | 15.5 | 1 O | |
| Thr230–Gly451 | 26,921.6 (G1) | 26,937.1 (G1) | 15.5 | 1 O | |
| LC | |||||
| 4 | Glu1–Cys215 | 23,679.3 | 23,710.0 | 30.7 | 2 O |
| 5 | Glu1–Cys215 | 23,679.3 | 23,678.4 | −0.9 | |
| HC | |||||
| pE1–Gly451 | 51,373.9 (G0) | 51,420.5 (G0) | 46.6 | 3 O | |
| pE1–Gly451 | 51,373.9 (G0) | 51,436.9 (G0) | 63.0 | 4 O | |
| pE1–Gly451 | 51,373.9 (G0) | 51,453.6 (G0) | 79.7 | 5 O | |
| C1 | |||||
| Fc | |||||
| 1 | Thr228–Gly451 | 26,997.6 (G0) | 27,045.2 (G0) | 47.6 | 3 O |
| Thr228–Gly451 | 27,159.8 (G1) | 27,206.2 (G0) | 46.4 | 3 O | |
| Lys227–Gly451 | 27,125.5 (G0) | 27,172.5 (G0) | 47.0 | 3 O | |
| Asp226–Gly451 | 27,240.6 (G0) | 27,288.1 (G0) | 47.5 | 3 O | |
| Asp226–Gly451 | 27,240.6 (G0) | 27,334.4 (G0) | 93.8 | 45-Da adduct + 3 O | |
| Asp226–Gly451 | 27,402.7 (G1) | 27,449.2 (G1) | 46.5 | 3 O | |
| 2 | Thr230–Gly451 | 26,759.4 (G0) | 26,806.6 (G0) | 47.2 | 3 O |
| Thr230–Gly451 | 26,921.6 (G1) | 26,968.4 (G1) | 46.8 | 3 O | |
| Thr228–Gly451 | 26,997.6 (G0) | 27,014.9 (G0) | 17.3 | 1 O | |
| Thr228–Gly451 | 27,159.8 (G1) | 27,177.3 (G1) | 17.2 | 1 O | |
| 3 | Thr230–Gly451 | 26,759.4 (G0) | 26,776.8 (G0) | 17.4 | 1 O |
| Thr230–Gly451 | 26,921.6 (G1) | 26,939.1 (G1) | 17.5 | 1 O | |
| Thr230–Gly451 | 26,759.4 (G0) | 26,877.9 (G0) | 118.6 | 71 Da adduct + 3 O | |
| Thr230–Gly451 | 26,921.6 (G1) | 27,040.0 (G1) | 118.4 | 71 Da adduct + 3 O | |
| LC | |||||
| 4 | Glu1–Cys215 | 23,679.3 | 23,709.8 | 30.5 | 2 O |
| 5 | Glu1–Cys215 | 23,679.3 | 23,677.4 | −1.8 | |
| HC | |||||
| 6 | pE1–Gly451 | 51,373.9 (G0) | 51,470.9 (G0) | 96.0 | 6 O |
| 7 | pE1–Gly451 | 51,373.9 (G0) | 51,436.9 (G0) | 63.0 | 4 O |
FIGURE 2.
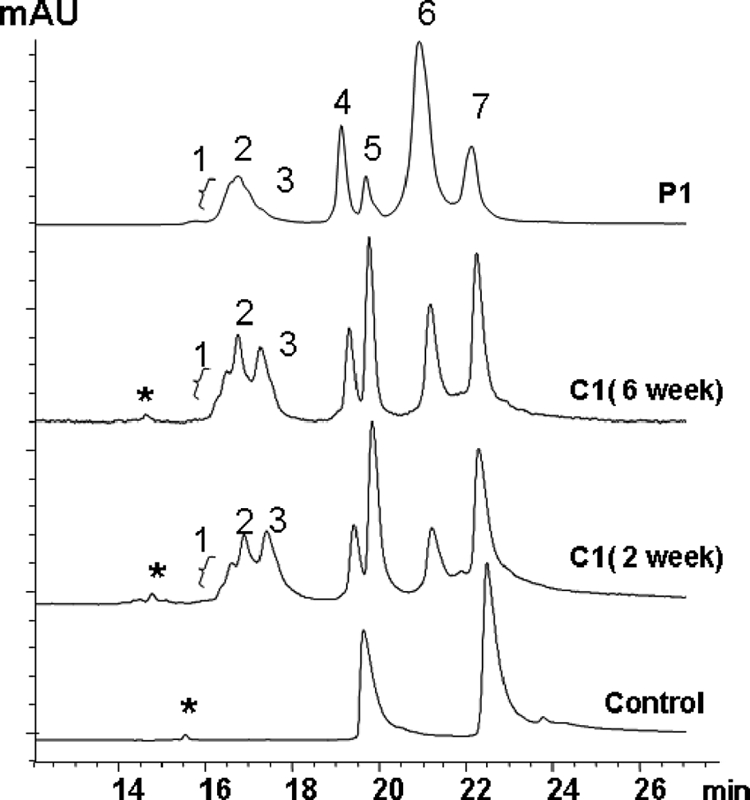
Comparative analysis of fragments P1 and C1 by RP-HPLC-TOF/MS. Purified P1 and C1 generated by incubation with H2O2 for 2 and 6 weeks were reduced and alkylated, analyzed by RP-HPLC, and characterized by in-line TOF/MS. Peaks 1–3 were found to be truncated HC of the Fc domain (Fc-HC), peaks 4 and 5 were identified as LC, and peaks 6 and 7 as intact HC. A minor peak shown in the untreated control sample and C1 profiles, labeled with an asterisk, was determined to be an oxidized Fc domain fragment generated by breaking the Asp-Pro (DP) peptide bond in HC of the Fc domain. The corresponding mass data are shown in Table 1. mAU, milliabsorbance unit.
H2O2-induced Fragmentation of the IgG
H2O2 is widely produced in healthy cells (16–19) and has been found to be involved in the oxidation and degradation of proteins (20–23); thus, the formation of the partial IgG1 molecule P1 and its heavily oxidized nature may suggest the involvement of H2O2 from the cell culture production. To test this hypothesis, we evaluated the effect of H2O2 on the stability of the IgG1 and employed SEC to measure any potential degradation products. SEC analysis of an IgG1 sample with a protein concentration of 10 mg/ml that was incubated at 25 °C for 8 h at pH 5.2 in the presence of less than 20 mm H2O2 did not detect any cleavage or other degradation products (data not shown). A H2O2 concentration of at least 20 mm, equal to a molar ratio of 1:300, was necessary to generate two smaller fragments (C1 and C2) with the retention times of 34.5 and 40 min, respectively (Fig. 1B). The fact that incubation for 21 days under the given conditions resulted in the time-dependent linear growth of only two distinct fragments (Fig. 1B), suggested that the cleavage reaction follows a specific mechanism.
