

Abstract
Expression of B7 costimulatory molecules represents an important compartment of immune response of epithelial cells following microbial infection. We reported here that the protozoan parasite Cryptosporidium parvum induced B7-H1 expression in cultured human cholangiocytes. Induced expression of B7-H1 was identified in cells after exposure to infective C. parvum parasite or parasite lysate. Interestingly, microRNA-513 (miR-513) level was reduced in cells after exposure to C. parvum, resulting in a relief of 3′-untranslated region-mediated translational suppression of B7-H1. Overexpression of miR-513 through transfection of miR-513 precursor inhibited C. parvum-induced B7-H1 protein expression. Moreover, enhanced apoptotic cell death was identified in activated human T cells following co-culture with C. parvum-infected cholangiocytes. The apoptosis of activated T cells was partially blocked by a neutralizing antibody to B7-H1 or transfection of cholangiocytes with miR-513 precursor. These data suggest a role of miR-513 in regulating B7-H1 expression by cholangiocytes in response to C. parvum infection.
Keywords: MicroRNAs, Cryptosporidium parvum, B7-H1, Epithelium, biliary, Immune response, Parasites, Apoptosis
INTRODUCTION
Epithelial cells are central participants in mucosal immunity against microbial infection in the gastrointestinal tract. The epithelial cells can produce antimicrobial molecules, proinflammatory cytokines and chemokines in response to microbial infection. Release of cytokines and chemokines will recruit other immune cells to the epithelium, resulting in enhanced adaptive anti-microbial immune response [1]. This host immune response is finely controlled and reflects a delicate balance between effector functions and their potential to cause unwanted damage to healthy tissues [1–2]. Disturbance of this balance will lead to devastating immunopathology, resulting in either an over-reacted immunoreaction or insufficient immunity to clear pathogenic agents [1–4].
The protozoan parasite, Cryptosporidium spp. is a causative agent of human gastrointestinal disease worldwide. Humans are infected by ingesting Cryptosporidium spp. oocysts. Once ingested, oocysts excyst in the gastrointestinal tract and release infective sporozoites. Mediated by specific ligands on the sporozoite surface and receptors on the host cell [5], the sporozoite attaches to the apical membrane of the host epithelial cell and forms a vacuole in which the organism remains intracellular but extracytoplasmic [5]. The intracellular sporozoite then matures and undergoes further development. C. parvum, the most common species in humans, is categorized as a “minimally invasive” mucosal pathogen. Epithelial immunity is an important compartment of host immune response against C. parvum infection [6,7].
MicroRNAs are a newly identified class of endogenous small regulatory RNAs that mediate either mRNA cleavage or translational suppression resulting in gene silencing [8]. Over 500 miRNAs have been identified in humans and are postulated to control 20–30% of human genes [8,9]. MicroRNAs can be envisioned as a mechanism to fine-tune the cellular responses to the environment and may be regulators of host anti-microbial immune response. Indeed, miRNAs have been implicated in the regulation of Toll-like receptor signaling, viral immune escape and anti-viral defense [10,11]. MicroRNA-155, which is induced during the macrophage inflammatory response, is hypothesized to participate in regulation of inflammation [12].
Cholangiocytes, epithelial cells lining the biliary tree, actively interact with other cells (e.g., T-cells and dendritic cells) in the liver via expression/release of adhesion molecules or immune mediators [3,4,13]. Recent studies indicate that cholangiocytes express several B7 costimulatory molecules in response to inflammatory stimuli [14]. Expression of B7-H1 (CD274 or PD-L1), a newly identified B7 member involved in regulation of cell-mediated immune responses [15,16], may help regulate the cholangiocyte response to ensure a controlled and balanced inflammatory reaction in the portal region of the liver [14,16,17]. We recently demonstrated that pro-inflammatory cytokines, such as IFN-γ, induces cholangiocyte B7-H1 expression. A cellular miRNA, miR-513, inhibits B7-H1 gene expression and relief of this miR-513-mediated gene suppression is involved in IFN-γ-induced B7-H1 expression in human cholangiocytes [18].
Infection of human cholangiocytes by C. parvum in vitro mimics parasitic apical invasion and epithelial innate immune responses in vivo [5,7]. In work described here, we show that C. parvum infection of cultured human cholangiocytes induces B7-H1 expression. C. parvum decreases miR-513 expression, a process that confers C. parvum-induced upregulation of B7-H1 in cholangiocytes. Consequently, miR-513-associated B7-H1 expression on cholangiocyte surface modulates apoptotic cell death of activated T cells in vitro. Thus, a novel miR-513-mediated regulatory pathway for B7-H1 expression induced by C. parvum has been identified in cholangiocytes, a process that may be relevant to biliary immune response associated with C. parvum infection.
