

Abstract
The enzyme CMP-Kdo synthetase (KdsB) catalyzes the addition of 2-keto-3-deoxymanno-octulonic acid (Kdo) to CTP to form CMP-Kdo, a key reaction in the biosynthesis of lipopolysaccharide. The reaction catalyzed by KdsB and the related CMP-acylneuraminate synthase is unique among the sugar-activating enzymes in that the respective sugars are directly coupled to a cytosine monophosphate. Using inhibition studies, in combination with isothermal calorimetry, we show the substrate analogue 2β-deoxy-Kdo to be a potent competitive inhibitor. The ligand-free Escherichia coli KdsB and ternary complex KdsB-CTP-2β-deoxy-Kdo crystal structures reveal that Kdo binding leads to active site closure and repositioning of the CTP phosphates and associated Mg2+ ion (Mg-B). Both ligands occupy conformations compatible with an Sn2-type attack on the α-phosphate by the Kdo 2-hydroxyl group. Based on strong similarity with DNA/RNA polymerases, both in terms of overall chemistry catalyzed as well as active site configuration, we postulate a second Mg2+ ion (Mg-A) is bound by the catalytically competent KdsB-CTP-Kdo ternary complex. Modeling of this complex reveals the Mg-A coordinated to the conserved Asp100 and Asp235 in addition to the CTP α-phosphate and both the Kdo carboxylic and 2-hydroxyl groups. EPR measurements on the Mn2+-substituted ternary complex support this model. We propose the KdsB/CNS sugar-activating enzymes catalyze the formation of activated sugars, such as the abundant CMP-5-N-acetylneuraminic acid, by recruitment of two Mg2+ to the active site. Although each metal ion assists in correct positioning of the substrates and activation of the α-phosphate, Mg-A is responsible for activation of the sugar-hydroxyl group.
Introduction
Lipopolysaccharide (LPS)4 is a major component of the outer leaflet of the outer membrane in Gram-negative bacteria and is essential for outer membrane integrity and cell viability (1–3). LPS has a well defined tripartite structure consisting of a hydrophobic moiety, lipid A, linked to an inner and outer core oligosaccharide that may itself be linked to a high molecular weight polysaccharide molecule or O antigen (1, 3). In the vast majority of Gram-negative bacteria, the minimum LPS structure for viability is lipid A that has been substituted with two residues of 2-keto-3-deoxymanno-octulonic acid (Kdo), referred to as Kdo2-lipid A (1, 3). The substitution of lipid A with Kdo requires that Kdo is first activated by addition to CTP to form CMP-Kdo (Fig. 1). The formation of CMP-Kdo is catalyzed by the enzyme Kdo cytidyltransferase (CMP-Kdo synthetase), which is known as KdsB in Escherichia coli (4–6). The enzyme is similar to CNS, which catalyzes the penultimate step in the addition of sialic acids to oligosaccharides in eukaryotes (7–9).
FIGURE 1.

Reaction catalyzed by KdsB. The reaction involves the addition of Kdo to CTP to form CMP-Kdo and pyrophosphate.
Kdo and N-acetylneuraminic acid are unique in as far as their activated forms are nucleoside monophosphate diesters as opposed to the more common nucleoside diphosphate diesters that are the usual product of sugar-activating enzymes. Furthermore, KdsB and CNS are unique in the sense that they directly couple the sugar to CTP unlike other sugar-activating enzymes that require a phosphorylated sugar substrate. Both CMP-Kdo synthetase and bacterial CMP-NeuAc synthetase have been postulated to be viable drug targets (10–12). The importance of CMP-Kdo synthetase as a target has been recognized by a number of groups and has resulted in the successful design of a series of potent in vitro inhibitors of the enzyme based on the structure of 2β-deoxy-Kdo (Fig. 2) (10). However, none of the 2β-deoxy-Kdo derivatives were capable of crossing the inner membrane and reaching the KdsB enzyme in the cytoplasm and consequently showed no in vivo activity (11, 12). The design of new, improved inhibitors of KdsB that are capable of crossing the cytoplasmic membrane has been limited by the lack of detailed kinetic and structural information on the enzyme. Although structural information is available for both the capsule-specific and lipopolysaccharide-specific isozymes of the E. coli CMP-Kdo synthetase (13, 14; and PDB code 1VH1) and various CMP-NeuAc synthetases (15, 16), no crystal structures are available for the catalytically relevant ternary complex formed with CTP and the respective sugars. Therefore, in this study we have developed a new continuous assay for the reaction, which has facilitated a complete kinetic characterization of the enzyme and subsequent inhibition studies with 2β-deoxy-Kdo. Furthermore, we have obtained a 1.9 Å crystal structure of a ternary complex of the LPS-specific isozyme of the E. coli enzyme with the 2β-deoxy-Kdo inhibitor and CTP bound in the active site in addition to a novel 2.5 Å ligand-free structure. This has allowed us to map the essential interactions made in the catalytically relevant ternary CTP-Kdo-KdsB complex and will be crucial for the design of more potent antimicrobial agents in the future.
FIGURE 2.

Synthesis of 2β-deoxy-Kdo. Kdo 3 was synthesized from d-arabinose 1 and oxaloacetic acid 2 and converted into 2β-deoxy-Kdo 7 as described under “Experimental Procedures.”
EXPERIMENTAL PROCEDURES
Materials
All chemicals were obtained from Sigma unless otherwise stated.