It was intriguing to observe that C1 and P1, originating from different preparations, elute with the same retention times. To investigate whether this phenomenon is possibly due to similar structural properties in regard to the nature and location of the cleavage, the IgG1 was incubated at 25 °C with H2O2, pH 5.2, for 2 and 6 weeks, and the generated C1 and C2 fragments were purified by SEC. The purified C1 and P1 samples were reduced by 5 mm DTT in the presence of 4 m guanidine hydrochloride, alkylated by 15 mm IAM, and analyzed by RP-HPLC-TOF/MS. Both C1 and P1 showed similar profiles that were composed of intact HC (peaks 6 and 7), intact LC (peaks 4 and 5), and a single truncated HC of the Fc domain (Fc-HC, peaks 1–3), whereas the untreated control contained only intact LC and HC (Fig. 2 and Table 1).
The molar ratio of the three components, Fc-HC, LC, and HC, in P1 and C1 was estimated to be 1:1:1 according to the integrated UV peak area, indicating that P1 and C1 represent a partial antibody lacking one Fab domain. In addition, each Fc-HC showed a heterogeneous RP-HPLC profile with at least three components, a front peak (peak 1), middle peak (peak 2), and back peak (peak 3). TOF/MS analysis (Fig. 3) suggested that peaks 1–3 were derived from single Fc-HC fragments starting from different N-terminal upper hinge residues (Asp226, Lys227, Thr228, and Thr230) and ending at the C-terminal residue Gly451. These Fc-HC fragments composed the typical distribution of Fc glycan structures with various degrees of oxidation (Table 1). In addition, two adducts of +45 and +71 Da were observed on some Fc-HC fragments. These are not commonly observed adducts and are not consistent with known modifications.
FIGURE 3.

RP-HPLC-TOF/MS analysis of the Fc-HC fragments. The deconvoluted mass spectra of peaks 1–3 from P1 are presented in A–C. The deconvoluted mass spectra of peaks 1–3 from C1 are presented in D–F. Each major peak is labeled with its mass, and the corresponding fragments, including modifications proposed by the mass analysis, are summarized in Table 1.
Mass analysis showed that peaks 4 and 5 for both P1 and C1 represented two LC components. One was consistent with the theoretical mass of intact LC (peak 5) and the other with oxidized intact LC because of a mass increase of 32 Da. Peaks 6 and 7 represented two HC components that were both oxidized. Peak 6 was characterized by molecular masses corresponding to a G0 glycan moiety (5)-containing species, which were 48 and 96 Da heavier than the calculated mass, suggesting the presence of oxidation in the HC. The HC component represented by peak 7 included various degrees of oxidation (+16 Da (P1) and +64 Da (C1) (see Table 1 and supplemental material for more details).
Analysis of C1 generated by a 6-week incubation at 25 °C in the presence of 20 mm H2O2, pH 5.2, demonstrated that longer H2O2 treatment did not result in the formation of new cleavage products but in a higher degree of the specific cleavage products C1 and C2 (see below) and higher oxidation levels, ∼25–40% for LC and ∼30–50% for HC (Fig. 2, data not shown), resulting from the oxidative condition induced by H2O2. Collectively, these results demonstrated that H2O2 was capable of inducing both oxidation of amino acid side chains and backbone fragmentation specifically in the IgG1 hinge region, and these results led us conclude that H2O2-induced hinge fragmentation is the likely cause for the formation of the partial IgG1 molecule P1 during cell culture.
Analysis of the Fragmentation Product (C2) by RP-HPLC- TOF/MS
The inability of isolating the missing Fab domain fragment from the cell culture prevented further evaluation of its structural properties. However, the available C2 fragment derived from the SEC fractionation (Fig. 1B) allowed further investigation of the mechanism underlying H2O2-induced hinge fragmentation. To this end, RP-HPLC-TOF/MS analysis of reduced and alkylated C2 was conducted to determine the structural properties of this species (Fig. 4). The C2 was found to represent a heavily oxidized Fab domain fragment, composed of the LC components (Fig. 4A, peaks 1 and 2) and HC component, the Fab portion of the heavy chain of the fragment (Fig. 4A, peaks 3 and 4). Among the LC components, one was consistent with the mass of intact unmodified LC (peak 2) and the other with the mass of intact LC that is heavier by 32 Da (peak 1), suggesting oxidization (data not shown). The Fd components (peak 3 and 4) were found to be heavily oxidized as well, with either one (+16 Da, peak 4, Fig. 4C) or three (+48 Da, peak 3, Fig. 4B) additional oxygen atoms. Furthermore, both Fd components contained a ladder of the C-terminal upper hinge residues, Cys225, Asp226, Lys227, Thr228, His229, and Thr230 (see supplemental Table S1 for more details). Combining these results with the results from the C1 and P1 analyses, it seems that H2O2 treatment resulted in upper hinge cleavage, which generated two fragments (C1 and C2) with complementary ladders of the C-terminal HC residues in the Fab domain fragment (C2) and N-terminal HC residues in the Fc domain (Fig. 5, C1). Collectively, these results suggested that the partial IgG1 antibody component P1 could be a degradation product of H2O2-mediated hinge fragmentation generated by a specific cleavage mechanism.
FIGURE 4.

RP-HPLC-TOF/MS analysis of the C2 fragment. A, UV profile of the purified, reduced, and alkylated C2 fragments after incubation at 25 °C with H2O2 for 2 and 6 weeks. mAU, milliabsorbance unit. B, deconvoluted mass spectrum of peak 3 in A. C, deconvoluted mass spectrum of peak 4 in A. Each major peak was labeled with its mass and the corresponding single letter amino acid of the N-terminal residue.
FIGURE 5.

Schematic illustration of the major cleavage sites in the IgG1. H2O2-mediated cleavage of the hinge region, as indicated by the arrows, resulted in the generation of complementary ladders of C- and N-terminal hinge residues (Asp226, Lys227, Thr228, His229, and Thr230) and the release of the C1 and C2 fragments via a one radical per molecule cleavage mechanism (see text for details). Solid lines indicate disulfide bonds.
