

Abstract
Porphobilinogen synthase (PBGS) catalyzes the first common step in tetrapyrrole (e.g. heme, chlorophyll) biosynthesis. Human PBGS exists as an equilibrium of high activity octamers, low activity hexamers, and alternate dimer configurations that dictate the stoichiometry and architecture of further assembly. It is posited that small molecules can be found that inhibit human PBGS activity by stabilizing the hexamer. Such molecules, if present in the environment, could potentiate disease states associated with reduced PBGS activity, such as lead poisoning and ALAD porphyria, the latter of which is associated with human PBGS variants whose quaternary structure equilibrium is shifted toward the hexamer (Jaffe, E. K., and Stith, L. (2007) Am. J. Hum. Genet. 80, 329–337). Hexamer-stabilizing inhibitors of human PBGS were identified using in silico prescreening (docking) of ∼111,000 structures to a hexamer-specific surface cavity of a human PBGS crystal structure. Seventy-seven compounds were evaluated in vitro; three provided 90–100% conversion of octamer to hexamer in a native PAGE mobility shift assay. Based on chemical purity, two (ML-3A9 and ML-3H2) were subjected to further evaluation of their effect on the quaternary structure equilibrium and enzymatic activity. Naturally occurring ALAD porphyria-associated human PBGS variants are shown to have an increased susceptibility to inhibition by both ML-3A9 and ML-3H2. ML-3H2 is a structural analog of amebicidal drugs, which have porphyria-like side effects. Data support the hypothesis that human PBGS hexamer stabilization may explain these side effects. The current work identifies allosteric ligands of human PBGS and, thus, identifies human PBGS as a medically relevant allosteric enzyme.
Introduction
Human porphobilinogen synthase (PBGS,2 EC 4.2.1.24, also known as 5-aminolevulinate dehydratase) exists as a quaternary structure equilibrium consisting of a high activity octamer, a low activity hexamer, and a dimer that can take on two distinct conformations, each of which dictates assembly to either the octamer or the hexamer (see Fig. 1a) (1). For the wild type protein at neutral pH, the octamer is the dominant assembly in the equilibrium (1). As PBGS catalyzes the first common step in the biosynthesis of the tetrapyrroles such as heme, low activity mutations in the human population are associated with the disease ALAD porphyria (2); all eight porphyria-associated PBGS mutations increase the propensity of the human protein to exist in the low activity hexameric assembly, thus establishing the physiologic relevance of the quaternary structure equilibrium to human health (3). In addition to ALAD porphyria, PBGS inhibition by divalent lead is a primary consequence of lead poisoning. Factors that stabilize the hexamer will further inhibit PBGS activity and, thus, potentiate the physiologic effects of lead poisoning.
FIGURE 1.

The structure and behavior of the alternate quaternary structure assemblies of PBGS. a, the dynamic equilibria between the quaternary structure assemblies of human PBGS are shown. The two predominant forms in solution are the high activity octamer (PDB code 1E51) and the low activity hexamer (PDB code 1PV8); interconversion between these forms requires dissociation to a dimer, which can take on alternate conformations. The interchange between dimer conformations involves alterations in a small number of ϕ/ψ angles. b, shown is a space-filling model of the human PBGS hexamer (PDB code 1PV8) highlighting the hexamer-specific cavity formed by chains A, B, and C, illustrated in dark blue, light blue, and gray, respectively. The remaining three subunits are shown in yellow. Small molecule binding to this cavity is proposed to stabilize the hexamer and shift the equilibrium toward this low activity form, inhibiting human PBGS activity. c, the small molecule docking boxes are illustrated at the interface of chains A, B, and C (colored as in b). The entire docked molecule must be contained in the 25-Å cube (purple); the center of the molecule must be contained in the 14 Å cube (green). d, shown is the pH activity profile of the wild type human PBGS (taken from Ref. 1). The basic portion of the curve reflects changes in the quaternary structure equilibrium, favoring hexamer with increasing pH (1). The Km value of 0.2 mm shown at pH 7 represents the Km of the octamer (at pH 7), which predominates at neutral pH. The Km value of 0.02 mm shown at pH 9 represents the Km of the octamer (at pH 9), whereas the Km value of 4.5 mm shown at pH 9 represents the Km of the hexamer.
The arrangement of the subunits in the PBGS hexamer creates a surface cavity that is not present in the octamer or the dimers (see Fig. 1b) (4). Ligand binding to this cavity is posited to stabilize the hexamer, draw the quaternary structure equilibrium toward the hexamer, and thus, inhibit function. The residues that comprise this surface cavity are phylogenetically variable. We have shown that small molecules that bind selectively to a plant PBGS hexamer, presumably by binding in this surface cavity, can act as species-specific PBGS inhibitors whose mechanism of action is stabilization of a low activity oligomer (4). Herein we apply these principles to the inhibition of human PBGS with the understanding that hexamer stabilizing molecules can potentiate diseases related to low PBGS activity. The approach used is computational docking of commercially available molecules to a hexameric crystal structure of the low activity human PBGS variant F12L (PDB code 1PV8 (5)). From the docking results we select a family of dissimilar structures for in vitro validation and discover three small molecules that inhibit human PBGS by stabilizing the low activity oligomer. Herein we use the term “morphlock” to describe a small molecule that stabilizes a specific functionally distinct alternate quaternary structure assembly, in this case the low activity hexamer.
Physiologically significant morphlocks can derive from environmental contaminants, natural products, or drugs in clinical use. A drug that functions as a morphlock toward an off-target protein could provide an unprecedented structural explanation for the side effects of the drug. One discovered morphlock, which we have called ML-3H2, is chemically similar to two amebicidal drugs currently in clinical use, clioquinol and iodoquinol. We address the ability of these drugs to stabilize the human PBGS hexamer and draw a putative correlation between these results and the neuropathic side effects of both drugs (6).
Most porphyric diseases are episodic, and physiologic mechanisms contributing to porphyric attacks are not fully understood. Morphlocks discovered to stabilize the wild type human PBGS hexamer are predicted to have increased potency against the naturally occurring ALAD porphyria-associated variants; we tested and confirmed this hypothesis with two such variants, E89K and A274T. The long range utility of this study is the identification of chemical structures that stabilize the human PBGS hexamer and that may potentiate diseases related to diminished PBGS activity.
EXPERIMENTAL PROCEDURES
Materials
The programs MACROMODEL, GLIDE (Version 3.5), LIGPREP, and QIKPROP and the graphical user interface MAESTRO were from Schrödinger, L.L.C. (New York, NY). The candidate inhibitors were from Life Chemicals, Inc. (Burlington, ON, Canada). All other chemicals were from Fisher or Sigma and were of the highest purity available. Electrophoresis equipment and reagents and chromatography equipment and resins were from GE Healthcare.