METHODS
C. parvum and Cholangiocytes
C. parvum oocysts of the Iowa strain were purchased from a commercial source (Bunch Grass Farm, ID). H69 cells are SV40 transformed normal human cholangiocytes originally derived from normal liver harvested for transplant and have been extensively characterized [7,19]. Jurkat cells were purchased from the ATCC.
In vitro Infection Model
An in vitro model of human biliary cryptosporidiosis using H69 cells was employed [18,19]. Before infecting cells, oocysts were treated with 1% sodium hypochlorite on ice for 20 min followed by extensive washing with DMEM-F12 medium. Oocysts were then added to the cell culture to release sporozoites to infect cells [18]. Whole C. parvum lysates were obtained as previously described [20]. Infection was done in a culture medium (DMEM-F12 with 100 U/ml penicillin and 100 μg/ml streptomycin) containing viable C. parvum oocysts (oocysts with host cells in a 10:1 ratio) or lysate from the same amount of oocysts.
Immunofluorescent Microscopy
Cells were fixed and permeabilized with ice cold 100% acetone for 20 min. Fixed cells were then incubated with a monoclonal B7-H1 antibody (clone 5H1-A3) [15,16] overnight at 4°C followed by Cy3-conjugated anti-mouse secondary antibody (Invitrogen). The slides were then counterstained with 4′,6-diamidino-2-phenylindole (DAPI, 5 μM) and assessed under a Zeiss LSM510 laser-scanning microscope.
Western Blot
Whole cell lysates were obtained using the M-PER Mammalian Protein Extraction Reagent (Pierce) plus several protease inhibitors (1 mM PMSF; 10 μg/ml leupeptin, 2μg/ml pepstatin). Cell lysates were then loaded (40 μg/lane) in a 4–12% SDS-PAGE gel to separate proteins and transferred to nitrocellulose membrane. Antibodies to B7-H1 (clone 5H1-A3) and β-actin (Sigma-Aldrich) were used for detection. Densitometric levels of B7-H1 signals were quantified and expressed as a ratio to actin.
Real-Time PCR
Total RNAs were extracted using Trizol reagent (Ambion) and PCR reactions were carried out in triplicate using the SYBR Green PCR master mix (Applied Biosystems) [18]. The primers were: 5′-GGTGCCGACTACAAGCGAAT-3′ (forward) and 5′-GGTGACTGGATCCACAACCAA-3′ (reverse) for human B7-H1; 5′-TGTGGAGACCATCAAGGAAGA-3′ (forward) and 5′-CGACAGTTCAGCCATCACTTG-3′ (reverse) for human IFN-γ; 5′-GCTGCACTTTGGAGTGATCG-3′ (forward) and 5′-GTTTGCTACAACATGGGCTACAG-3′ (reverse) forhuman TNF-α, and 5′-TGCACCACCAACTGCTTAGC-3′ (forward) and 5′-GGCATGGACTGTGGTCATGAG-3′ (reverse) for human GAPDH. The Ct values were analyzed using the comparative Ct (ΔΔCt) method. The amount of target was obtained by normalizing to the endogenous reference (GAPDH) and relative to control (untreated cell) [21].
For analysis of miR-513, total RNA was isolated from cells with the mirVana™ miRNA Isolation kit (Ambion). Comparative real-time PCR was performed by using the Taqman Universal PCR Master Mix (Applied Biosystems). Specific primers and probes for mature miR-513 and snRNA RNU6B were obtained from Applied Biosystems. All reactions were run in triplicate. The amount of miR-513 was obtained by normalizing to snRNA RNU6B and relative to control (untreated cell) as previously reported [18,22,23].
Bead-based Multiplex Sandwich Immunoassays
A bead-based multiplex sandwich immunoassay, read with a Luminex 200 system (Luminex), was used to measure the concentrationsof TNF-α, IFN-γ, and miR-513 in H69 cells. For measurement of TNF-α and IFN-γ, total cell lysates were used and concentrations were measured by using the antibody-conjugated beads directed against TNF-α or IFN-γ (Millipore) as previously reported by others [24]. For measurement of miR-513, total cellular RNAs are isolated using the mirVana™ miRNA Isolation Kit (Ambion). An amount of 0.5 μg of total RNAs was used for Biotin-labeling using the FlexmiR MicroRNA Labeling Kit for miR-513 (Luminex). Signal for miR-513 was recorded and standardized to the standard bead according to the manufacturer’s instructions.