Hydroxyl Radical-induced Hinge Fragmentation
The similar profiles and the same cleavage sites between the P1 and C1 suggested that the partial form that was purified from the bioreactor CHO cell culture could be mimicked in vitro by incubation of the molecule with H2O2. It has been shown that H2O2 can regulate the biological function of proteins through a radical-induced oxidation pathway. Hydroxyl radicals (HO•) can be generated from H2O2 and are involved in various chemical reactions that result in the degradation of proteins (20–23). To examine whether hydroxyl radicals are involved in the observed hinge fragmentation, and to evaluate the factors that may influence cleavage, the IgG1 was subjected to incubation with H2O2 under various conditions that were designed to determine the root cause of the hinge cleavage in an IgG1. As shown in Fig. 6, catalase completely blocked cleavage, indicating that hydroxyl radicals were involved in the cleavage reaction. The total amount of free thiol (–SH) groups was determined to be ∼0.22 mol/mol antibody under denatured conditions in the presence of 4 m guanidine hydrochloride using Ellman's reagent 5,5′-dithiobis-(2-nitrobenzoic acid). Complete alkylation of the free thiol groups by incubation of NEM with the IgG1 for 3 h at 37 °C, pH 5.0, only decreased the cleavage by ∼7%, suggesting that unpaired Cys residues do not play a critical role in the cleavage reaction.
FIGURE 6.

Hydroxyl radicals mediate hinge fragmentation. The IgG1 was treated with H2O2 (20 mm) at 25 °C for 5 days in 10 mm glutamic acid, pH 5.2, in the presence of either catalase (200 units/ml), EDTA (20 mm), or with and without FeCl2 (10 μm) and analyzed by SEC. The integrated percent peak area of cleaved IgG1 as measured by SEC for each condition is reported on the y axis. For the NEM-blocking assay, the IgG1 was incubated with 40 mm NEM for 3 h at 37 °C and then buffer-exchanged into the glutamic acid buffer, prior to the addition of H2O2. Other conditions are the same as described in Fig. 1.
Preincubation with EDTA inhibited ∼90% of the H2O2-induced cleavage, suggesting an involvement of metal ions in the reaction. However, EDTA did not completely block the cleavage, as H2O2 was still capable of cleaving the IgG1, despite a much slower reaction rate (data not shown). Treatment with H2O2 in the presence of 1–10 μm of FeCl2 resulted in ∼5 times more cleavage than treatment with H2O2 alone (Fig. 6), whereas 10 μm FeCl2 in the absence of H2O2 produced little cleavage during a 5-day incubation. Similar results were obtained using 10 μm copper acetate (data not shown). These results suggested that hydroxyl radicals are responsible for the hinge fragmentation and that the reaction can be accelerated by the addition of transition metal ions. Because trace amounts of transition metals ions are always present in solvents or may be bound to proteins, their concentration could be high enough to function as catalysts for the generation of radicals to induce fragmentation. This hypothesis is consistent with previous observations that transition metal ions play an important role in site-selective radical attack either by binding to a protein (34–37) or staying free in solution (37, 38, 40). In both cases, the metal ions accelerated the reaction by catalyzing the generation of hydroxyl radicals through the Fenton chemistry reaction (41). Collectively, these facts suggested the possibility that the hinge fragmentation is due to hydroxyl radical attack.
Characterization of the Hinge Fragmentation Products
The oxidation of proteins by reactive oxygen species, as described in Refs. 24–27, results in both the modification of the target molecule by the incorporation of oxygen atoms into the side chains of the residues and its fragmentation via hydroxyl radical-induced backbone cleavage. The radical-induced cleavage of peptide bonds has been shown by these authors (24–27) to follow two routes, described as the diamide and α-amidation pathways that generate typical adducts at a new N terminus of the C-terminal fragment product. These adducts can be identified and characterized by peptide mapping. To this end, the C1 and C2 fragments were subjected to Lys-C peptide map analysis. As expected, the Lys-C peptide maps (Fig. 7A) confirmed the presence of oxidations that were responsible for the observed mass increases in the C1 and C2 fragments detected by RP-HPLC-TOF/MS analyses (Table 1 and refer to supplemental S2.1 for more details).
FIGURE 7.

LC-MS/MS of the Lys-C peptide maps for the control, C1, and C2. The IgG1 and its fragments were reduced, alkylated, and digested by Lys-C as described under “Experimental Procedures.” The digest was analyzed on a Phenomenex Proteo column (2.0 × 150 mm) and monitored at 214 nm. A, stacked view of Lys-C peptide maps of the control, C1, and C2. Major oxidized peptides, revealed by a shift in retention time and increase in mass in the C1 chromatogram, are labeled by arrows (for more detail see supplemental S2). B, expanded view of the hinge region of the control and C1 samples, which shows the hinge peptide (H12, residues 228–253) and its derivatives (H12a–f). Peak a, full-length H12 peptide (residues 228–253) with a sulfonic acid (SO3H) at Cys231; peak b, truncated H12 peptide (residues 230–253); peak c, truncated H12 peptide (residues 231–253) with SO3H at Cys231; peak d, truncated H12 peptide (residues 230–253) with a +71-Da adduct at Thr230; peak e, other hinge peptide derivatives of which only two peptides were identified as follows: full-length H12 peptide (residues 228–253) with His229 converted to Asp 229 and full-length H12 peptide (residues 228–253) with a +45-Da adduct at the N terminus; peak f, truncated H12 peptides (residues 232–253 and 233–253) with +16-Da adduct at Pro in the positions of 232 and 233. mAU, milliabsorbance unit.
The Lys-C peptide maps also revealed significant differences between the C1 fragment after H2O2 treatment and in the untreated control in the H12 Lys-C peptide, representing the sequence of the hinge region (Fig. 7B). In addition to the intact H12 hinge peptide (228THTCPPCPAPELLGGPSVFLFPPKPK253) that was found in both C1 and the untreated control, several variants of the hinge peptide (H12a–f) were only found in C1 (Fig. 7). Peptide sequencing by tandem mass spectrometry (MS2) showed that H12a–f were modified or truncated hinge peptides containing either oxidations or adducts located at or near the N terminus of the peptides (Fig. 8 and Table 2). Both H12a (residues 228–253) and H12c (a truncated form, residues 231–253), representing ∼12% of all the H12 peptides, contained a cysteine residue (Cys231) that was oxidized to sulfonic acid (–SO3H, Fig. 8A, and refer to supplemental Fig. S2.2.1 for more details). Interestingly, among the several derivatives of the hinge peptide that were coeluting into peak H12e, one contained an Asp residue instead of His229, most likely due to oxidative conversion of histidine to aspartic acid (Table 2 and refer to supplemental Fig. S2.2.2) (27). In addition, +45- and +71-Da adducts were found on the N-terminal residues of Thr228 (Fig. 8B) and Thr230 (Fig. 8C), respectively.
FIGURE 8.