Preparation of KdsB
The gene encoding KdsB from E. coli was cloned into the pET15b expression vector (Novagen). His-tagged protein was produced by overexpression in BL21(DE3) cells (Novagen) at 37 °C for 4 h. Cells were resuspended in binding buffer (50 mm Tris, pH 7.5, 500 mm NaCl, 5 mm imidazole) and lysed by sonication (four times for 30 s). The enzyme was purified to >95% purity by using a single step purification procedure on a nickel-Sepharose column (Prochem). Proteins were loaded onto the column in binding buffer, washed with binding buffer containing 50 mm imidazole, and eluted with 250 mm imidazole. After elution, the imidazole was removed by dialysis into 10 mm Tris/HCl, pH 7.5, and the protein was concentrated using stirred concentrator cells (Amicon). Protein concentrations were determined using the Bio-Rad DC protein assay with bovine serum albumin as standard. Typical protein yields were ∼60 mg from a 1-liter culture.
Synthesis of 2β-Deoxy-Kdo
The synthesis of 2β-deoxy-Kdo is outlined schematically in Fig. 2. First, the Cornforth synthesis of Kdo 3 from d-arabinose 1 and oxaloacetic acid 2 (17, 18) was repeated. During this procedure, it was necessary to control the reaction pH very closely, and the addition of NiCl2 (19) proved beneficial. The crude Kdo 3 product was progressed by acetylation and esterification to the mixture of fully protected α/β anomers 4, which was then converted into 2β-deoxy-Kdo 7 using a slight modification of a published procedure (20). In brief, the anomeric esters were converted to the anomeric chlorides 5 using TiCl4, and the halogen was removed by hydrogenolysis in the presence of pyridine. It was easiest to separate the α/β anomers at this stage; the desired fully protected 2β-deoxy compound 6 was obtained crystalline and in high purity. Deacetylation of 6 under Zemplen conditions followed by hydrolysis of the methyl ester afforded inhibitor 2β-deoxy-Kdo 7, which was isolated by ion exchange as its crystalline ammonium salt; this form was used in all subsequent experiments (see supplemental material for characterization of the final product).
Steady-state Enzyme Assays
The concentration of CTP was determined using an extinction coefficient in aqueous solution of 9 mm−1 cm−1 at 271 nm. The activity of KdsB was measured by following the formation of pyrophosphate as it is released during the reaction using the EnzChek pyrophosphate assay kit (Invitrogen). In these assays the pyrophosphate is first split into two molecules of phosphate by inorganic pyrophosphatase, which is then utilized by the enzyme purine ribonucleoside phosphorylase in a reaction that has an accompanying absorbance change at 360 nm. The amount of pyrophosphate produced during the assay was quantified by comparison with a pyrophosphate standard curve. A Cary 50 UV-visible spectrophotometer (Varian) was used to measure the initial rates of pyrophosphate production of 10 nm KdsB in 10 mm Tris/HCl, pH 7.5, 5 mm MgCl2 over a range of substrate concentrations. The Km and Vmax values were obtained by fitting the initial rates of pyrophosphate synthesis against the concentrations of each substrate to Equation 1 by nonlinear regression analysis using the SigmaPlot program (SPSS Inc),
where v is the initial rate; [CTP] and [Kdo] are the substrate concentrations; Vmax is the initial rate achieved as both [CTP] and [Kdo] approach ∞; KKdo is the value of [Kdo] giving Vmax/2 as [CTP] approaches ∞; KCTP is the value of [CTP] giving Vmax/2 as [Kdo] approaches ∞; and c is a constant.
For inhibition assays with 2β-deoxy-Kdo, the initial rates of pyrophosphate production of 10 nm KdsB in 10 mm Tris/HCl, pH 7.5, 5 mm MgCl2 were measured over a range of Kdo concentrations and at variable concentrations of 2β-deoxy-Kdo. Data for the inhibition studies were fitted to Equation 2 for competitive inhibition, and standard errors were calculated by nonlinear regression analysis using the SigmaPlot program (SPSS Inc),
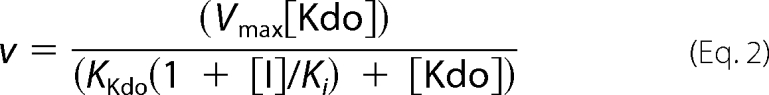 |
where v is the initial rate;[Kdo] is the concentration of Kdo; Vmax is the initial rate achieved as [Kdo] approaches ∞; KKdo is the value of [Kdo] giving Vmax/2; [I] is the concentration of the 2β-deoxy-Kdo inhibitor; and Ki is the inhibition constant. All of the steady-state rates were measured in duplicate.