MicroRNA-513 Precursor and Antisense Oligonucleotide
To manipulate cellular function of miR-513, we utilized an antisense approach to inhibit miR-513 function and a precursor transfection approach to increase miR-513 expression in H69 cells. Cells were grown up to 80% confluence and treated with miR-513 antisense 2-methoxy oligonucleotide (Ambion) or the miR-513 precursor (Ambion) using the lipofectamine™ 2000 reagent (Invitrogen). Experiments were typically performed 48 h after transfection.
Luciferase Reporter Constructs and Luciferase Assay
Complementary 34 bp DNA oligonucleotides containing the putative miR-513 target site within the 3′-untranslated region (UTR) of human B7-H1 were synthesized and cloned into the multiple cloning sites of the pMIR-REPORT Luciferase vector (Ambion) [18]. Another pMIR-REPORT Luciferase construct containing mutant 3′-UTR (GTGAC to TGGAC) was generated as a control. We then transfected cholangiocytes with each reporter construct and exposed to C. parvum 48 h after transfection. Luciferase activity was measured 24 h later and normalized to the control β-gal level [18,25].
Co-culture of H69 Cells with T Cells
A co-culture model of H69 and Jurkat cells was utilized as previously reported [18]. Briefly, H69 cells were grown to 70% confluence in 24 well plates and were harvested and washed after exposure to C. parvum in the presence or absence of miR-513 precursor for 48 h. The adherent H69 cells (1 × 105) were then co-cultured with 1×105 Jurkat cells in 96 well plates in the presence of 20 nM TPA (12-o-tetradecanoylphorbol 13-acetate, Sigma-Aldrich). In parallel experiments, neutralizing antibodies to B7-H1 (5 μg/ml) [15,16,26] or Fas (5 μg/ml) [27] were added to the co-culture media. After co-culture for 24 h, non-adherent Jurkat cells were collected from the medium and apoptotic cell death of the Jurkat cells measured by DAPI staining [18,28].
Statistical Analysis
Data were compared using the ANOVA test. p < 0.05 was considered to represent statistical significance.
RESULTS
C. parvum induces B7-H1 expression in cultured human cholangiocytes
We first tested whether C. parvum infection can induce B7-H1 expression in human cholangiocytes using our in vitro model of human biliary cryptosporidiosis [7]. Consistent with our previous results [18], we detected expression of B7-H1 mRNA in H69 cells under control conditions. A time-dependent increase of B7-H1 mRNA was identified in cells after exposure to viable C. parvum oocysts (Fig. 1A). Interestingly, cells incubated with C. parvum lysate for 8 h also showed an increase of B7-H1 mRNA expression (Fig. 1A). Compared with cells exposed to viable C. parvum oocysts for 12 h and 24 h, a greater induction of B7-H1 mRNA was detected in cells after incubation with C. parvum lysate from the same amount of oocysts (Fig. 1A). In contrast, B7-H1 protein was barely detectable in non-infected control cells by Western blot (Fig. 1B) or immunofluorescence (Fig. 1C), suggesting a posttranscriptional suppression of B7-H1 expression as previously reported [18,26]. Accordingly, B7-H1 protein became detectable in cells after exposure to viable C. parvum or C. parvum lysate for 12 h and 24 h as assessed by Western blot (Fig. 1B). By immunoconfocal microscopy, expression of B7-H1 protein was observed not only in the directly infected cells (Fig. 1C) but also in the bystander non-infected cells (Fig. 1C).
FIGURE 1.

C. parvum infection induces B7-H1 expression in cultured cholangiocytes. (A and B) Time-dependent expression of B7-H1 at the message (A) and protein (B) levels in H69 cells following C. parvum infection. H69 cells were exposed to culture medium with viable C. parvum oocysts (oocysts with host cells in a 10:1 ratio) or C. parvum lysate from the same amount of oocysts for up to 24 h followed by real-time PCR and Western blot analyses. PCR reactions were performed in triplicate. A representative Western blot from three independent experiments is shown in B. Actin was blotted as a loading control. (C and D) Expression of B7-H1 expression in H69 cells induced by C. parvum as assessed by immunofluoscent staining. Cells were exposed to viable C. parvum oocysts for 24 h followed by immunoconfocal microscopy for B7-H1. Arrows indicate parasites stained in red. *p < 0.05 vs. the non-infected control; #, p < 0.05, vs. cells exposed to C. parvum oocysts.