MS2 analysis of the hinge peptide derivatives H12a, -d, and -e from Lys-C and Asp-N peptide maps. A, H12a, hinge peptide (2,837.3 Da) comprising sulfonic acid-oxidized Cys231 (228THTC231(–SO3H)PPCAPELLGGPSVFLFPPKPK). B, H12e-1, hinge peptide (2,893.8 Da) with a +45-Da adduct (–COOH) at the Thr228 residue of 228THTCPPCAPELLGGPSVFLFPPKPK. C, H12d, hinge peptide (2,679.9 Da) with a +71-Da adduct at the Thr230 residue of 230TCPPCAPELLGGPSVFLFPPKPK. D, peptide 225CDKTHTCPPCAPELLGGPSVFLFPPKPK (3,241 Da) from the Asp-N peptide map with Cys225–SO3H and ∼RC=O rather than ∼RNH2 at the N terminus. Some ions were always present in each figure, such as y5, y6, y8, y13, y14, y15, y17, and y19, and thus these peaks were only labeled in Fig. 6A and were not labeled in the following figures.
TABLE 2.
Hinge peptide derivatives determined by Lys-C peptide map
The estimation of abundance for each peptide was based on the individual integrated percent peak area relative to the total integrated peak area of all H12 peptide derivatives. A few H12 peptide derivatives were detected in lower quantities, coeluting with the H12e peptides. However, due to poor b-ion signals in the corresponding MS2 spectra, the identification of the N terminus of some peptides was not possible. Therefore, only two peptides could be identified and are listed in the table. The total integrated peak area of the H12e peptide was ∼8%. However, the quantities of the two identified peptides eluting in peak e could not be determined (ND). The abundance of the intact H12 peptide was estimated at ∼56%. Since the amino acid sequence (222PKSCDKTHTCPPC-AP) in the hinge region contain two Lys-C cleavage sites, some truncated hinge peptides with adducts at the N terminus may not be retained on the reversed-phase column because of their small size and were therefore not detectable.
| Peptide | Lys-C peak no. | Adduct location | % |
|---|---|---|---|
| 228THTCPPCAPELLGGPSVFLFPPKPK | a | SO3H at Cys231 | ∼12 |
| 230TCPPCAPELLGGPSVFLFPPKPK | b | ∼5 | |
| 231CPPCAPELLGGPSVFLFPPKPK | c | SO3H at Cys231 | ∼11 |
| 230TCPPCAPELLGGPSVFLFPPKPK | d | 71 Da at Thr230 | ∼8 |
| 228THTCPPCAPELLGGPSVFLFPPKPK | e | 45 Da at Thr228 | ND |
| 228THTCPPCAPELLGGPSVFLFPPKPK | e | Asp229 | ND |
| 232PPCAPELLGGPSVFLFPPKPK | f | 16 Da at Pro232 | Trace |
| 233PCAPELLGGPSVFLFPPKPK | f | 16 Da at Pro233 | Trace |
Based on previously published work (24–27), the +45-Da adduct may be explained by the formation of an isocyanate group (O=C=N–, +28 Da) at the N terminus of the C-terminal portion of one of the Fc heavy chains because of radical cleavage of the upper hinge region (DKTHT) through the diamide pathway. With the unstable nature of the isocyanate group, the reaction with hydroxyl yields the formation of N-terminal carboxylic acid (–COOH, +45 Da). The complementary N-terminal cleavage product would yield a carbonyl/aldehyde derivative at the C terminus of the Fab heavy chain (Fd). Hydrolysis of this unstable intermediate derivative would yield the formation of an amide at the new C terminus in the N-1 (for more details see under “Discussion”). The +71-Da adduct, on the other hand, could be explained by cleavage via the α-amidation pathway. This mode of fragmentation has been shown to lead to the formation of an N-terminal N-α-ketoacyl/N-pyruvyl group (+71 Da) at the C-terminal cleavage product of the peptide backbone (for more details see under “Discussion”). Following these events, according to the mechanisms described in Refs. 24–27, it is expected that hydrolysis of these unstable intermediates leads to the formation of truncated IgG1 HC fragments of which the C-terminal Fc-HC fragments contain a regular N terminus and the N-terminal Fd fragments contain at its C terminus, instead of a carboxyl group (–COOH), a C-terminal residue with an amide group (–CONH2, −1 Da).
Further data supporting the diamide pathway as the mechanism for cleavage were obtained by RP-HPLC analysis from two other peptides, 232PPCPAPELLGGPSVFLFPPKPK253 and 233PCPAPELLGGPSVFLFPPKPK253, with retention times of ∼95 min (as indicated by “f” in Fig. 8B). Both peptides in a trace amount were +16 Da heavier than their theoretical mass. The +16-Da modification was determined to be located at the N-terminal proline (Pro) residue by MS2 sequencing. Based on the N-terminal location of the +16-Da modification and in agreement with the diamide pathway, we propose that the +16-Da modification was the result of the hydrolysis of the isocyanate intermediate at the N terminus of the N-terminal Pro residue. Hydrolysis of the O=C=N group yielded breakage of the N–Cα bond of the Pro side chain and the additional mass of +16 Da at the N terminus (see supplemental Figs. S2.2.3 and S2.2.4). Observation of these two peptides provided crucial information regarding the mechanism for the radical cleavage (see under “Discussion”).
Because the amino acid sequence of the hinge region (222PKSCDKTHTCPPCAP) includes a number of Lys-C and Asp-N proteolytic cleavage sites, a second peptide map using Asp-N as an orthogonal method maximizes the detection of potential modifications of hinge region residues. The Asp-N map not only confirmed the observed modifications obtained by the Lys-C map (data not shown), but also detected new modifications on a peptide that was not obtainable using Lys-C. The peptide, 225CDKTHTCPPCPAPELLGGPSVFLFPPKPK253, was detected in C1 and contained a sulfonic acid group (–SO3H) in the side chain of Cys225 (Fig. 8D). In addition, the measured mass of 3,241.0 Da for this peptide was 2 Da less than the calculated theoretical value. The MS2 spectrum showed evidence for the presence of an N-terminal RC=O group rather than a RCH-NH2 group, suggesting a hydroxyl radical induced cleavage reaction via the α-amidation pathway, which resulted in the formation of an N-α-ketoacyl group at a new N terminus that was 2 Da lighter than a regular N terminus.
Knowing that Cys225 and Cys215 form the interchain disulfide bond between HC and LC (Fig. 5) and the fact that Cys225 was found to be oxidized to sulfonic acid suggested the presence of an unpaired and oxidized Cys215. This expectation was confirmed by the observation that ∼25% of Cys215 was oxidized to sulfonic acid in C2 and ∼10% in C1 (see supplemental Fig. S2.3 for more detail). This finding was supported by additional data from the analysis of a nonreduced peptide map, which detected ∼4% of unpaired Cys215 in the LC of the untreated control (see supplemental Figs. S23.1 and Fig. S3.1 for more details). It is known that the LC and HC stay strongly associated and maintain a functional Fab domain without the interchain disulfide bond. The association constant between the LC and HC was estimated to be ∼1010 m−1 (42). Therefore, even though oxidization of the unpaired Cys residues results in the formation of LC Cys215–SO3H and HC Cys225–SO3H, it would not disrupt the structural integrity of the Fab domain.