In Silico Library of Compounds
Two-dimensional representations of the Life Chemicals kinase-targeted and G-protein-coupled receptor-targeted libraries of compounds (69,593 compounds) were provided in SD format by the vendor. The structures were converted to MAESTRO format, and entries that contained metal ions or atom types other than carbon, hydrogen, nitrogen, oxygen, phosphorus, and sulfur and halogens were discarded. Hydrogens were added as appropriate for the structures, generating a single uncharged stereoisomer per compound. The structures were energy-minimized using MACROMODEL with the MMFF force field (7) and then expanded to include all forms likely to be present in the pH range 5–9 using the LIGPREP utility, resulting in a total of ∼111,000 structures. The structures were again energy-minimized using MACROMODEL, which yielded output files that were suitable for docking with GLIDE.
In Silico Docking
Docking was performed using the Fox Chase Cancer Center computer cluster running GLIDE (32 licenses) (8). The docking target was a hexamer-specific surface cavity of the human PBGS crystal structure of the F12L variant (PDB code 1PV8 (5)) shown in Fig. 1b. The F12L hexamer comprises three identical asymmetric dimers, and the hexamer-specific surface cavity is located at the interface of three subunits. Thus, the docking boxes contained components from three subunits (see Fig. 1c). A cube (25 Å dimension) was used to define a region where the entire molecule must fit to be scored by GLIDE. Within this box a smaller cube (14 Å dimension) was defined to restrict the location of the center of the docked compounds. The asymmetry of the crystallographic dimer produces binding sites in the hexamer that are identical in sequence but structurally distinct. In the crystal structure of the F12L variant the subunits are labeled alphabetically as A–F. One of the inhibitor binding sites is formed by subunits A, B, and C (ABC), and the other is formed by subunits B, A, and F (BAF). Three of the six binding sites in the hexamer are equivalent to the ABC site, and three are equivalent to the BAF site. Each compound library was docked both to the ABC site and the BAF site. The positions of the docking boxes at the ABC interface are shown (see Fig. 1c). All compounds were docked in Standard Precision mode, and the docked pose of each compound was given a score based on a proprietary modification of the CHEMSCORE program. Standard Precision docking in GLIDE mainly evaluates goodness of fit based on geometries. The highest ranking compounds (the best 10%) in the Standard Precision mode were reevaluated in Extra Precision mode, where polarity and hydrophobicity are taken into account. Again, compounds with the highest ranking 10% of scores in this round were considered for purchase based on several additional criteria as follows. We asked if the docked molecule made van der Waals contacts or hydrogen bonds with all three subunits and had a predicted solubility (log S) estimate of at least −6 (calculated using QIKPROP (9)). To limit cost and yield a manageable number of compounds for in vitro analysis, the purchased set was further limited to molecules of dissimilar structure that docked at various locations within the relatively large docking box.
Compound Solutions
Compounds purchased from Life Chemicals were dissolved in dimethyl sulfoxide (DMSO) to yield a concentration of 10 mm. Solutions were stored in darkness at room temperature.
Expression and Purification of PBGS
PBGS from human (N59/C162A wild type variant, E89K, and A274T) were expressed and purified as previously reported (3, 10).
Native Gel Electrophoresis
Electrophoresis was performed using a PhastSystem with PhastGel native buffer strips. Either 6-lane (4 μl per lane) or 8-lane (1 μl per lane) sample applicators were used to load the samples. Separations were performed using 12.5% polyacrylamide gels. After separation, gels were developed on the PhastSystem using Coomassie Blue or activity stain as previously described (4, 11). For the gel shift screen of putative morphlocks with human PBGS, samples were prepared by mixing 8 μl of protein (0.3 mg/ml, 8.3 μm subunits) in 0.1 m Bis-Tris propane-HCl, pH 8.0, 10 mm β-mercaptoethanol, and 10 μm ZnCl2 with 2 μl of 10 mm compound in DMSO. The resultant samples, which contained 20% DMSO and 2 mm compound, were incubated at 37 °C in for 30 min, 6 h, and 24 h before loading and running the gels. Compounds that substantially stabilize the human PBG hexamer in 6 h were then incubated under the screening conditions for 0, 2, 4, and 6 h. These compounds were also evaluated for dose-response relationships, where human PBGS (0.3 mg/ml) was incubated under the screening conditions for 6 h, but with varied compound concentrations as indicated. In all cases the final concentration of DMSO was maintained at 20%. Hexamer stabilization was also evaluated as a function of pH, where the samples were incubated as described for the screening conditions for 6 h but at varied pH as indicated. For the native PAGE experiments containing the PBGS substrate 5-aminolevulinic acid (ALA), the gel samples were prepared as for the dose-response evaluations, and the ALA and compounds were both present for the duration of the incubation. Where indicated, gels were stained for PBGS activity as previously described (4).
Quantification of PAGE results by densitometry was carried out using the program ImageJ (12). Three separate determinations were made to quantify the density of each gel band. The quantified data for each gel lane is presented as % hexamer, which was defined as the amount of protein present in the hexamer band relative to the total protein in the hexamer and octamer bands. For the dose response by gel shift evaluations, the data were fitted either to a simple hyperbolic binding equation (Equation 1) or, when appropriate, to an offset hyperbolic binding equation (Equation 2) or to the Hill equation (Equation 3),
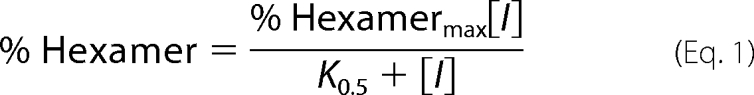 |
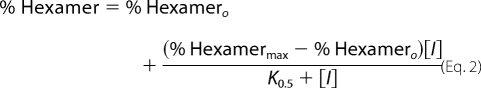 |
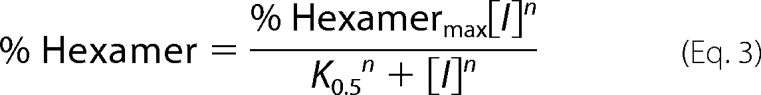 |
where % Hexamermax is derived from the fit as the highest fraction of hexamer, [I] is the concentration of the inhibitor, K0.5 is the concentration of inhibitor at the midpoint of the curve, % Hexamero is the fraction of hexamer in the starting sample, and n is the Hill coefficient.