Isothermal Calorimetry (ITC)
ITC experiments were performed using a VP-ITC microcalorimeter (Microcal Inc.) at 25 °C. Protein samples were dialyzed into 10 mm Tris/HCl, pH 7.5. Aliquots (10 μl) of 100 μm 2β-deoxy-Kdo were titrated into 1.28 ml of 100 μm KdsB and 100 μm CTP at 420-s intervals with a stirring speed of 350 rpm. Parallel experiments were performed by injecting the inhibitor into the buffer. The heats of dilution were negligible and subtracted from their respective titrations prior to data analysis. Thermodynamic parameters n (stoichiometry), Kd (1/Ka, the association constant), and ΔH0 (enthalpy change) were obtained by nonlinear least squares fitting of experimental data using the single-site binding model of the Origin software package (version 7.0) provided with the instrument. The free energy of binding (ΔG0) and entropy change (ΔS0) were obtained using Equations 3 and 4,
Structural Studies
KdsB was crystallized in the presence and absence of ligands by the sitting drop technique at 4 °C by adding 2 μl of mother liquor to 2 μl of KdsB (14.5 mg ml−1). For the ligand-free enzyme, crystals were produced using 100 mm sodium acetate, pH 4.6, 250 mm sodium sulfate, 15% (v/v) glycerol as a reservoir solution. Ligand-bound crystals were produced in the presence of 5 mm CTP and 5 mm 2β-deoxy-Kdo, using 85 mm HEPES, pH 7.5, 17% (w/v) polyethylene glycol 10000, 6.8% ethylene glycol, 15% (v/v) glycerol as a reservoir solution. Crystals were mounted on a nylon loop and flash-cooled in liquid nitrogen. Data were collected from single cryofrozen crystals of the unbound (2.5 Å) and ligand complexed (1.9 Å) forms of KdsB at Diamond. The data were scaled and integrated using XDS (21) and subsequently handled using the CCP4 suite (22). The structures were solved by molecular replacement using Phaser (23). Refinement and model building were carried out using either Refmac 5 (24) or Phenix (25) and COOT (26). Data and final refinement statistics are given in Table 1.
TABLE 1.
Crystallographic data collection and refinement statistics.
| Unbound (3K8E) | Liganded (3K8D) | |
|---|---|---|
| Space group | C2 | P31 |
| Unit cell | a = 124.47 b = 77.18 c = 143.63 β = 90.03 Å | a = b = 94.77 c = 153.14 Å |
| Resolution | 2.5 Å (47.0 Å) | 1.9 Å (45.3 Å) |
| Highest resolution shell | 2.57–2.51 Å | 1.92–1.90 Å |
| Total reflections | 171,980 | 634,356 |
| Unique reflections | 46,074 | 120,795 |
| Completeness | 97.8% (97.0%) | 99.7% (99.1%) |
| Redundancy | 3.7 (3.8) | 5.3 (4.9) |
| Rmerge | 6.0 (34.6) | 7.9 (33.0) |
| I/σI | 16.03 (4.97) | 16.0 (5.3) |
| Rwork | 19.9 | 17.3 |
| Rfree | 23.4 | 20.0 |
| r.m.s.d.a angle | 1.317° | 1.109° |
| r.m.s.d. length | 0.077 Å | 0.007 Å |
| Overall B | 54.9 Å2 | 27.1 Å2 |
| HOH B | 54.0 Å2 | 44.2 Å2 |
| CTP B (Å2) | 17.7 Å2 | |
| Kdo B (Å2) | 18.7 Å2 | |
| Mg-B (Å2) | 13.0 Å2 |
a r.m.s.d., root mean square deviation.
EPR Spectroscopy
Continuous wave EPR spectra were run at X-band using a Bruker ELEXSYS E500 spectrometer in conjunction with an Oxford Instruments ESR900 cryostat and ITC503 temperature controller. The microwave power was 50 microwatts; the modulation frequency was 100 kHz, and the modulation amplitude was 5 G. Each spectrum presented is the sum of four scans accumulated at a temperature of 20 K. 250-μl samples of 280 μm KdsB in 10 mm Tris/HCl, pH 7.5, in the presence of either 280 or 560 μm MnCl2 were used for EPR measurements. Various combinations of 5 mm CTP and 5 mm 2β-deoxy-Kdo were also included when indicated.
Computational Modeling
Chain A of the KdsB-CTP-2β-deoxy-Kdo crystal structure was used as a starting point for all molecular dynamics simulations and energy minimizations. A second Mg2+ cation was added in the proposed Mg-A position. The models were immersed in a TIP3 water box (27), and charge parameters and topology files of ligands were obtained using the Antechamber program (28) using AM1-BCC charges (29). NAMD2 software (30) and Amber forcefield (31) were used to perform the simulations. The cutoff parameter for the computation of nonbonded interactions was set to 12 Å, and the electrostatic forces were softened by defining a relative dielectric constant of 2 for the system. Several cycles (up to five) of energy minimization (1000 steps, conjugate gradient), followed by molecular dynamics simulations (200 ps at 200 K) were performed to stabilize the enzyme-ligand complex and obtain the final models.
RESULTS AND DISCUSSION
Kinetic Characterization of KdsB
A major limitation of previous studies with KdsB and the related enzyme, KpsU, has been the inability to measure steady-state enzyme activity using continuous assay methods. We have now developed a new linked assay for the enzyme, which allows us to monitor the continuous formation of the pyrophosphate product as it is released during the reaction (supplemental Fig. S1). Consequently, this has enabled us to determine the true kinetic parameters for the enzyme by measuring the initial rate of pyrophosphate formation over a range of CTP and Kdo concentrations. The dependence of the initial rate on substrate concentration, shown graphically in Fig. 3A, followed Michaelis-Menten kinetics and reveals a ternary complex steady-state mechanism (supplemental Fig. S2). Under the conditions used, the Vmax was determined to be 2.3 ± 0.01 μm min−1, which corresponds to a kcat value of 3.8 ± 0.02 s−1 (assuming that the enzyme is 100% active). The Km for Kdo was calculated to be 97.9 ± 10.4 μm, and the Km for CTP was calculated to be 4.8 ± 0.5 μm. These values differ slightly to those obtained in an earlier study where Km values were calculated to be 290 μm for Kdo and 200 μm for CTP, although it should be noted that the previous parameters were measured by using stopped assays at an elevated pH of 9.5 (4). In addition, previous studies have also shown that in solution Kdo is a mixture of pyranose and furanose forms with ∼2% in the β-pyranose form (32) that is accepted by KdsB (33). Therefore, the Km value that has been calculated for Kdo is significantly higher than the true value, which is likely to be ∼2 μm.