Oxidation of Cys Residues in the Hinge
Finding the side chain of Cys225 oxidized to sulfonic acid in the HC suggested that the oxidation was due to the reaction with H2O2. Given the fact that the Cys231 residues from each HC are expected to form an interchain disulfide bond and that the concentration of free thiol (–SH) groups was estimated at 0.22 mol/mol antibody by Ellman's assay, the hinge peptide was further examined by nonreducing Lys-C peptide mapping after labeling potentially free, unpaired cysteine residues with NEM (31). The result from the 5,5′-dithiobis-(2-nitrobenzoic acid) assay showed no detectable free –SH groups after NEM treatment. Meanwhile, the peptide mapping analysis did not identify any NEM-labeled Cys residues in the intact or truncated hinge peptides of both the control and C1 samples. Instead, some peptides with NEM-labeled Cys were detected at the C terminus of the LC (see supplemental Fig. S 3.1 for more details) and in the CH3 domain (data not shown). This result suggested the complete absence of unpaired cysteine residues in the hinge region. The observation of similar amounts of Cys231–SO3H in both the intact and truncated hinge peptide in C1 after incubation with H2O2 suggested that Cys231 is becoming oxidized by H2O2 after the disulfide bond is broken (reduced) because of attack by hydroxyl radicals.
Once the interchain disulfide bond between the two Cys231 residues is broken, the initial reaction step will be one of the two Cys231 residues that becomes oxidized to sulfenic acid (Cys231–SOH, Reaction 2), and the other would form a thiyl radical (Cys231–S•, Reaction 2) in agreement with previously described radical chemistry (43, 44). Sulfenic acid is unstable and capable of reacting with any accessible thiol group to form a disulfide bond or undergoing further oxidation to sulfinic acid (Cys231–SO2H) or sulfonic acid (Cys231–SO3H) (45, 46). Once oxidized to sulfonic acid, Cys231 has lost its ability to form an interchain disulfide bond with its counterpart on the other HC. Because of the presence of H2O2, the highly reactive thiyl radical oxidizes to sulfonic acid and simultaneously transfers its radical along the peptide backbone to the residues of the upper hinge region. In addition, the interchain disulfide bond between the Fab LC and HC (Fd) could be broken by radical attack as a post-cleavage event that would generate additional sulfonic acid at LC Cys215, which explains why more LC Cys215 was observed in C2 (∼25%) than in C1 (∼10%). It should be noted that the failure to detect Cys-SO3H in the reduced and alkylated HC and LC by RP-HPLC-TOF/MS was due to poor ionization efficiency (for details see supplemental S3).
Identification of Radical Formation and Location
The data above suggest Cys231 as the site of initial radical attack that subsequently leads to the observed cysteine oxidation and various hinge fragmentation products. In support of this suggestion, the spin trap 5,5-dimethyl-1-pyrroline N-oxide (DMPO), a reagent widely used to provide evidence for the involvement of HO• radicals in many biological reactions (36, 48–50), was utilized to track the formation of radicals and the specific location of the initial radical attack. With the spin trapping approach, radicals react with DMPO, thereby making a covalent bond with the DMPO adduct at the site of radical formation detectable by mass spectrometric analysis. The IgG1 was treated at 25 °C with H2O2, pH 5.2, in the presence of DMPO, and fragmentation was monitored by SEC. DMPO, in a molar ratio with H2O2 of 5:1, completely blocked fragmentation over a time course of 2 weeks (data not shown), suggesting that the binding of DMPO to the site(s) of radical residence completely inhibited hinge fragmentation.
To determine the site(s) of radical formation (DMPO binding), the IgG1 was subjected to Lys-C peptide mapping and RP-HPLC-MS2 analysis. The generated MS2 spectra provided clear evidence that only Cys231, located in the H12 hinge peptide, was found to contain the DMPO adduct (Fig. 9). This conclusion was supported by the observation of the (M + H)2+ ion of m/z 1,451.98, which corresponds in mass to the addition of DMPO (+113 Da) into the hinge peptide (2,789 Da, corresponding to the mass with one Cys modified by IAA). The mass differences between y22, y23, and y24 correspond to the mass of a DMPO-modified Cys231 residue, and the presence of the b-ions (b6 and b9) confirmed the assignment. The most intensive signal in the MS2 spectrum was a double charged y-ion (m/z 1202) that corresponded to y23 comprising Cys231 (Fig. 9) but lacking the side chain and the DMPO adduct because of a neutral loss of 160 Da (47 Da + 113 Da = 160 Da), which was confirmed by MS3 sequencing (data not shown). These results confirmed the hypothesis that the initial site of radical attack is located at Cys231 because trapping the radical at this site by DMPO completely inhibited the hinge fragmentation. Furthermore, besides the observed DMPO adduct formation at Cys231, Cys231 was also identified to be oxidized to sulfonic acid (SO3H; data not shown), confirming the radical chemistry (35, 36) that resulted in the reduction of the disulfide bond and led to the formation of the two different products at the two Cys residues.
FIGURE 9.

MS2 determination of the site of initial radical formation in the IgG1. The IgG1 was incubated at 25 °C in the presence of H2O2 (20 mm) and DMPO (100 mm) at pH 5.2. The sample was buffer-exchanged, digested with Lys-C, and analyzed by RP-HPLC-MS2. Hinge peptide H12 (2,789 Da, corresponding to the mass with one Cys modified by IAA) was found modified by the DMPO adduct (2,902 Da). The MS2 spectrum of the 1,451.98-Da ion (2+) showed the DMPO adduct to be located at Cys231. For clarity, not all identified fragment ions are labeled.
The observation of the initial radical formation at Cys231 is in agreement with previously published reaction rate constants (27). The reaction rate constant of a hydroxyl radical with Cys (3.4 × 1010 m−1 s−1) is much faster than with His (1.3 × 1010 m−1 s−1), Thr (5.1 × 108 m−1 s−1), Asp (7.5 × 107 m−1 s−1), and Lys (3.5 × 107 m−1 s−1) (27), and therefore, Cys231 is kinetically the primary target for hydroxyl radical attack. Although the cleavage of peptide bonds by the radical eventually occurs at an α-carbon position of the protein backbone, the initial radical formation in the side chain of Cys231 is not contradicting the occurring backbone cleavage in the upper hinge region because electron transfer between the side chains of residues and between a side chain and the backbone has been known to play an important role in the radical-driven reaction (25, 26). The formation of Cys-S•, the conversion of His to Asp, and the oxidative degradation of His in the hinge indicated an attack on the side chain by the radical and suggested an important role of the side chain in radical cleavage. It was determined that an electron has a comparable reaction rate constant with His (6.4 × 107 m−1 s−1), Thr (2.0 × 10 7 m−1 s−1), Lys (2.0 × 107 m−1 s−1), and Asp (1.8 × 107 m−1 s−1) (27), indicating that these upper hinge residues are capable of localizing an electron, leading to backbone cleavage in subsequent reaction steps (for more detail see under “Discussion”). Thus, the electron localization at an upper hinge residue would initiate one radical cleavage per molecule that generates the complementary ladders of the C- and N-terminal heavy chain residues of the Fab and Fc domains, respectively.