Activity Assays
The activity of human PBGS was assayed by monitoring the production of the product, porphobilinogen. The concentration of porphobilinogen was determined based on the absorbance at 555 nm (ϵ555 = 60,200 m−1 cm−1) of its complex with ρ-dimethylamino-benzaldehyde that is formed upon treatment with modified Ehrlich's reagent. The assay volume was 1 ml, and all assays were performed at 37 °C. The assay buffer was 0.1 m Bis-Tris propane-HCl, pH 8.0, 10 mm β-mercaptoethanol, and 10 μm ZnCl2. Reactions were initiated by the addition of 100 μl of 0.1 m ALA-HCl to 900 μl of assay buffer containing PBGS and allowed to proceed for 5 min before the addition of 500 μl of STOP reagent (20% trichloroacetic acid, 0.1 m HgCl2). The stopped reactions were vortexed and centrifuged, the supernatants were treated with modified Ehrlich's reagent, and the absorbance at 555 nm was measured as described previously (13).
Inhibition
Inhibition was assessed using the activity assay after incubation of 90 μl of enzyme in the appropriate assay buffer with 10 μl of compound solution (stock 10 mm in DMSO) or DMSO alone at 37 °C for specific time periods. After this preincubation, 800 μl of the appropriate assay buffer was added, and the mixture was allowed to equilibrate at 37 °C for 15 min before the addition of 100 μl of 0.1 m ALA-HCl. The compound concentrations reported in the text are those in the final 1-ml assay volume. Inhibition data were plotted as fractional activity relative to the absence of compound, and inhibition curves were fitted to a hyperbolic function, Equation 4.
 |
where FA is the fractional activity, FAmax is the fractional activity in the absence of compound (set as 1), [I] is the concentration of compound, and IC50 is the [I] at 50% inhibition. All kinetic data were fit using the program SigmaPlot® (Systat Software, Inc., San Jose, CA).
Chemical Characterization of Hexamer-stabilizing Compounds
The purity and identity of ML-3A9, ML-3F2, and ML-3H2, as purchased from Life Chemicals, were assessed using a Waters liquid chromatography-mass spectrometry system and 1H NMR as detailed in supplemental Table 1.
Identification of the ML-3H2 Analogs in Clinical Use
The chemical structures of both ML-3A9 (1,6-dihydro-N-(3-methoxyphenyl)-1-methyl-6-oxo-2-[(2-pyridinylmethyl)thio]-5-pyrimidinecarboxamide, see Fig. 2a) and ML-3H2 (5-chloro-7-(diethylaminomethyl)quinolin-8-ol, see Fig. 2b) were used as entries in the search engine CAS Scifinder. Two drugs currently in clinical use, clioquinol (5-chloro-8-hydroxy-7-iodoquinoline) and iodoquinol (5,7-diiodo-8-hydroxyquinoline), were found to be chemically similar to ML-3H2. These compounds were purchased for analysis and evaluated using methods identical to the compounds from Life Chemicals.
FIGURE 2.

The structures and docked poses of ML-3A9 and ML-3H2 at the ABC interface. a, the structure of ML-3A9 is shown. b, the structure of ML-3H2 is shown. c, the docked pose of ML-3A9 is displayed as a stick model with the carbons in green; other atoms are colored using the Corey-Pauling-Koltun convention. The three subunits of the ABC interface are represented as ribbons with subunits A, B, and C colored dark blue, light blue, and gray, respectively. The side chains within 4 Å of the docked compound are shown as sticks with the carbon atoms colored in the same manner as their respective ribbon representations and all other atoms colored using the Corey-Pauling-Koltun convention. Hydrogen bonds between ML-3A9 and the protein are shown as dashed lines. d, the docked pose of ML-3H2 is illustrated as described in c. Note that the protein is oriented differently in c and d to effectively illustrate planar elements of the respective compounds. Residues in common between c and d are labeled in each figure for reference. This figure was produced using Maestro (Schrödinger, LLC, New York, NY) and MarvinSketch (Chem Axon Kft., Budapest, Hungary).
RESULTS
Docking and Compound Selection
Computational docking was used as a prescreening method on large libraries of “drug-like” compounds available from Life Chemicals. The prescreening allows selection of compounds for in vitro analysis that have an increased probability of binding to the target relative to a random selection of compounds. However, one cannot know whether the optimally docked stereoisomer is actually contained in the commercially available compound, as stereochemistry is often not specified.
The structure of the hexameric F12L variant of human PBGS (PDB code 1PV8 (5)) was used as the target for in silico docking (see Fig. 1, b and c). Docking of the ∼111,000 structures derived from the Life Chemicals “G-protein-coupled receptor-targeted” and “kinase-targeted” libraries using GLIDE in SP mode resulted in a distribution of scores such that 10% of the compounds had good scores, 80% scored similarly to each other but worse than the top 10%, and 10% scored badly. Docking of the top 10% of compounds from the SP screen using GLIDE in XP mode gave a similar distribution of scores. The compounds with the top 10% of GLIDE XP mode scores (∼1100 structures) were further processed to select diverse docking positions and diverse structures. This resulted in the selection of ∼100 compounds for purchase and in vitro evaluation as stabilizers of the human wild type PBGS hexamer. Only a portion of those selected, 77 compounds, were readily available from the vendor. supplemental Table 2 includes the structures of the compounds that were purchased and evaluated.
Native Gel Mobility Shift Screening of Purchased Compounds
The docking process described above was designed to provide a selection of compounds likely to perturb the quaternary structure equilibrium of human PBGS toward the hexameric assembly. Such a perturbation can be evaluated using a native PAGE mobility shift assay, as this method separates the octameric, hexameric, and dimeric assemblies (4, 13). Coomassie staining has proven the most reliable means of visualizing gel shift results; however, the sensitivity of this staining technique requires protein concentrations (0.3–1 mg/ml) that are higher than what would be found physiologically in erythrocytes (1–5 μg/ml) or that are used for in vitro kinetic assays (5–10 μg/ml). If the desired mechanism of inhibition is operative, high concentrations of protein will disfavor hexamer formation and increase the apparent K0.5 of hexamer-stabilizing compounds apparent from the gel shift assay relative to an IC50 based on activity assays. Thus, compound concentrations used in the gel shift assay are higher than those that might be used for a screen based upon inhibition of PBGS activity.