FIGURE 3.

Steady-state kinetic characterization of KdsB and inhibition studies with 2β-deoxy-Kdo. A, dependence of the initial rate of pyrophosphate formation catalyzed by 0.01 μm KdsB on the concentration of CTP at a series of fixed concentrations of Kdo as follows: 10 μm (♦), 25 μm (□), 50 μm (■), 100 μm (△), 250 μm (▲), 500 μm (○), and 1000 μm (●). The points represent the experimental data, and the lines represent the predicted values obtained by fitting the data points to Equation 1 by nonlinear regression analysis (see under “Experimental Procedures”). B, dependence of the initial rate of pyrophosphate formation catalyzed by 0.01 μm KdsB on the concentration of Kdo at a series of fixed concentrations of the 2β-deoxy-Kdo inhibitor as follows: 0 μm (♦), 1 μm (□), 2 μm (■), 5 μm (△), 10 μm (▲), 25 μm (○), and 50 μm (●). The points represent the experimental data, and the lines represent the predicted values obtained by fitting the data points to the equation for competitive inhibition (Equation 2) by nonlinear regression analysis (see “Experimental Procedures”). C, binding isotherms for the titration of 10 μm KdsB with 2β-deoxy-Kdo. All titrations were performed in 10 mm Tris/HCl, pH 7.5, at 25 °C, and the data were fitted to a one-site model as described under “Experimental Procedures.” Inset shows the raw data for ITC experiments.
Inhibition Studies with 2β-Deoxy-Kdo
Previous studies have identified a number of inhibitors of the enzyme that are based on the structure of 2β-deoxy-Kdo (Fig. 2) (10–12). Consequently, we have carried out detailed kinetic inhibition studies with this compound using the new continuous assay that we have developed. The initial rates of pyrophosphate formation have been measured over a range of Kdo and 2β-deoxy-Kdo concentrations, which is shown graphically in Fig. 3B. These studies reveal that 2β-deoxy-Kdo acts as a potent competitive inhibitor with the Kdo substrate of the enzyme (supplemental Fig. S3) with a Ki of 2.03 ± 0.08 μm. In addition, ITC experiments have been used to measure the binding of the 2β-deoxy-Kdo inhibitor to the enzyme. Fig. 3C represents the ITC binding isotherms resulting from the titration of KdsB with 2β-deoxy-Kdo in the presence of CTP. The binding process was exothermic, and the isotherms could be fitted to a single-site model, yielding a complex dissociation constant of 1.7 ± 0.53 μm, which is very similar to the Ki value calculated from the earlier competitive inhibition measurements. The thermodynamic binding parameters were calculated using Equations 3 and 4, yielding an enthalpy change, ΔH0, of −1.29 ± 0.08 kJ mol−1 and an entropy change, ΔS0, of 22.1 ± 1.8 J K−1.
Additional kinetic inhibition studies at a range of CTP concentrations have shown that 2β-deoxy-Kdo acts a noncompetitive inhibitor against the CTP substrate (supplemental Fig. S3). Moreover, similar measurements using the CTP analogue, CMP, revealed that this compound acted as a competitive inhibitor against CTP, with a Ki of 1.30 ± 0.58 mm, and as a noncompetitive inhibitor against the Kdo substrate (supplemental Fig. S4). This pattern of inhibition is consistent with a compulsory order ternary complex mechanism rather than a random order mechanism.
Crystal Structures of Free and Ligand-bound KdsB
The crystal of ligand-free KdsB contains two dimers in the asymmetric unit (see supplemental Fig. S6). The individual monomers are highly similar in fold to the structure of the capsule-specific KdsB (13, 14) and consists of a central 7-stranded β-sheet surrounded by α-helices (Fig. 4A). The structure has a distinct cleft that separates the CTP-binding domain from the dimer interface domain, which is proposed to be involved in sugar binding. An overlay of the four individual monomers reveals that they all adopt an “open” conformation with little difference in backbone conformation, except for the Asp163–Phe178 loop at the dimer interface, which has a conformation that is dependent on the crystal packing environment. Significantly higher B-factors are observed for sections of the CTP-binding domain, including the phosphate-binding PP-loop Arg10–Lys19, the C-terminal α-helix Gln227–Ala245, and the Arg69–Ser74 loop region. A 2.6 Å structure for the ligand-free E. coli lipopolysaccharide-specific KdsB in a different space group has been deposited in the PDB data base (code 1VH1) by a structural genomics consortium. This structure reveals an essentially identical conformation of the dimer interface, with minor deviation in the position of the CTP-binding domain indicating a high degree of flexibility in the relative position of the CTP-binding and dimerization domains. Furthermore, due to higher average B-factors, several loops in the CTP-binding domain have not been modeled.
FIGURE 4.

Crystal structures of E. coli KdsB. A, overlay of the ligand-free and CTP-2β-deoxy-Kdo-KdsB crystal structures. The dimerization domain and CTP-binding domains are colored blue and yellow, respectively, for the CTP-2β-deoxy-Kdo-KdsB structure, and the ligand-free structure is depicted in gray. The bound CTP and 2β-deoxy-Kdo are shown in atom-colored sticks with green and purple carbons, respectively. B, electron density obtained for the bound ligands in the CTP-2β-deoxy-Kdo-KdsB crystal structure. The σA weighted 2FoFc map is contoured at levels 1.5 and 7 σ (pinpointing the heavier phosphor and magnesium atoms) and contoured respectively in blue and red. Associated water molecules are shown as red spheres.