DISCUSSION
The data presented here provide compelling evidence for a radical-induced hinge-specific cleavage mechanism observed for an IgG1 mAb. The fragmentation profile observed in the untreated control samples purified from cell culture could be mimicked in vitro by incubation of the antibody with H2O2. The reaction seems to be initiated by the formation of a hydroxyl radical via a Fenton-like reaction of a transition metal ion with H2O2. In the next step of the reaction, the hydroxyl radical specifically attacks the hinge region by breaking the interchain disulfide bond yielding radical formation in one of the Cys231 side chains (thiyl radical) and oxidation to sulfonic acid in the other. Subsequent radical transfer and localization at the hinge region residues lead to backbone cleavage producing one free Fab domain fragment with a ladder of the C-terminal heavy chain residues complementary to the N-terminal ladder of one of the heavy chains of the Fc domain in the truncated IgG1 mAb. To the best of our knowledge, this is the first report demonstrating specific backbone fragmentation in the hinge of an IgG1 mAb as a result of H2O2-mediated radical cleavage. Published three-dimensional structures of the full-length human b12 IgG1 antibody provided the evidence that the two separate IgG1 hinges may assume different conformations to accommodate the vastly different placements of the two Fab domains relative to the Fc domain (51, 52). It remains unclear in the literature whether tension in the two hinges caused by their orientation may facilitate the fragmentation (53), and which of the two Fab domains was cleaved off. However, our results indicate that hydroxyl radicals generated by a Fenton-like reaction mechanism seem to be responsible for the initiation of the IgG1 hinge fragmentation. We found that the presence of transition metal ions accelerated the backbone cleavage of the IgG1 by catalyzing the generation of hydroxyl radicals. The initial site of radical attack was at the interchain disulfide bond connecting the two heavy chains at Cys231 and yielded a thiyl radical at one of the Cys231 side chains, which initiated an electron/radical transfer to the hinge residues and yielded protein backbone fragmentation. This mechanism is consistent with a number of studies that have shown that preferential cleavage can be induced selectively by radicals at specific sites within a protein (32, 54, 55).
It has been recently demonstrated that antibodies have the ability to catalyze the generation of H2O2 in a highly efficient manner and by a mechanism that involves the oxidation of H2O by singlet oxygen (1O2) molecules (9, 10). Some polyoxide radicals (HO2• and HO3•), which bear the chemical signature of hydroxyl radicals (HO•), were observed (9–12). These additional sources of HO•, different from the Fenton reaction, may explain why EDTA failed to completely inhibit H2O2-mediated radical cleavage of the IgG1. Therefore, the radical reaction mechanism described here is different from the β-elimination mechanism (56) or the hypothetical direct hydrolysis of the peptide bond in the upper hinge region (53, 57) proposed in other studies, where similar hinge fragmentation of other IgG1 molecules was observed.
Proposed Backbone Cleavage Mechanism
We are proposing a mechanism for H2O2-mediated radical-induced hinge fragmentation of an IgG1 that includes three major reaction steps. In the first reaction step, a hydroxyl radical is mainly generated by the Fenton-like reaction in which the transition metal ions such as Fe3+/Fe2+ and Cu2+/Cu+ react with H2O2 (Reaction 1).
In the second step, the hydroxyl radical attack breaks the hinge region interchain disulfide bond between the Cys231 residues (Cys231–S—S-Cys231, shown as RS-SR in Reaction 2). The fact that in our experiments DMPO completely blocked the hinge fragmentation and was found to only react with Cys231 demonstrated that the interchain disulfide bond between Cys231 residues is broken by the attack of a hydroxyl radical, which yields the formation of a thiyl radical (Cys231–S•, see Reaction 2) on one cysteine and sulfenic acid (Cys231–SOH, see Reaction 2) on the other cysteine residue. Due to the presence of H2O2, sulfenic acid is further oxidized to sulfinic acid (Cys231–SO2H) and sulfonic acid (Cys231–SO3H) as the principal products (see Reaction 2).
Following the breakage of the disulfide bond, two events may take place in parallel. The first one is the radical-induced cleavage of the hinge, and the second one is the reformation of the Cys231–S—S-Cys231 disulfide bond. In the first event, the radical on the thiyl sulfur of Cys231 (Cys231–S•) transfers upstream to become localized at a hinge residue to initiate backbone fragmentation in the hinge region (Reaction 2). In the second event, reformation of the disulfide bond between Cys231–S• on one heavy chain and Cys231–SOH on the other heavy chain is possible, because the thiyl radical would convert back to a regular thiol group (Cys231–SH) after electron transfer. The disulfide bond reformation in the hinge region is chemically preferred, and it leads to the reformation of the disulfide bond between one truncated Fc heavy chain and one intact full-length heavy chain of the IgG1. However, if Cys231–SOH has been oxidized to SO3H by H2O2 (Reaction 2), Cys231–SH located in the truncated hinge region of Fc-HC would be eventually oxidized to Cys231–SO3H as well.
In the third reaction step, protein backbone fragmentation can be initiated via two major pathways, the diamide and α-amidation pathways. In the diamide pathway (Reaction 3), the cleavage at the α-carbon of the peptide bond yields an isocyanate derivative (III) at the heavy chain N terminus of the C-terminal (Fc-HC) and a ketone carbonyl function (II) at the heavy chain C terminus of the N-terminal backbone fragment (Fd). The α-amidation pathway (Reaction 4) is characterized by the formation of an N-terminal α-ketoacyl derivative (V) at the C-terminal backbone fragment (Fc-HC) and a diamide function (IV) at the C terminus of the N-terminal backbone fragment (Fd).
Hydrolysis of the isocyanate derivative could yield a carboxylic acid function (–COOH), which would explain the observed +45-Da adduct at the N terminus of the C-terminal portion of the heavy chain backbone fragment (Fc-HC). The carbonyl derivatives at the C terminus of the N-terminal heavy chain fragment (Fd) is hydrolytically labile as well, and mild differential hydrolysis will give rise to an amide function at N-1 of the peptide (Reaction 5) (23).
On the other hand, the electron attack at the γ-carbon position in the side chain of certain amino acids in the presence of oxygen could result in the oxidative degradation that leads to the formation of an unsaturated product of dehydropeptides, which only retains a β-CH2 group as a side chain (Reaction 6) (24, 26). This compound can be easily hydrolyzed to yield amide and keto acid functions (Reaction 7). Thus, the +71-Da adduct at the N terminus of the Thr230 residue could result from the oxidative degradation of His229. Further hydrolysis of these intermediate derivatives results in the formation of a regular N-terminal residue (24–27).