For human PBGS, the gel shift assay involved incubation of 2 mm compound with 0.3 mg/ml enzyme (8.3 μm subunit) at 37 °C for a fixed period of time and subsequent separation of the assemblies by 12.5% native PAGE. No significant change in PBGS oligomer distribution was observed for any of the compounds when using a 2-h incubation at pH 7. Although the high activity octameric assembly dominates the wild type human PBGS quaternary structure equilibrium at neutral pH, at higher pH values the hexamer is more highly favored (1, 10). Thus, the gel shift screen was repeated at pH 8 where the intrinsic population of the wild type hexamer is measurable, and the probability of trapping this species is, thus, increased. Screening at pH 8 was carried out using incubation times of 30 min, 6 h, and 24 h. Minimal effects were observed at 30 min, and significant conversion to hexamer in the presence of solvent DMSO alone was observed at 24 h. Consequently, the 6 h gel shift assay was used as a first criterion for selection of hexamer-stabilizing compounds. Under these conditions we identified 12 compounds that promoted some degree of hexamer formation, most of which were at a level of 5 -30% conversion to hexamer (see supplemental Fig. 1). However, three compounds, herein called ML-3A9, ML-3F2, and ML-3H2, significantly shifted the wild type human PBGS quaternary structure equilibrium 90–100% toward the hexamer and were selected for further characterization. 1H NMR and liquid chromatography-mass spectrometry analyses of the three compounds confirmed the chemical identity and purity of ML-3A9 and ML-3H2 (as detailed in supplemental Table 1); however, liquid chromatography-mass spectrometry of ML-3F2 revealed multiple peaks suggesting an impure sample, and 1H NMR data were not fully consistent with the reported identity of this compound. Further analyses of the hexamer-stabilizing compounds, therefore, focused on ML-3A9 and ML-3H2. The structures of ML-3A9 and ML-3H2 and their docked poses in the putative hexamer-specific small-molecule binding site are shown in Fig. 2.
Native PAGE Analyses of the Effects of ML-3A9 and ML-3H2 on the Quaternary Structure Equilibrium of PBGS
To determine the time required for these small molecules to perturb the quaternary structure equilibrium of wild type human PBGS toward the hexamer in the gel shift assay, samples were incubated with 2 mm compound for 0, 2, 4, and 6 h at pH 8.0 as illustrated and quantified in Fig. 3a. We observe that 2 h is insufficient to reach complete conversion to hexamer; however, 4–6 h is sufficient. Thus, we used a 6-h incubation time for further evaluation of the compound-PBGS interactions.
FIGURE 3.

Native PAGE evaluation of morphlocks that stabilize the human wild type PBGS hexamer. The upper and lower bands of the gels note the mobility of PBGS octamers and hexamers, respectively (5). Quantitative densitometry results are shown to the right of the gels. Error bars represent the S.D. of three densitometry quantifications; in most cases the errors do not exceed the size of the data point. a, shown are time courses for hexamer stabilization by ML-3A9 and ML-3H2. Human PBGS was incubated at pH 8.0 with either DMSO or 2 mm inhibitor in DMSO for the indicated times at 37 °C. b, dose-response evaluation of hexamer stabilization by ML-3A9 (○, dotted line) and ML-3H2 (■, solid line) at pH 8.0 are shown. Human PBGS was incubated with the inhibitors at the indicated concentrations for 6 h at 37 °C. Data for ML-3A9 fit best to a hyperbolic binding equation, and data for ML-3H2 fit best to the Hill equation. c, dose-response evaluations of hexamer stabilization by ML-3A9 and ML-3H2 at pH 7.35 (top row) and 9.05 (bottom row) are shown. Samples were prepared, and results are quantified and described as for b. d, shown are dose-response evaluations of hexamer stabilization by ML-3A9 and ML-3H2 in the presence of the PBGS substrate ALA. Samples were prepared as for b but also included ALA at a final concentration of 1 mm. The data for both compounds fit best to the Hill equation.
A dose-response analysis was carried out at pH 8.0 and a PBGS concentration of 0.3 mg/ml for ML-3A9 and ML-3H2 using the native PAGE gel shift assay after a 6-h incubation time with compound concentrations of 30 μm, 100 μm, 300 μm, 1 mm, and 2 mm (Fig. 3b). No bands indicative of species smaller than hexamer (e.g. dimer) were present. The dose-response gel shift results for ML-3A9 fit to a hyperbolic equation from which we estimate the inhibitor concentration at which the protein is 50% hexamer (K0.5) to be 140 ± 10 μm. Interestingly, the conversion of octamers to hexamers induced by ML-3H2 fits better to the Hill equation, from which we extract a K0.5 of 120 ± 10 μm and a Hill coefficient of 1.7 ± 0.1. These K0.5 values are significantly higher than we expect to see in an enzyme inhibition assay, which is carried out at significantly lower protein concentration, as observed previously (4).
We have previously established that the hexameric assembly of wild type human PBGS is rare at pH 7 but becomes a substantial part of the equilibrium ensemble at higher pH and provides a structural basis for reduced activity at basic pH (see Fig. 1d) (1). This predicts that the apparent K0.5 as analyzed by the gel shift dose-response analysis will also be pH-dependent. The dose response by gel shift evaluation was, therefore, repeated at pH 7.35 and 9.05 for both compounds (Fig. 3c). At pH 7.35, the dose response of ML-3A9 was hyperbolic, yielding a K0.5 value of 260 ± 50 μm, whereas the dose response of ML-3H2 again fit better to the Hill equation with a K0.5 of 200 ± 30 μm and a Hill coefficient of 2.5 ± 0.7. At pH 9.05, the analysis became more complicated, as the protein is ∼50% hexamer in the absence of compound. As with the lower pH data, the dose response of ML-3A9 was hyperbolic, yielding a K0.5 value of 770 ± 450 μm, whereas the dose response of ML-3H2 fit to the Hill equation with a K0.5 of 100 ± 10 μm and a Hill coefficient of 3.7 ± 1.7. Note that the highest concentration of ML-3H2 shown on the gel was not included in the fitting analysis, as this condition induced formation of higher order assemblies of unknown structure. Although all of the data are generally well described by the fit equations, the error relative to the absolute value of the derived parameters is large. As such, the reported K0.5 values should be considered estimates. A more clear indication of the pH-dependent phenomenon derives from the apparent end point of each of the fits included in Figs. 3, b and c. At the pH values 7.35, 8.0, and 9.05, these endpoints with ML-3A9 are, respectively, 71 ± 4, 83 ± 2, and 100 ± 10% hexamer for ML-3A9. For ML-3H2, these respective values are 61 ± 4, 95 ± 2, and 97 ± 2% hexamer. In general these data indicate that trapping the hexameric assembly is more facile at the high pH values, as was predicted by the higher equilibrium concentration of hexamer at basic pH in the absence of compound. We have also varied the pH of the incubation while holding each compound at its apparent K0.5 at pH 8.0 and observed the expected increase in the amount of hexamer present as a function of pH (not shown).
We note that prior studies consistently demonstrate that incubation of PBGS (from many sources) with the substrate ALA serves to stabilize the octameric assembly (4, 13). Substrate addition promotes a closed conformation of the active site lid, which in turn stabilizes a subunit interaction that is found in the octamer and not the hexamer. Thus, the opposing effects of the substrate on hexamer stabilization by ML-3A9 and ML-3H2 were examined by native PAGE. The dose-response experiment at pH 8.0 (shown in Fig. 3b) was repeated with the inclusion of 1 mm ALA in the samples. As shown in Fig. 3d, the octamer-stabilizing effect of ALA impairs the hexamer-stabilizing ability of both compounds, although the magnitude of this effect differs. In the presence of ALA, the titration curves for both compounds fit poorly to a hyperbolic function, and the data for both were fit to the Hill equation (see Fig. 3d). For ML-3A9, the fit yielded a K0.5 of 190 ± 10 μm and a Hill coefficient of 2.7 ± 0.2. The data for ML-3H2 (for which only three of the gel lanes presented detectable hexamer) yielded a K0.5 of 1150 ± 20 μm and a Hill coefficient of 2.2 ± 0.1.