The 1.9 Å crystal structure of the KdsB-CTP-2β-deoxy-Kdo ternary complex also contains two dimers in the asymmetric unit. No significant difference can be observed between the individual monomers that all adopt a “closed” conformation (Fig. 5 A). This structure is related to the open conformation by an ∼13° rotation of the CTP-binding domain around hinge regions 80–81, 95–96, 106–107, and 225–233 (as determined by DynDom (34)) leading to a maximal shift in position of ∼9 Å for PP-loop residue Arg15. In addition, the PP-loop changes conformation to establish a network of polar contact with the CTP phosphates. Electron density is clearly visible for both the bound CTP as well as 2β-deoxy-Kdo within the active site cleft (Fig. 4B). In addition, a Mg2+ ion can be readily identified bound to the CTP phosphates. 2β-Deoxy-Kdo is bound at the interface between the dimerization domain and the CTP-binding domain through predominantly polar interactions. Many of the interactions with residues from the dimerization domain (e.g. Arg157, His181, Tyr185, Gln210, and Glu211) do not appear to significantly alter the KdsB structure when compared with the ligand-free KdsB structure (Fig. 5). However, interactions made with residues from the CTP-binding domain are clearly formed concomitant with conversion from the open to the closed conformation. The strictly conserved Gln98 establishes a double hydrogen bond with the sugar ligand that requires a shift in the Gln98 backbone position, leading to extended β-sheet interactions between strands β6 and β10 from the dimerization domain. In addition, a putative charged interaction is established between the PP-loop Arg15 and the 2β-deoxy-Kdo carboxylate that similarly requires the open to closed transformation to occur. In addition to the direct interactions between the CTP-binding domain residues and 2β-deoxy-Kdo itself, a complex network of (water-mediated) polar interactions is established between 2β-deoxy-Kdo and the bound CTP and Mg2+ ion.
FIGURE 5.

Crystallographic snapshots along the KdsB reaction path. Key residues from the active site of four different E. coli CMP-Kdo synthetase crystal structures are displayed with residues derived from the dimerization domain in blue carbons and those derived from the CTP-binding domain in yellow carbons. Mg2+ ions (when present) are shown as gray spheres. The positions of the bound ligands from the CTP-2β-deoxy-Kdo-KdsB crystal structure are shown as thin lines in the other crystal structures for easy comparison. The ligand-free enzyme structure represents E (this work). although the CTP-bound E. coli capsule-specific CMP-Kdo synthetase structure (PDB code 1GQ9, see Ref. 14) represents the CTP-E complex. The ternary enzyme complex E-CTP-Kdo is mimicked by the CTP-2β-deoxy-Kdo-KdsB crystal structure (this work), and the E-CMP-Kdo product-bound enzyme structure is represented by the CMP-Kdo-bound E. coli capsule-specific CMP-Kdo synthetase structure (PDB code 1GQC, see Ref. 14).
CTP is predominantly bound through interactions with residues from the CTP-binding domain, with exception of a charged interaction between the γ-phosphate and Arg164 from the dimerization domain of the opposite monomer (Fig. 5). The cytosine base and ribose moiety are bound in a highly similar manner to previously determined CTP or CMP complexes with the related capsule-specific KdsB isozyme (homology 46% identity over 249 amino acids; PDB codes 1GQ9 and 1GQC) or CMP-acylneuraminate synthase (homology 29% identity over 117 amino acids; PDB codes 1QWJ, 1EYR, and 1EZI) with Arg78 largely responsible for cytosine specificity (13–16). In contrast, the conformation of the phosphates is markedly different from that observed in the CTP-KdsB complex (13, 14). This appears to be a direct consequence of the open to closed transition that brings the α-phosphate in direct contact with the bound 2β-deoxy-Kdo. Closure of the active site cleft in the absence of a reorientation of the CTP phosphates would lead to several steric clashes between both ligands. The putative position of the Kdo hydroxyl with respect to the new CTP α-phosphate position (at a distance of ∼3.0 Å) is ideal for an Sn2-type substitution reaction. The reorientation of the α-phosphate through rotation along the phosphate-ribose bond also leads to a distinct position of both β- and γ-phosphate groups. Most importantly, this reorganization is accompanied by a displacement of the associated Mg2+ ion from the position observed in the CTP-KdsB complex (13, 14) (Fig. 5). In the ternary KsdB-CTP-2β-deoxy-Kdo complex, the Mg2+ ion is located at the interface between both ligands and, unusually, establishes contacts with all three CTP phosphate groups in addition to a water-mediated contact with the 2β-deoxy-Kdo carboxylate. The position vacated by the Mg2+ upon active site closure is occupied by one of the α-phosphate oxygen atoms, placing the latter within hydrogen bonding contact of the conserved residues Lys19, Asp100, and Asp235.
Modeling of a Second Mg2+-binding Site in KdsB
The initial step in the Sn2-type substitution catalyzed by KdsB and the related CNS enzymes is the deprotonation of the β-anomeric hydroxyl group, requiring a general base in the active site of these enzymes. However, previous crystal structures of both KdsB and CNS did not reveal the presence of a protein-derived base in the vicinity of bound product molecules (13–16). It was suggested that a water molecule bound to basic protein residues and/or the single associated Mg2+ ion (possibly as a hydroxyl-ion) could act as a general base. However, the ternary KsdB-CTP-2β-deoxy-Kdo complex structure reveals the enzyme adopts a closed conformation prior to catalysis that is unlike the more open conformations observed when the enzyme is associated with the CMP-sugar product or CTP alone (Fig. 5) (13, 14). Previously proposed reaction schemes, based on the latter structures, are therefore unlikely to reflect the correct mechanism. In contrast, the KsdB-CTP-2β-deoxy-Kdo structure reveals that neither protein-derived nor bound water molecules are close enough to the putative position of the hydroxyl group of Kdo to allow direct proton abstraction.