It should be pointed out that if an electron is transferred to a Pro residue, the lack of a typical side chain structure prevents the Pro from following the reaction pathways like other amino acid residues. Although fragmentation at Pro residues via the diamide pathway has been described (24), there is no evidence for a cleavage mechanism at Pro residues that follow the α-amidation pathway. It is reasonable to believe that the cleavage at the α-carbon of the peptide bond (N–Cα) via the α-amidation pathway would not generate two separated fragments of the IgG1 because the ring side chain of Pro would still be connecting the two fragments. This may explain why most of the observed backbone cleavages occurred at residues in the upper hinge region (DKTHT) and only very little in the core region of the hinge (CPPC). Reactions 1–7 are as follows.
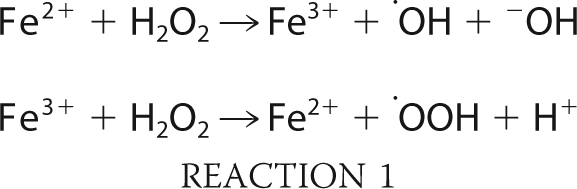 |

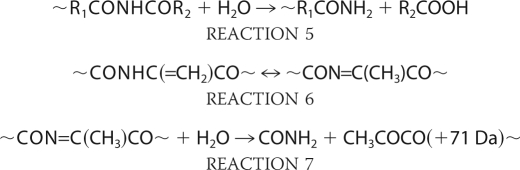 |
The observation that partial IgG1 molecules are present in an IgG1 isolated from a CHO cell culture does provide evidence that partial molecules can be generated in vivo. Many different physiological and environmental processes can lead to the formation of reactive oxygen species such as H2O2 (25), and it is well established that mitochondria are the main source of reactive oxygen species generation in cells (39, 47, 58, 59). The rate of H2O2 production in isolated mitochondria is 0.6–1.0 nmol/mg−1 min−1 (47), and the H2O2 concentration in liver cells has been estimated to be 10−7–10−9 mol/liter (58). Given these facts and the intrinsic capacity of an IgG to generate H2O2 (8–13), the results presented here offer new insights into a possible degradation pathway for IgG1 antibodies through the specific degradation of the hinge region. The highly conserved core sequence of the hinge CPXPC (X = Pro for IgG1 and IgG2, X = Arg for IgG3, and X = Ser for IgG4) may imply that other classes of antibodies also could undergo similar radical-induced degradation reactions, particularly for the IgG3 subtype that has an upper hinge sequence similar to IgG1; whether or not such radical reaction or cleavage products or both would apply to IgG2 and IgG4 remains to be determined. Further investigation of the factors that influence the radical-induced hinge cleavage will provide more insights into mechanisms to prevent it. Based on such knowledge, designing a new generation of IgG1 antibodies with a new upper hinge sequence that is more resistant to radical attack could not only improve the yield of production but also potentially enhance the stability and efficacy profile in vivo.
Acknowledgments
We are grateful to Dr. Michael Treuheit for the critical comments on the manuscript; Drs. Bruce Kerwin and David Hambly for valuable discussions; Drs. Randall Ketchem and Guna Kannan for their assistance in IgG1 structure analysis; and Danielle Pace and Theresa Martinez for critical reading of the manuscript.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S2.2.1–S2.2.4, S2.3.1–S2.3.2, S3.1, S3.2.1, and S3.2.2 and Tables S1 and S3.3.
- mAb
- monoclonal antibody
- CHO
- Chinese hamster ovary
- DMPO
- 5,5-dimethyl-1-pyrroline N-oxide
- SEC
- size-exclusion chromatography
- RP-HPLC
- reversed-phase high performance liquid chromatography
- TOF
- time-of-flight
- MS
- mass spectrometry
- LC
- light chain
- HC
- heavy chain
- DTT
- dithiothreitol
- IAM
- iodoacetamide
- NEM
- N-ethylmaleimide.
REFERENCES
- 1.Carter P. J. (2006) Nat. Rev. Immunol. 6, 343–357 [DOI] [PubMed] [Google Scholar]
- 2.Reichert J. M., Dewitz M. C. (2006) Nat. Rev. Drug Discov. 5, 191–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schrama D., Reisfeld R. A., Becker J. C. (2006) Nat. Rev. Drug Discov. 5, 147–159 [DOI] [PubMed] [Google Scholar]
- 4.Dube D. H., Bertozzi C. R. (2005) Nat. Rev. Drug Discov. 4, 477–488 [DOI] [PubMed] [Google Scholar]
- 5.Jefferis R. (2009) Nat. Rev. Drug Discov. 8, 226–234 [DOI] [PubMed] [Google Scholar]
- 6.Walsh G., Jefferis R. (2006) Nat. Biotechnol. 24, 1241–1252 [DOI] [PubMed] [Google Scholar]
- 7.Frokjaer S., Otzen D. E. (2005) Nat. Rev. Drug Discov. 4, 298–306 [DOI] [PubMed] [Google Scholar]
- 8.Wentworth A. D., Jones L. H., Wentworth P., Jr., Janda K. D., Lerner R. A. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 10930–10935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wentworth P., Jr., Wentworth A. D., Zhu X., Wilson I. A., Janda K. D., Eschenmoser A., Lerner R. A. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 1490–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Datta D., Vaidehi N., Xu X., Goddard W. A., 3rd (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 2636–2641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu X., Muller R. P., Goddard W. A., 3rd (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 3376–3381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu X., Wentworth P., Jr., Wentworth A. D., Eschenmoser A., Lerner R. A., Wilson I. A. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 2247–2252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang P. X., Sanders P. W. (2007) J. Am. Soc. Nephrol. 18, 1239–1245 [DOI] [PubMed] [Google Scholar]
- 14.Rhee S. G., Chang T. S., Bae Y. S., Lee S. R., Kang S. W. (2003) J. Am. Soc. Nephrol. 14, S211–S215 [DOI] [PubMed] [Google Scholar]