Hexamer Stabilization of Human PBGS Porphyria-associated Variants by ML-3A9 and ML-3H2
We propose that chemical entities that stabilize the hexameric assembly of human PBGS can potentiate the symptoms of ALAD porphyria. To further test this hypothesis, we evaluated ML-3A9 and ML-3H2 with two porphyria-associated human PBGS variants, E89K and A274T. Although the wild type protein purifies as >95% octamer, the porphyria-associated variants, E89K and A274T, are associated with an increased percentage of hexamer (3). This suggests that, relative to wild type, these variants accumulate more easily in the inactive hexameric form and would be more sensitive to hexamer stabilizing inhibition. The percentages of octamer in the E89K and A274T starting samples used in this study were 54 and 78%, respectively. Dose-response evaluations for ML-3A9 and ML-3H2 (Fig. 4, a and b) were set up under conditions identical to the wild type pH 8.0 gels in Fig. 3b. Consistent with our hypothesis, the K0.5 values for both compounds are lower for the variants relative to the wild type. The dose-response curve for ML-3A9 fit to a hyperbolic equation for both variants, yielding K0.5 values of 90 ± 20 and 80 ± 20 μm for E89K and A274T, respectively. As we had observed for the wild type protein, the dose response for ML-3H2 fit to the Hill equation for both variants, yielding for E89K a K0.5 of 70 ± 10 μm and Hill coefficient of 3.1 ± 0.7 and for A274T a K0.5 of 80 ± 10 μm and Hill coefficient of 3.1 ± 0.1.
FIGURE 4.

Native PAGE evaluation of morphlocks on porphyria-associated human PBGS variants E89K and A274T, which are known to stabilize the human wild type PBGS hexamer. The upper and lower bands of the gels note the mobility of PBGS octamers and hexamers, respectively (5). a, dose-response evaluations of hexamer stabilization of the E89K variant by ML-3A9 (○, dotted line) and ML-3H2 (■, solid line) at pH 8.0 are shown. The E89K variant was incubated with the inhibitors at the indicated concentrations for 6 h at 37 °C. Gels are shown to the left of the figure, and quantification by densitometry is shown to the right. Error bars representing the S.D. of three quantifications are shown where they exceed the size of the data point. Data for ML-3A9 fit best to a hyperbolic binding equation, and data for ML-3H2 fit best to the Hill equation. b, dose-response evaluations of hexamer stabilization of the A274T variant by ML-3A9 (○, dotted line) and ML-3H2 (■, solid line) at pH 8.0 are shown. Samples were prepared, and results are quantified and presented as for a.
Kinetic Evaluation of ML-3A9 and ML-3H2 as Inhibitors of Human PBGS
Stabilization of the low activity hexamer as demonstrated by the gel shift analyses is predicted to correlate with inhibition of human PBGS. Accordingly, ML-3A9 and ML-3H2 were evaluated as inhibitors of wild type human PBGS activity at a final protein concentration of 10 μg/ml and a preincubation time of 6 h. The dose-response curves in Fig. 5 are fitted to a simple hyperbolic decay and correspond to IC50 values of 58 ± 6 and 10 ± 1 μm for ML-3A9 and ML-3H2, respectively. Based on time course and pH studies of the gel shift phenomenon as shown in Fig. 3, the apparent IC50 values are predicted to be lower than observed if a longer protein-inhibitor preincubation time or a higher pH for the preincubation and assay had been used. Correspondingly, a shorter preincubation time and/or low incubation or assay pH would be expected to result in a larger apparent IC50 value. Thus, these IC50 values are a relative measure of how well each of the active compounds draws the human PBGS equilibrium toward the hexamer under assay conditions. As it is established that substrate serves to stabilize the octameric assembly (Fig. 3d and Refs. 4 and 13), the quaternary structure equilibrium within the assay mixture is a result of competing forces to stabilize the inactive hexamer by the small molecule inhibitors and to stabilize the octamer by substrate.
FIGURE 5.

Inhibition of PBGS catalytic activity by ML-3A9 and ML-3H2. Dose-response inhibition curves are illustrated for human PBGS, which was preincubated with ML-3A9 (○, dotted line) or ML-3H2 (■, solid line) for 6 h before the addition of substrate.
Our ability to inhibit PBGS activity by trapping the inactive hexameric assembly reinforces our prior deduction that the hexameric assembly of human PBGS is a native assembly state in dynamic equilibrium with the octameric assembly (1). The behavior of the hexamer-trapping inhibitors, as shown by the pH dependence in native PAGE gel mobility analyses, further validates our hypothesis that the pH dependence of the oligomeric equilibrium is responsible for diminished activity of human PBGS at basic pH (Fig. 1d).
Evidence That the Discovered Morphlocks Bind Specifically to PBGS Hexamers
To establish that the inhibitors promote hexamer formation by binding preferentially to and stabilizing the hexamer, samples treated with both inhibitors were analyzed by native PAGE gels stained for PBGS activity as detailed in supplemental Fig. 2. We have previously demonstrated that the hexameric component of PBGS (in the absence of hexamer-stabilizing inhibitors) will stain for PBGS activity because the substrate-mediated conversion to the active octamer occurs within the gel matrix (4, 11). The absence of PBGS activity in the hexamer bands in the samples treated with ML-3A9 and ML-3H2 demonstrates that these compounds bind specifically to the hexamer, remain bound during the electrophoresis, and inhibit the in-gel substrate-mediated hexamer to octamer conversion.
Native PAGE Analyses of the Effects of Clioquinol and Iodoquinol on the Quaternary Structure Equilibrium of PBGS
The amebicidal clioquinol (Fig. 6a) and iodoquinol (Fig. 6b) are structural analogs of ML-3H2 and were evaluated to determine whether human PBGS hexamer stabilization is an off-target effect of these drugs. Dose-response analyses for both of these compounds versus wild type human PBGS and the porphyria-associated variants E89K and A274T were carried out at pH 8.0 (Fig. 6c) under the same conditions used for ML-3A9 and ML-3H2. Both compounds induced dose-dependent hexamer formation that fit to a simple hyperbolic equation (Equation 1) for wild type PBGS and both porphyria-associated variants, with more potent effects observed for the variants. Clioquinol and Iodoquinol induced hexamer formation in wild type human PBGS with K0.5 values of 70 ± 40 and 100 ± 30 μm, in E89K PBGS with K0.5 values of 30 ± 10 and 40 ± 10 μm, and in A274T PBGS with K0.5 values of 40 ± 30 and 70 ± 30 μm, respectively. These data support the hypothesis that inhibition of human PBGS is an off-target side effect of these drugs and may contribute to clinically observed side effects.