A remarkable similarity exists between Kdo and related sugar-activating enzymes and DNA/RNA polymerases in terms of overall chemistry catalyzed (34, 35). In both cases, the enzymes catalyze the formation of a sugar-phosphate linkage concomitant with release of pyrophosphate. The mechanism of the latter enzymes has been well characterized and depends on the binding of two distinct Mg2+ ions. The Mg-A ion facilitates the nucleophilic attack of the 3′-OH group, whereas the Mg-B ion coordinates NTP α-, β-, and γ-phosphates ensuring correct orientation. The Mg-A is coordinated by a series of conserved aspartates, whereas Mg-B is largely bound by the NTPs and presumably recruited to the active site simultaneously with NTP binding.
The ternary KsdB-CTP-2β-deoxy-Kdo shows a remarkable similarity with respect to the respective positions of the sugar and NTP substrate as well as the NTP-associated Mg2+ ion despite the obvious lack of any similarly in overall folding (Fig. 6). In contrast, a second Mg2+ ion could not be reliably detected within the KsdB active site, despite extensive soaking with higher Mg2+ concentrations (data not shown). Instead, weak electron density could sometimes be observed to occupy a position similar to Mg-A in the DNA/RNA polymerases that was modeled as a water molecule. To assess the hypothesis of a second magnesium-binding site, a model of KdsB containing this ion, in complex with Kdo, was generated and energetically optimized. The orientation of the substrate was similar to the crystal structure, although small variations occur, due to the force field simulation. The second Mg2+ ion is octahedrally coordinated by three carboxylate groups (derived from the conserved Asp100 and Asp235 and the Kdo substrate), the CTP α-phosphate, and the Kdo 2-hydroxy group (Fig. 6). The lack of the latter ligand in the crystals might contribute to the lack of binding of a second magnesium ion in the crystal structures. The fact that a single metal ion is observed to bind in a position similar to that of Mg-A in both the CMP and the CMP-Kdo-bound E. coli capsule-specific CMP-Kdo synthetase structure (PDB codes 1GQ9 and 1GQC, see Ref. 14) further supports this model.
FIGURE 6.

Comparison between KdsB and RNA polymerase actives sites. A juxtaposition of the active site of T. thermophilus RNA polymerase (PDB code 2OJ5) with a model for the ternary CTP-Kdo-KdsB complex (modeled with an additional Mg2+ ion present in site A). The NTPs are colored with green carbons in both cases, and the attacking substrate is rendered with purple carbons (Kdo or RNA, respectively). Amino acids involved in binding the Mg2+ ions or in stabilizing the transitions state (e.g. His1242 and Lys19) are shown with yellow carbons.
Metal Ion Binding to KdsB
The substitution of Mn2+ for Mg2+ allows EPR spectroscopy to be employed to study metal ion binding stoichiometries and environments. Mn2+ has a high spin d5 configuration, S = 5/2, and the stable Mn isotope, 55Mn, has nuclear spin I = 5/2. Because the high spin d5 configuration has low angular momentum, the resulting spectrum consists of multiple overlapping degenerate transitions at g = 2 and is dominated by the six-line hyperfine splitting resulting from I = 5/2. The line shapes of the six lines are determined by the zero field splitting, which lifts the degeneracy of the overlapping transitions. The zero field splitting is dependent on the environment of the metal ion, particularly the symmetry of that environment, and thus the EPR spectra of Mn2+ ions directly reflect the environments of such ions (36). Fig. 7 shows a selection of Mn2+ EPR spectra illustrating the effect of Mn2+ binding to KdsB (the remaining spectra are shown in the supplemental material). Spectrum A arises from the binding of 2 eqs of Mn2+ to KdsB in the presence of 2β-deoxy-Kdo and CTP. This spectrum is essentially identical to spectrum B, which was constructed in silico by the addition of spectra C and D. Spectrum C arises from 1 eq of Mn2+ bound to KdsB, in the presence of 2β-deoxy-Kdo and CTP, whereas spectrum D arises from 1 eq of Mn2+ bound to KdsB in the presence of CTP. Although spectra C and D are clearly differentiated, for example by the feature marked 1 in spectrum C, they can both be distinguished from the spectrum of Mn2+ in buffer, spectrum E, for example at the fields marked 2 and 3. Thus, these data show that KdsB can bind two Mn2+ ions, one associated with 2β-deoxy-Kdo binding and the other associated with CTP binding. That these Mn2+ ions bind in those sites occupied by Mg2+ in the active complex is shown by competition experiments in which Mn2+ is expelled from these sites by an excess of Mg2+ (supplemental Fig. S5).
FIGURE 7.

EPR measurements confirm the presence of two metal-binding sites in KdsB. X-band continuous wave EPR spectra of Mn2+ ions in the following environments and stoichiometries: A, 2 eq of Mn2+ bound to KdsB with 2β-deoxy-Kdo and CTP; B, sum of the following two spectra which are as follows: C, 1 eq of Mn2+ bound to KdsB with 2β-deoxy-Kdo and CTP, and D, 1 eq of Mn2+ bound to KdsB with CTP; E, 1 eq of Mn2+ in the experiment buffer. Experimental conditions are given under “Experimental Procedures,” spectra are further discussed under “Results and Discussion.”