- 15.Poole L. B., Karplus P. A., Claiborne A. (2004) Annu. Rev. Pharmacol. Toxicol. 44, 325–347 [DOI] [PubMed] [Google Scholar]
- 16.Rhee S. G., Bae Y. S., Lee S. R., Kwon J. (2000) Sci. STKE 53, PE1. [DOI] [PubMed] [Google Scholar]
- 17.Nathan C. (2003) J. Clin. Invest. 111, 769–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reth M. (2002) Nat. Immunol. 3, 1129–1134 [DOI] [PubMed] [Google Scholar]
- 19.DeYulia G. J., Jr., Cárcamo J. M., Bórquez-Ojeda O., Shelton C. C., Golde D. W. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 5044–5049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stamler J. S., Hausladen A. (1998) Nat. Struct. Biol. 5, 247–259 [DOI] [PubMed] [Google Scholar]
- 21.Salmeen A., Andersen J. N., Myers M. P., Meng T. C., Hinks J. A., Tonks N. K., Barford D. (2003) Nature 423, 769–773 [DOI] [PubMed] [Google Scholar]
- 22.Claiborne A., Mallett T. C., Yeh J. I., Luba J., Parsonage D. (2001) Adv. Protein Chem. 58, 215–276 [DOI] [PubMed] [Google Scholar]
- 23.Paget M. S., Buttner M. J. (2003) Annu. Rev. Genet. 37, 91–121 [DOI] [PubMed] [Google Scholar]
- 24.Garrison W. M. (1987) Chem. Rev. 87, 381–398 [Google Scholar]
- 25.Stadtman E. R., Levine R. L. (2003) Amino Acids 25, 207–218 [DOI] [PubMed] [Google Scholar]
- 26.Berlett B. S., Stadtman E. R. (1997) J. Biol. Chem. 272, 20313–20316 [DOI] [PubMed] [Google Scholar]
- 27.Davies M. J., Dean R. T. (1997) Radical Mediated Protein Oxidation, pp. 50–120, Oxford University Press, Oxford [Google Scholar]
- 28.Shen H. M., Pervaiz S. (2006) FASEB J. 20, 1589–1598 [DOI] [PubMed] [Google Scholar]
- 29.Dröge W. (2002) Physiol. Rev. 82, 47–95 [DOI] [PubMed] [Google Scholar]
- 30.Martindale J. L., Holbrook N. J. (2002) J. Cell. Physiol. 192, 1–15 [DOI] [PubMed] [Google Scholar]
- 31.Shukla A. A., Hubbard B., Tressel T., Guhan S., Low D. (2007) J. Chromatogr. B 848, 28–39 [DOI] [PubMed] [Google Scholar]
- 32.Yan B., Valliere-Douglass J., Brady L., Steen S., Han M., Pace D., Elliott S., Yates Z., Han Y., Balland A., Wang W., Pettit D. (2007) J. Chromatogr. A 1164, 153–161 [DOI] [PubMed] [Google Scholar]
- 33.Martinez T., Guo A., Allen M. J., Han M., Pace D., Jones J., Gillespie R., Ketchem R. R., Zhang Y., Balland A. (2008) Biochemistry 47, 7496–7508 [DOI] [PubMed] [Google Scholar]
- 34.Dean R. T., Wolff S. P., McElligott M. A. (1989) Free Radic. Res. Commun. 7, 97–103 [DOI] [PubMed] [Google Scholar]
- 35.Zhao Y., Wang Z. B., Xu J. X. (2003) J. Biol. Chem. 278, 2356–2360 [DOI] [PubMed] [Google Scholar]
- 36.van der Wijk T., Overvoorde J., den Hertog J. (2004) J. Biol. Chem. 279, 44355–44361 [DOI] [PubMed] [Google Scholar]
- 37.Davies K. J., Delsignore M. E. (1987) J. Biol. Chem. 262, 9908–9913 [PubMed] [Google Scholar]
- 38.Stadtman E. R. (1993) Annu. Rev. Biochem. 62, 797–821 [DOI] [PubMed] [Google Scholar]
- 39.Turrens J. F., Alexandre A., Lehninger A. L. (1985) Arch. Biochem. Biophys. 237, 408–414 [DOI] [PubMed] [Google Scholar]
- 40.Berlett B. S., Chock P. B., Yim M. B., Stadtman E. R. (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 389–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stadtman E. R., Berlett B. S. (1991) J. Biol. Chem. 266, 17201–17211 [PubMed] [Google Scholar]
- 42.Horne C., Klein M., Polodoulis I., Dorrkingtin K. J. (1982) J. Immunol. 129, 660–664 [PubMed] [Google Scholar]
- 43.Purdie J. W. (1967) J. Am. Chem. Soc. 89, 226–238 [DOI] [PubMed] [Google Scholar]
- 44.Poole L. B., Claiborne A. (1989) J. Biol. Chem. 264, 12330–12338 [PubMed] [Google Scholar]
- 45.Kice J. L. (1980) Adv. Phys. Org. Chem. 17, 65–70 [Google Scholar]
- 46.Claiborne A., Yeh J. I., Mallett T. C., Luba J., Crane E. J., 3rd, Charrier V., Parsonage D. (1999) Biochemistry 38, 15407–15416 [DOI] [PubMed] [Google Scholar]
- 47.Chance B., Sies H., Boveris A. (1979) Physiol. Rev. 59, 527–605 [DOI] [PubMed] [Google Scholar]
- 48.Witting P. K., Mauk A. G. (2001) J. Biol. Chem. 276, 16540–16547 [DOI] [PubMed] [Google Scholar]
- 49.Deterding L. J., Ramirez D. C., Dubin J. R., Mason R. P., Tomer K. B. (2004) J. Biol. Chem. 279, 11600–11607 [DOI] [PubMed] [Google Scholar]
- 50.Witting P. K., Douglas D. J., Mauk A. G. (2000) J. Biol. Chem. 275, 20391–20398 [DOI] [PubMed] [Google Scholar]
- 51.Stanfield R. L., Fieser T. M., Lerner R. A., Wilson I. A. (1990) Science 248, 712–719 [DOI] [PubMed] [Google Scholar]
- 52.Saphire E. O., Stanfield R. L., Crispin M. D., Parren P. W., Rudd P. M., Dwek R. A., Burton D. R., Wilson I. A. (2002) J. Mol. Biol. 319, 9–18 [DOI] [PubMed] [Google Scholar]
- 53.Cordoba A. J., Shyong B. J., Breen D., Harris R. J. (2005) J. Chromatogr. B 818, 115–121 [DOI] [PubMed] [Google Scholar]
- 54.Kato Y., Uchida K., Kawakishi S. (1992) J. Biol. Chem. 267, 23646–23651 [PubMed] [Google Scholar]
- 55.Wolff S. P., Dean R. T. (1987) Biochem. J. 245, 243–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cohen S. L., Price C., Vlasak J. (2007) J. Am. Chem. Soc. 129, 6976–6977 [DOI] [PubMed] [Google Scholar]
- 57.Gaza-Bulseco G., Liu H. (2008) Pharmacol. Res. 25, 1881–1890 [DOI] [PubMed] [Google Scholar]
- 58.Boveris A., Chance B. (1973) Biochem. J. 134, 707–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Turrens J. F., Boveris A. (1980) Biochem. J. 191, 421–427 [DOI] [PMC free article] [PubMed] [Google Scholar]