FIGURE 6.

Native PAGE analysis of the clinical drugs clioquinol and iodoquinol with wild type human PBGS and the porphyria-associated variants, E89K and A274T. The chemical structures and formulae of clioquinol and iodoquinol are presented in panels a and b, respectively. Panel c illustrates the dose-response of hexamer stabilization by clioquinol (▴, dotted line) and iodoquinol (♦, solid line) of wild type PBGS (top) and the variants E89K (middle) and A274T (bottom). PBGS (0.3 mg/ml) was incubated with the inhibitors at the indicated concentrations for 6 h at 37 °C before resolution on 12.5% acrylamide native PAGE. Gels are shown to the left of the figure, and quantification by densitometry is shown to the right. Error bars representing the S.D. of three quantifications are shown where they exceed the size of the data point. All data fit to hyperbolic binding equations.
Comparative Analyses of Compounds and Docking Sites in Human Versus Pea PBGS
The same set of ∼111,000 structures that were docked against the human PBGS crystal structure were previously docked against the analogous region of a pea PBGS homology model. From this we discovered a hexamer-stabilizing compound that shifted the quaternary structure equilibrium of pea PBGS, which we called morphlock-1 and whose chemical identity is 2-oxo-1,2-dihydro-benzo(cd)indole-6-sulfonic acid[2-hydroxy-2-(4-nitro-phenyl)-ethyl]-amide (4). It is important to note that only 3% of the top 100 compounds ranked by the GLIDE scoring function are common between docking of these ∼111,000 structures to the pea versus the human PBGS (4). Furthermore, neither ML-3A9 nor ML-3H2 was ranked by the GLIDE scoring function in the top 1000 compounds docked to the pea PBGS, and morphlock-1 was not ranked in the top 1000 compounds docked to human PBGS. Herein we provide a comparison among the docked poses of the structures of ML-3A9, ML-3H2, and morphlock-1, which are all structurally dissimilar. There is some spatial overlap between the docked poses of ML-3H2 and morphlock-1, whereas neither of these compounds overlaps the docked pose of ML-3A9 (Fig. 7, a and b). A sequence alignment of residues within the docking box used for pea and human PBGS shows that there is significant sequence variation (Fig. 7c), particularly with regard to residues that make contact with each of the compounds as docked, thus providing a rationale for why each compound is effective only for PBGS from the targeted species. For a related application, we have considered sequence variations between human PBGS and that of human pathogens within the residues that compose the morphlock docking box. This variation predicts species-selective inhibition of heme biosynthesis by hexamer trapping inhibitors as an approach to development of antimicrobial agents (4).
FIGURE 7.

Comparison of the predicted binding sites for ML-3A9, ML-3H2, and morphlock-1. a, a ribbon diagram of the A, B, and C subunits of hexameric human PBGS (PDB code 1PV8) is shown in gray, and the subunits are labeled A, B, and C, respectively. A purple box indicates the search region that was used for docking. The molecule positions and orientations are illustrated as predicted by docking and are indicated by stick representations of ML-3A9 (orange), ML-3H2 (green), and morphlock-1 (magenta). Morphlock-1 was positioned by overlapping the hexameric assemblies of pea PBGS on human PBGS and transferring the docked location and orientation of morphlock-1. b, a close-up view of a in the same orientation is provided; in this case the ribbons are colored according to subunit (A in blue, B in light blue, and C in gray). c, a sequence alignment of human PBGS and pea PBGS includes only those residues within the targeted docking boxes. The criterion for a “contact” between an atom of the morphlock and an atom of the protein is a ratio of the distance between the two atoms divided by the sum of the van der Waals radii of the two atoms where that ratio does not exceed 1.30. The sequence alignment is colored (as shown in the key) to indicate contacts between the morphlock and its targeted PBGS and sequence conservation between human and pea PBGS for residues that are involved in side-chain contacts.
Evaluation of in Silico Docking as a Library Refinement Tool
In silico methods are becoming increasingly utilized in the identification of protein ligands such as drugs. The percentage of lead compounds identified from a library that is prescreened by in silico docking is estimated to increase some 10-fold compared with a high throughput only screen (14). In the case of this study, in silico docking was used as part of the process used to select a reasonable number of compounds for purchase and in vitro analysis from a library of 110,000 structures derived from nearly 70,000 commercially available compounds. The goal of the docking was to enrich the compounds selected for purchase with molecules likely to inhibit PBGS by the hypothesized hexamer-stabilizing mechanism. In vitro gel shift evaluation of 77 purchased compounds identified 12 as hits. Thus, in silico docking appears to provide an efficient means to target oligomer-specific sites in alternate quaternary structure assemblies of proteins.
A question that arises is this: Are we more likely to find hits among compounds selected from the docking than we would from a set of compounds purchased randomly? Although we do not have a random set of compounds, we have a set of 76 compounds that had previously been purchased as potential hexamer-stabilizing inhibitors of pea PBGS in a screen of the same commercial library to the analogous hexamer-specific surface cavity. The phylogenetic variation in the amino acids that make up this surface cavity is substantial when comparing human PBGS to pea PBGS (see Fig. 7c and Ref. 4). We evaluated the ability of the compounds selected to stabilize the pea PBGS hexamer for their ability to stabilize the human hexamer using the 6-h preincubation protocol described above. With one exception, compounds selected for pea PBGS hexamer stabilization did not perturb the human PBGS quaternary structure equilibrium toward the hexamer greater than a few percent (not shown). The single exception revealed that one of the compounds purchased for in vitro testing with pea PBGS was chemically identical to a compound also purchased for in vitro testing with human PBGS. This compound (coincidentally, the impure ML-3F2) was cataloged by the vendor under two different catalogue numbers with a difference of 1 dalton in molecular mass. The results of this important control suggest that we are, indeed, more likely to find compounds that stabilize the human PBGS hexamer among the compounds selected by docking to the human PBGS hexamer than to the pea PBGS hexamer.
DISCUSSION
Human PBGS Quaternary Structure Equilibrium and Human Health
Human PBGS exists in an equilibrium of alternate quaternary structure assemblies, as illustrated in Fig. 1a (1, 3, 5, 10, 13). The physiologic relevance of this equilibrium is established by a correlation between human PBGS variants and the disease ALAD porphyria (3). All ALAD porphyria-associated variants shift the distribution of PBGS quaternary structural forms toward the lower activity hexamer, providing a structural basis for why PBGS activity is reduced in patients carrying these alleles. Herein we provide evidence that small molecules can stabilize the low activity hexameric assembly of human PBGS using both wild type protein and ALAD porphyria-associated variants. We posit that such small molecules will inhibit PBGS activity and potentiate disease states associated with diminished PBGS activity. Although ALAD porphyria is rare, diminished PBGS activity also results from the much more common condition of lead poisoning. In both disease states symptoms are related to the toxic effects of increased concentrations of the PBGS substrate ALA, which accumulates as a result of the lowered PBGS activity.