Mechanism for KdsB
The proposed direct coordination of the Kdo β-anomeric hydroxyl group by Mg-A activates this group for a nucleophilic attack on the CTP α-phosphate (Fig. 8). The preceding deprotonation of the attacking hydroxyl group could occur via a water molecule coordinating Mg-A, which is in contact with the solvent via a series of water molecules. The interaction of the CTP α-phosphate with the conserved Lys19 as well as both the associated Mg2+ ions would allow for stabilization of the negative pentavalent transient intermediate following attack of the Kdo β-anomeric hydroxyl group. The coordination of Mg-B by both CTP β- and γ-phosphates will assist the leaving of the pyrophosphate group, as observed for the DNA/RNA polymerases.
FIGURE 8.

Schematic overview of the proposed KdsB reaction. For clarity, Kdo is represented with R = C7H2OHC8H2OH.
It is interesting to note that the axis of the rotation relating the open and closed conformation is roughly perpendicular to the α-phosphor-hydroxyl-Kdo bond formed. Hence, the inherent mobility of the CTP-binding domain is likely to lead to transient compression of the α-phosphor-hydroxyl-Kdo distance, possibly assisting catalysis. Following breakage of the α-β-phosphate bond and formation of the CMP-Kdo product, the enzyme adopts a more open, “intermediate” conformation, concomitant with the reorientation of Gln98 and altered ligation of the Mg-A ion as revealed by the CMP-Kdo crystal structure (14). This presumably allows the release of the pyrophosphate and associated Mg-B and eventually the CMP-Kdo product itself.
Comparison with the Related CNS Enzymes
KdsB is related in both fold and in the reaction catalyzed to the CNS found in all eukaryotes (except plants) and certain bacteria. Crystal structures are available for the bacterial Neisseria meningitidis CNS in the presence and absence of the analogue CDP as well as the murine CNS (catalytic domain only) in complex with the product CMP-5-N-acetylneuraminic acid (15, 16). Comparison between KdsB and CNS reveals that the CTP-binding domain is highly similar, whereas significant differences are observed between the respective dimerization domains (Fig. 9). Although the stems of the β-strands from the central β-sheet coincide, the connecting loop regions are vastly different in size and fold. As a result, in CNS the sugar binding occurs in part through residues from the opposite monomer as opposed to the situation that occurs in KdsB. Nevertheless, a similar mechanism to that proposed for KdsB is possible for CNS, given the similarity between both enzymes. As for KdsB, previous studies on CNS have not focused on the ternary complex but have based reaction mechanism proposals on product or CDP-bound structures (15, 16). However, for CNS enzymes, a sugar-binding induced reorganization of the CTP-binding domain, as observed for KdsB, is a distinct possibility. In this respect, we note that the CNS residues Lys59, Thr143, and Asp247 in concert with the substrate 5-N-acetylneuraminic acid hydroxyl and carboxylate groups (similar to KdsB Lys19, Asp100, and Asp235 and the Kdo substrate, respectively) could act in a similar manner to activate the CTP α-phosphate and bind a second Mg-A ion following such active site closure. Furthermore, despite the large differences in the respective dimerization domain structure, a conserved Arg residue (KdsB Arg164/CNS Arg202) from the opposite monomer is located close to the CTP phosphate that is involved in binding the γ-phosphate in KdsB. Therefore, we propose that CNS uses a similar Mg2+-dependent mechanism to that postulated for KdsB in analogy to the DNA/RNA polymerases. Crystal structures of CNS in complex with CTP and appropriate 5-N-acetylneuraminic acid based inhibitors will be essential to establish whether this is indeed the case.
FIGURE 9.

Comparison of E. coli KdsB and the related murine CNS structure. A, KdsB and the related murine CNS structure (PDB code 1QWJ, see Ref. 16) are shown side by side in similar orientations. One monomer is colored coded according to Fig. 4, and the second monomer is shown in gray. Bound ligands and key active side residues are shown in atom-colored sticks (color coding as for Figs. 5–7; murine CNS (mCNS) contains the bound product CMP-5-N-acetylneuraminic acid). B, overlay of the KdsB and murine CNS active site structures. Murine CNS key active site residues and bound ligand are shown in atom-colored sticks, although the corresponding KdsB residues are shown as thin lines for comparison. Active site residues for murine CNS are labeled.
Conclusions
It has long been recognized that the KdsB enzyme is a possible target in developing new antimicrobial agents against Gram-negative bacteria (10–12). In this study, we have developed a thorough kinetic and structural understanding of this enzyme. On the basis of the crystallographic data, modeling, and EPR studies, and in analogy to the DNA/RNA polymerases, we propose that KdsB and related enzymes recruit two magnesium ions to the active site. Both magnesium ions are directly coordinated by the KdsB substrates, and although each serves to activate the α-phosphate, Mg-A is responsible for activation of the Kdo-hydroxyl group. The activation of the α-phosphate could potentially significantly promote wasteful CTP hydrolysis. In KdsB, this side reaction is avoided by the fact that CTP is not activated by a coordinated set of interactions with Lys19 and Mg-B until Kdo binding leads to the formation of the closed complex, concomitant with recruitment of the Mg-A ion and hence activation of both the CTP α-phosphate group and Kdo substrate. It is clear that the major aim now is to develop new inhibitors that are capable of crossing the inner membrane of the cell and reaching the enzyme in the cytoplasm.
This work was supported by the Biotechnology and Biological Sciences Research Council, United Kingdom.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S6.