C. parvum infection does not induce TNF-α and IFN-γ production in cholangiocytes in vitro
C. parvum-associated expression of TNF-α and IFN-γ was previously demonstrated in vivo in intestinal cryptosporidiosis [29]. To test the potential involvement of TNF-α and IFN-γ in C. parvum-induced B7-H1 expression, we assessed the production of these soluble mediators in H69 cells following C. parvum infection. No alteration of TNF-α mRNA (Fig. 2A) or protein (Fig. 2B) was detected in H69 cells after exposure to viable C. parvum oocysts or lysate. No IFN-γ mRNA and protein was detected in the non-infected H69 cells and no induction of IFN-γ at the message and protein levels was identified in cells after exposure to C. parvum oocysts or lysate (data not shown). In contrast, a significant increase of TNF-α mRNA (Fig. 2A), but not TNF-α protein (Fig. 2B), was detected in cells after stimulation with E. coli lipopolysaccharide (LPS, 1 μg/ml, Sigma-Aldrich), consistent with the previous results [30].
FIGURE 2.
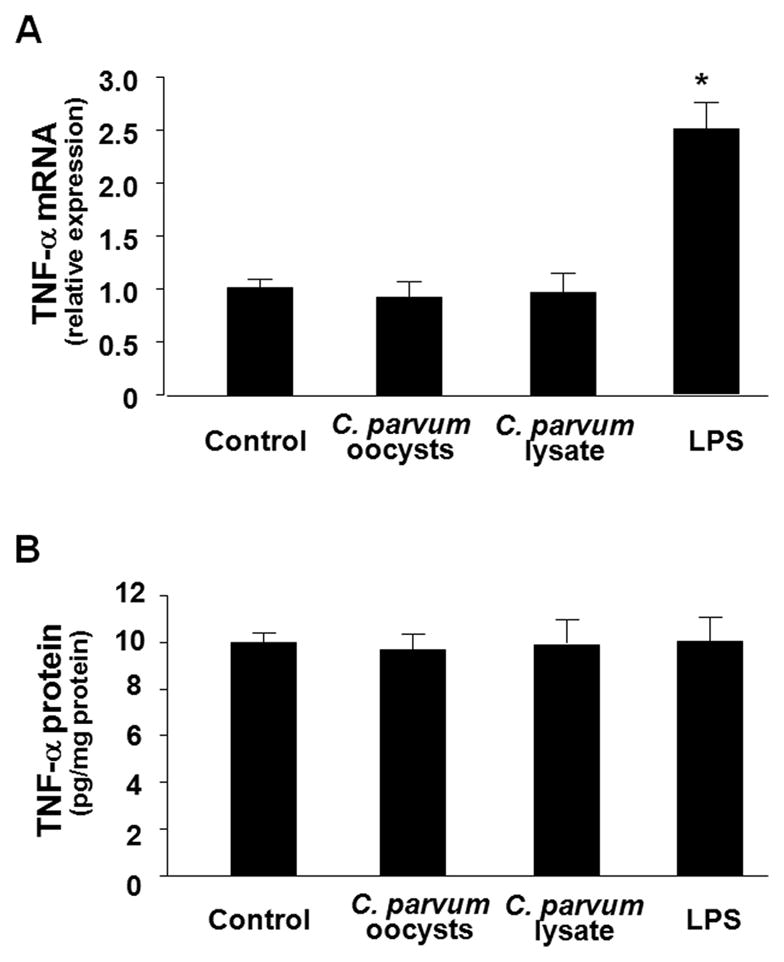
C. parvum infection does not induce TNF-α production in cholangiocytes in vitro. H69 cells were exposed to viable C. parvum oocysts followed by analysis for TNF-α at message level by real-time PCR and at protein level by bead-based multiplex sandwich immunoassay. No increase of TNF-α expression either at the message level (A) or at protein level (B) was detected in H69 cells after exposure to viable C. parvum oocysts or lysate for 8 h or 24 h, respectively. As the positive control, a significant increase of TNF-α mRNAs (A), but not at the protein level (B), was detected in H69 cells after exposure to LPS. * p < 0.05, vs. the non-stimulated control.