The structural similarity between ALA and the neurotransmitter 4-aminobutyric acid is related to the neurologic sequelae of porphyrias and lead poisoning. The fact that patients experience debilitating symptoms only sporadically throughout life is one poorly understood aspect of porphyria. It is possible that exposure to compounds like the morphlocks discovered herein could contribute to the sporadic nature of the disease. It is also possible that established drugs that act as morphlocks toward human PBGS could raise metabolic ALA levels and cause neurologic side effects. Thus, it is intriguing that the discovered morphlock ML-3H2 has a history in drug development (15). ML-3H2 was synthesized in the 1950s in an effort to improve the effectiveness and solubility of the halogenated quinolinols, which are a principal class of amebicidal agents. An iodide at position 7 of an existing amebicidal agent was replaced with a diethylaminomethyl group to produce ML-3H2. This modification did not produce a more potent amebicidal agent, and the original iodinated compound and a diiodinated derivative remain in use under the names clioquinol and iodoquinol (Fig. 6).
Most intriguing is that side effects associated with clioquinol and iodoquinol include deleterious effects on the nervous system (6). Our demonstration that both drugs do indeed stabilize the low activity hexameric assembly of human PBGS, as depicted in Fig. 6c, supports a relationship between the side effects of these drugs and inhibition of human PBGS. Both compounds induced dose-dependent hexamer formation and were more potent against the porphyria-associated variants than wild type human PBGS. The concentration of clioquinol in the plasma of patients using topical preparations of the drug has been measured at 1–2 μm (16), which is substantially lower than the experimentally determined K0.5 values derived from our native PAGE assay (40–100 μm). However, the concentration of PBGS used in the gel shift assays (0.3 mg/ml) is also much higher than the 1–5 μg/ml that can be estimated for erythrocytes (17, 18), where PBGS expression is enhanced over the normal housekeeping levels (19). We have demonstrated that increased protein concentration causes an inflated apparent K0.5 for inhibitors that function by stabilizing PBGS hexamers (4). If it is determined that clioquinol is able to induce PBGS hexamer formation (and concomitant inhibition of PBGS activity) in vivo, this would constitute an unprecedented explanation for a drug side effect. It is possible that other known drugs, toxins, or environmental contaminants could have a similar hexamer-stabilizing inhibitory effect on human PBGS.
Significant Differences between Plant and Human PBGS
The current study follows the discovery of hexamer trapping agents for plant PBGS, but the required time for in vitro hexamer stabilization was found to be significantly longer for human PBGS. This difference stems from a phylogenetic variation in the use of an allosteric magnesium ion, used in plant PBGS, which binds at a subunit interface that is present in the octamer but not present in the hexamer (5). Magnesium binding to this site stabilizes the octamer and increases activity by increasing the mole fraction of octamer in the quaternary structure equilibrium. In contrast, human PBGS does not contain the amino acids that compose the allosteric magnesium binding site. Instead, there is a guanidinium group of an arginine residue that is spatially equivalent to the magnesium of plant PBGS and that we have shown acts to stabilize the human PBGS octamer (3, 10). Presumably because the allosteric magnesium can dissociate (Kd ∼ 2 mm) and the guanidinium group cannot, the interchange of quaternary structure assemblies of human PBGS is much less facile than for plant PBGS. We have shown that human PBGS assemblies are metastable and interconvert only under specific circumstances, with relatively long half-times on the order of 1–3 h (10, 13). The slow interchange of quaternary structure assemblies for human PBGS dictated considerably longer preincubation times for the in vitro analysis of putative morphlocks relative to pea PBGS.
The Morpheein Model of Allosteric Regulation
In the current understanding of protein dynamics, proteins are viewed as existing in an ensemble of conformations (20–26). Within this context, quaternary structure dynamics as a fundamental basis for allosteric control of function is an extension of what we already understand about how protein motion is related to function. Although the behavior of PBGS was initially unexpected, it is now well established that the octamer can come apart, the dissociated form can change conformation, and the altered conformation can reassociate to a functionally distinct hexamer (or vise versa) (1, 4, 5, 10, 13). In the case of PBGS this equilibrium forms the basis for allosteric regulation, and thus, we introduced the morpheein model of allostery (27, 28) whose kinetic characteristics are distinct from the classic models of allostery (29–31). Regardless of the model put forward, the common theme in allostery is the binding of a ligand at one location of a protein (e.g. not the active site) affecting the behavior of the protein at another location (e.g. the active site). For plant and many microbial PBGS, the magnesium ligand that binds at an octamer-stabilizing site distinct from the active site, defines these proteins as allosteric. For human PBGS, the current study, which shows for the first time that small molecules can bind at a hexamer-stabilizing site distinct from the active site, defines human PBGS as allosteric.
Although PBGS is the first protein that has been unequivocally established to exist as a dynamic, fully reversible equilibrium of oligomers that can come apart, change shape, and reassemble differently, there are other proteins whose published characteristics are consistent with using the morpheein mechanism of allostery. We have suggested morpheein as a general term to describe homo-oligomeric proteins that can dissociate, change conformation, and reassociate into structurally and functionally distinct assemblies (4, 27, 28). The implications of identifying a protein as a morpheein are significant. Small molecule stabilization of alternate oligomers of proteins that exist in an equilibrium of quaternary structure assemblies (morpheein forms) provides a largely untapped resource for discovery of new drugs (4) and for understanding off-target side effects. A morpheein-trapping mechanism is distinct from a mechanism that simply interferes with the process of assembly. In the case of PBGS and perhaps also for other morpheeins, the targeted oligomer-stabilizing surface cavity has a large volume and structurally complex protein components, allowing ligands to bind in a variety of locations. Thus, unlike for an enzyme active site, one can consider diverse structures as ligands. We have previously suggested the proteins human immunodeficiency virus integrase, tumor necrosis factor α, and mammalian ribonucleotide reductase as morpheein drug targets (4); for each of these proteins there is evidence of functionally distinct alternate quaternary structure assemblies (32–34). The results presented herein suggest that morpheeins be considered as a possible explanation for off-target side effects of drugs in clinical use.
Acknowledgment
We acknowledge the contributions of Cynthia Myers of the Fox Chase Cancer Center Organic Synthesis Facility for the chemical and spectral analyses reported.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 ES003654 (to E. K. J.), R21 AI063324 (to E. K. J.), P30 CA006927 (to the Fox Chase Cancer Center), and T32 CA009035 (to the Institute for Cancer Research).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1 and 2 and Tables 1 and 2.