The atomic coordinates and structure factors (codes 3K8D and 3K8E) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- LPS
- lipopolysaccharide
- Kdo
- 2-keto-3- deoxymanno-octulonic acid
- CNS
- CMP-5-N-acetylneuraminic acid synthase
- ITC
- isothermal calorimetry
- PDB
- Protein Data Bank.
REFERENCES
- 1.Raetz C. R., Whitfield C. (2002) Annu. Rev. Biochem. 71, 635–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nikaido H. (2003) Microbiol. Mol. Biol. Rev. 67, 593–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raetz C. R., Reynolds C. M., Trent M. S., Bishop R. E. (2007) Annu. Rev. Biochem. 76, 295–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ray P. H., Benedict C. D., Grasmuk H. (1981) J. Bacteriol. 145, 1273–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldman R. C., Kohlbrenner W. E. (1985) J. Bacteriol. 163, 256–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldman R. C., Bolling T. J., Kohlbrenner W. E., Kim Y., Fox J. L. (1986) J. Biol. Chem. 261, 15831–15835 [PubMed] [Google Scholar]
- 7.Bravo I. G., García-Vallvé S., Romeu A., Reglero A. (2004) Trends Microbiol. 12, 120–128 [DOI] [PubMed] [Google Scholar]
- 8.Münster A. K., Eckhardt M., Potvin B., Mühlenhoff M., Stanley P., Gerardy-Schahn R. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 9140–9145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kean E. L. (1991) Glycobiology 1, 441–447 [DOI] [PubMed] [Google Scholar]
- 10.Claesson A., Luthman K., Gustafsson K., Bondesson G. (1987) Biochem. Biophys. Res. Commun. 143, 1063–1068 [DOI] [PubMed] [Google Scholar]
- 11.Goldman R., Kohlbrenner W., Lartey P., Pernet A. (1987) Nature 329, 162–164 [DOI] [PubMed] [Google Scholar]
- 12.Hammond S. M., Claesson A., Jansson A. M., Larsson L. G., Pring B. G., Town C. M., Ekström B. (1987) Nature 327, 730–732 [DOI] [PubMed] [Google Scholar]
- 13.Jelakovic S., Schulz G. E. (2001) J. Mol. Biol. 312, 143–155 [DOI] [PubMed] [Google Scholar]
- 14.Jelakovic S., Schulz G. E. (2002) Biochemistry 41, 1174–1181 [DOI] [PubMed] [Google Scholar]
- 15.Mosimann S. C., Gilbert M., Dombroswki D., To R., Wakarchuk W., Strynadka N. C. (2001) J. Biol. Chem. 276, 8190–8196 [DOI] [PubMed] [Google Scholar]
- 16.Krapp S., Münster-Kühnel A. K., Kaiser J. T., Huber R., Tiralongo J., Gerardy-Schahn R., Jacob U. (2003) J. Mol. Biol. 334, 625–637 [DOI] [PubMed] [Google Scholar]
- 17.Cornforth J. W., Firth M. E., Gottschalk A. (1958) J. Biochem. 68, 57–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hershberger C., Davis M., Binkley S. B. (1968) J. Biol. Chem. 243, 1585–1588 [PubMed] [Google Scholar]
- 19.Ogura H., Shirai R. (1989) Tetrahedron Lett. 30, 2263–2264 [Google Scholar]
- 20.Luthman K., Orbe M., Wåglund T., Claesson A. (1987) J. Org. Chem. 52, 3777–3784 [Google Scholar]
- 21.Kabsch W. (1993) J. Appl. Crystallogr. 26, 795–800 [Google Scholar]
- 22.Collaborative Computational Project, No. 4 (1994) Acta Crystallogr. Sect. D 50, 760–76315299374 [Google Scholar]
- 23.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta Crystallogr. Sect. D 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 25.Adams P. D., Grosse-Kunstleve R. W., Hung L.-W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C. (2002) Acta Crystallogr. Sect. D 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 26.Emsley P., Cowtan K. (2004) Acta Crystallogr. Sect. D 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 27.Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L. (1983) J. Chem. Phys. 79, 926–935 [Google Scholar]
- 28.Wang J., Wang W., Kollman P. A., Case D. A. (2006) J. Mol. Graph Model. 25, 247–260 [DOI] [PubMed] [Google Scholar]
- 29.Jakalian A., Jack D. B., Bayly C. I. (2002) J. Comput Chem. 23, 1623–1641 [DOI] [PubMed] [Google Scholar]
- 30.Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. (2005) J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cornell W. D., Cieplak P., Bayly C. I., Gould I. R., Merz K. M., Ferguson D. M., Spellmeyer D. C., Fox T., Caldwell J. W., Kollman P. A. (1995) J. Am. Chem. Soc. 117, 5179–5197 [Google Scholar]
- 32.Brade H., Zähringer U., Rietschel E. T., Christian R., Schulz G., Unger F. M. (1984) Carbohydr. Res. 134, 157–166 [Google Scholar]
- 33.Kohlbrenner W. E., Fesik S. W. (1985) J. Biol. Chem. 260, 14695–14700 [PubMed] [Google Scholar]
- 34.Nudler E. (2009) Annu. Rev. Biochem. 78, 335–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rothwell P. J., Waksman G. (2005) Adv. Protein Chem. 71, 401–440 [DOI] [PubMed] [Google Scholar]
- 36.Reed G. H., Markham G. D. (1984) in Biological Magnetic Resonance (Berliner L. J., Reuben J. eds) Vol. 6, pp. 73–142, Plenum Press, New York [Google Scholar]