C. parvum infection decreases miR-513 expression in cultured human cholangiocytes
We recently demonstrated that a cellular miRNA, miR-513, targets B7-H1 3′-UTR resulting in translational suppression in human cholangiocytes [18]. To test whether cellular expression of miR-513 is altered by C. parvum infection, we exposed H69 cells to viable C. parvum oocysts or lysate and then measured cellular miR-513 levels. As shown in Fig. 3A, H69 cells showed a significant decrease of miR-513 expression after exposure to either viable C. parvum oocysts or lysate as assessed by real-time PCR. Decrease of miR-513 in H69 cells after exposure to viable C. parvum oocysts was further confirmed by using the bead-based Immunoassay for miR-513 (Fig. 3B).
FIGURE 3.

C. parvum infection decreases expression of miR-513 in cholangiocytes. Cells were exposed to C. parvum for 12 h followed by real-time PCR (A) and Luminex FlexmiR Select (B) analyses for miR-513. Experiments were performed in triplicate. *, p < 0.05 ANOVA versus the control.
Decreased miR-513 expression impacts C. parvum-induced B7-H1 protein expression in cholangiocytes
To test whether decreased miR-513 levels are involved in C. parvum-induced B7-H1 protein expression in cholangiocytes, we measured miR-513-mediated translational suppression of B7-H1 in H69 cells following C. parvum infection using a luciferase reporter assay as previously reported [18,25]. A pMIR-REPORT luciferase construct that contains the 3′-UTR of B7-H1 with the putative miR-513 binding site (Fig. 4A) was generated to transfect H69 cells. Transfected cells were then exposed to viable C. parvum oocysts or lysate for 24 h followed by measurement of luciferase activity. Consistent with our previous results [18], a significant decrease of luciferase activity was detected in cells transfected with the B7-H1 3′-UTR construct compared with the empty vector control (Fig. 4B), suggesting endogenous translational repression of the construct with the B7-H1 3′-UTR. In contrast, the pMIR-REPORT luciferase construct that contains the mutant B7-H1 3′-UTR (GTGAC to TGGAC) showed no significant change in luciferase activity, confirming the specificity of the B7-H1 3′-UTR-mediated translational suppression. Importantly, cells exposed to viable C. parvum oocysts or lysate showed a significant increase of B7-H1 3′-UTR-mediated luciferase activity (Fig. 4B), suggesting relief of B7-H1 3′-UTR-mediated translational suppression in cells following C. parvum infection.
FIGURE 4.

Relief of miR-513-mediated B7-H1 translational suppression occurs in cholangiocytes following C. parvum infection. (A) The schematic of B7-H1 mRNA shows a potential binding site in the B7-H1 3′-UTR for miR-513. (B) The complementary miR-513-binding site in the B7-H1 3′-UTR was inserted downstream of a luciferase reporter on the pMIR-Report plasmid. A control plasmid with the mutant 3′-UTR sequence was also generated. (C) H69 cells were transfected with the reporter constructs and were harvested 24 h after transfection. Luciferase activities were measured and normalized to the control β-gal level. A scrambled antisense oligonucleotide and a non-specific precursor were used as controls. These data are representative of three independent experiments. *, p < 0.05, vs. the empty vector; #, p < 0.05, vs. the B7-H1 3′-UTR reporter construct.
To further test the role of miR-513 in C. parvum-induced B7-H1 protein expression in cholangiocytes, we transfected H69 cells with increasing doses of miR-513 precursor for 48 h and then exposed those cells to infective C. parvum oocysts for 24 h followed by Western blot for B7-H1. As shown in Fig. 5A, miR-513 precursor significantly blocked C. parvum-induced B7-H1 protein expression in H69 cells in a dose-dependent manner. In contrast, transfection with a control RNA sequence did not alter B7-H1 expression. Moreover, no significant change in B7-H1 mRNA levels was found in miR-513 precursor-treated cells following C. parvum infection compared with cells treated with the control precursor or the non-treated control cells (Fig. 5B). Coupled with the data demonstrating downregulation of cellular miR-513 levels following C. parvum infection, these data suggest that relief of miR-513-mediated translational repression of B7-H1 is involved in C. parvum-induced B7-H1 protein expression in cholangiocytes.
FIGURE 5.