- PBGS
- porphobilinogen synthase
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- ALA
- 5-aminolevulinic acid.
REFERENCES
- 1.Selwood T., Tang L., Lawrence S. H., Anokhina Y., Jaffe E. K. (2008) Biochemistry 47, 3245–3257 [DOI] [PubMed] [Google Scholar]
- 2.Maruno M., Furuyama K., Akagi R., Horie Y., Meguro K., Garbaczewski L., Chiorazzi N., Doss M. O., Hassoun A., Mercelis R., Verstraeten L., Harper P., Floderus Y., Thunell S., Sassa S. (2001) Blood 97, 2972–2978 [DOI] [PubMed] [Google Scholar]
- 3.Jaffe E. K., Stith L. (2007) Am. J. Hum. Genet. 80, 329–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lawrence S. H., Ramirez U. D., Tang L., Fazliyez F., Kundrat L., Markham G. D., Jaffe E. K. (2008) Chem. Biol. 15, 586–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Breinig S., Kervinen J., Stith L., Wasson A. S., Fairman R., Wlodawer A., Zdanov A., Jaffe E. K. (2003) Nat. Struct. Biol. 10, 757–763 [DOI] [PubMed] [Google Scholar]
- 6.Kauffman R. E., Banner W., Jr., Blumer J. L., Gorman R. L., Lambert G. H., Snodgrass W. (1990) Pediatrics 86, 797–7982146587 [Google Scholar]
- 7.Halgren T. A. (1999) J. Comput. Chem. 20, 730–748 [DOI] [PubMed] [Google Scholar]
- 8.Halgren T. A., Murphy R. B., Friesner R. A., Beard H. S., Frye L. L., Pollard W. T., Banks J. L. (2004) J. Med. Chem. 47, 1750–1759 [DOI] [PubMed] [Google Scholar]
- 9.Jorgensen W. L., Duffy E. M. (2000) Bioorg. Med. Chem. Lett. 10, 1155–1158 [DOI] [PubMed] [Google Scholar]
- 10.Tang L., Breinig S., Stith L., Mischel A., Tannir J., Kokona B., Fairman R., Jaffe E. K. (2006) J. Biol. Chem. 281, 6682–6690 [DOI] [PubMed] [Google Scholar]
- 11.Jaffe E. K., Ali S., Mitchell L. W., Taylor K. M., Volin M., Markham G. D. (1995) Biochemistry 34, 244–251 [DOI] [PubMed] [Google Scholar]
- 12.Abramoff M. D., Magelhaes P. J., Ram S. J. (2004) Biophotonics International 11, 36–42 [Google Scholar]
- 13.Tang L., Stith L., Jaffe E. K. (2005) J. Biol. Chem. 280, 15786–15793 [DOI] [PubMed] [Google Scholar]
- 14.Shekhar C. (2008) Chem. Biol. 15, 413–414 [DOI] [PubMed] [Google Scholar]
- 15.Burckhalter J. H., Edgerton W. H. (1951) J. Am. Chem. Soc. 73, 4837–4839 [Google Scholar]
- 16.Stohs S. J., Ezzedeen F. W., Anderson A. K., Baldwin J. N., Makoid M. C. (1984) J. Invest. Dermatol. 82, 195–198 [DOI] [PubMed] [Google Scholar]
- 17.Anderson P. M., Desnick R. J. (1979) J. Biol. Chem. 254, 6924–6930 [PubMed] [Google Scholar]
- 18.Despaux N., Comoy E., Bohuon C., Boudène C. (1979) Biochimie 61, 1021–1028 [DOI] [PubMed] [Google Scholar]
- 19.Kaya A. H., Plewinska M., Wong D. M., Desnick R. J., Wetmur J. G. (1994) Genomics 19, 242–248 [DOI] [PubMed] [Google Scholar]
- 20.Benkovic S. J., Hammes G. G., Hammes-Schiffer S. (2008) Biochemistry 47, 3317–3321 [DOI] [PubMed] [Google Scholar]
- 21.Goodey N. M., Benkovic S. J. (2008) Nat. Chem. Biol. 4, 474–482 [DOI] [PubMed] [Google Scholar]
- 22.James L. C., Tawfik D. S. (2003) Trends Biochem. Sci. 28, 361–368 [DOI] [PubMed] [Google Scholar]
- 23.Dill K. A., Chan H. S. (1997) Nat. Struct. Mol. Biol. 4, 10–19 [DOI] [PubMed] [Google Scholar]
- 24.Linderstrom-Lang K., Schellman J. A. (1959) in The Enzymes (Boyer P. D., Lardy H., Myrback K. eds) Vol. 1, 2nd Ed., pp. 443–510, Academic Press Inc., New York [Google Scholar]
- 25.Whitten S. T., Kurtz A. J., Pometun M. S., Wand A. J., Hilser V. J. (2006) Biochemistry 45, 10163–10174 [DOI] [PubMed] [Google Scholar]
- 26.Hilser V. J., García-Moreno E. B., Oas T. G., Kapp G., Whitten S. T. (2006) Chem. Rev. 106, 1545–1558 [DOI] [PubMed] [Google Scholar]
- 27.Jaffe E. K. (2005) Trends Biochem. Sci. 30, 490–497 [DOI] [PubMed] [Google Scholar]
- 28.Lawrence S. H., Jaffe E. K. (2008) Biochem. Mol. Biol. Educ. 36, 274–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koshland D. E., Jr., Némethy G., Filmer D. (1966) Biochemistry 5, 365–385 [DOI] [PubMed] [Google Scholar]
- 30.Monod J., Changeux J. P., Jacob F. (1963) J. Mol. Biol. 6, 306–329 [DOI] [PubMed] [Google Scholar]
- 31.Monod J., Wyman J., Changeux J. P. (1965) J. Mol. Biol. 12, 88–118 [DOI] [PubMed] [Google Scholar]
- 32.Hayouka Z., Rosenbluh J., Levin A., Loya S., Lebendiker M., Veprintsev D., Kotler M., Hizi A., Loyter A., Friedler A. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 8316–8321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He M. M., Smith A. S., Oslob J. D., Flanagan W. M., Braisted A. C., Whitty A., Cancilla M. T., Wang J., Lugovskoy A. A., Yoburn J. C., Fung A. D., Farrington G., Eldredge J. K., Day E. S., Cruz L. A., Cachero T. G., Miller S. K., Friedman J. E., Choong I. C., Cunningham B. C. (2005) Science 310, 1022–1025 [DOI] [PubMed] [Google Scholar]
- 34.Kashlan O. B., Cooperman B. S. (2003) Biochemistry 42, 1696–1706 [DOI] [PubMed] [Google Scholar]