Decreased miR-513 expression impacts C. parvum-induced B7-H1 protein expression in cholangiocytes. (A) Transfection of miR-513 precursor reduces C. parvum-induced B7-H1 protein expression. H69 cells were transfected with miR-513 precursor or a control non-specific precursor for 48 h, exposed to C. parvum oocysts for 24 h followed and then assayed for B7-H1 by Western blot. A representative Western blot from three independent experiments and densitometric levels of B7-H1 signals are also shown. (B) MicroRNA-513 precursor transfection does not affect C. parvum-induced B7-H1 mRNA expression. H69 cells were transfected with miR-513 precursor or a control non-specific precursor for 48 h and then exposed to viable C. parvum oocysts for 24 h followed by real-time PCR analysis for B7-H1 mRNA. *, p < 0.05, vs. non-infected control cells.
B7-H1 expression on cholangiocytes following C. parvum infection induces apoptotic cell death in activated T cells
B7-H1 ligation has been reported to induce apoptotic cell death in activated T cells [15,26,31]. Therefore, we examined the level of apoptotic cell death in Jurkat cells after co-culture with H69 cells that had or had not been exposed to C. parvum. Because Jurkat cells do not express programmed death receptor-1 (PD-1) [32], the receptor for B7-H1 [15], Jurkat cells were first treated with phorbol 12-myristate 13-acetate (PMA, 20 nM, Sigma-Aldrich, St. Louis, MO) to induce PD-1 expression as previously reported [32]. As shown in Fig. 6, a significant increase of apoptosis was detected in Jurkat cells after co-culture with C. parvum-infected H69 cells (Fig. 6). Apoptosis in co-cultured Jurkat cells was partially blocked by pretreatment of H69 cells with a B7-H1 neutralizing antibody or miR-513 precursor (Fig. 6). In addition, a neutralizing antibody to Fas also showed a significant inhibitory effect on the associated apoptosis (Fig. 6), consistent with our previous results demonstrating that Fas/Fas-ligand pathway is involved in the apoptosis in the co-cultured Jurkat cells [28,33]. Thus, besides the activation of the Fas/Fas-ligand pathway, miR-513-mediated expression of B7-H1 also contributes to the apoptotic cell death in T-cells induced by co-culture with C. parvum-infected cholangiocytes.
FIGURE 6.

MicroRNA-513 influences B7-H1-associated apoptotic cell death in co-cultured Jurkat cells. H69 cells were first exposed to C. parvum oocysts for 24 h and then co-cultured with Jurkat cells in the presence of PMA with or without neutralizing antibodies to B7-H1 or Fas for 24 h. Jurkat cells were then harvested for apoptosis assay by DAPI staining. These data are representative of three independent experiments. Ab = antibody; *, p < 0.05, vs. Jurkat cells co-cultured with non-C. parvum infected H69 cells as the control; #, p < 0.05, vs. Jurkat cells co-cultured with C. parvum oocysts-treated H69 cells without antibody or precursor treatment.
DISCUSSION
The key findings in this report are: i) C. parvum infection induces B7-H1 expression in cholangiocytes; ii) B7-H1 expression induced by C. parvum involves relief of miR-513-mediated translational suppression of B7-H1; and iii) B7-H1 expression on cholangiocytes following C. parvum infection induces apoptotic cell death in activated T cells. These data suggest that miR-513 regulates C. parvum-induced expression of B7-H1 in cholangiocytes and is involved in biliary epithelial reactions in response to C. parvum infection.
B7-H1 is a key member of the B7 family of costimulatory molecules important in regulation of immune response, in particular, T cell homeostasis. Expression of B7-H1 by epithelial cells in response to microbial infection is tightly regulated to ensure an appropriate anti-microbial immune response. Upregulation of B7-H1 has recently been reported in intestinal and airway epithelial cells in response to Helicobacter pylori and human rhinovirus infection [21,34]. In this study, we found that C. parvum infection induces B7-H1 expression in human cholangiocytes. Whereas cholangiocytes normally only express B7-H1 mRNA, not B7-H1 protein, we found significant B7-H1 protein expression in cells after exposure to C. parvum. By immunofluorescent staining, we detected an increase of B7-H1 expression not only in cells directly infected by C. parvum but also in non-infected bystander cells. Furthermore, cells exposed to C. parvum lysate also showed B7-H1 protein expression, indicating that direct infection by viable C. parvum is not required to induce B7-H1 expression. The greater induction of B7-H1 mRNA by C. parvum lysate compared to C. parvum oocysts after incubation for 12 h and 24 h may reflect a decreased stimulation to the host cells from the organism after cellular internalization, because a similar induction of B7-H1 mRNA was detected at an early time point. Taken together, we postulate that either molecules from the parasite or host-induced pro-inflammatory cytokines can trigger B7-H1 expression. Pro-inflammatory cytokines, such as IFN-γ and TNF-α, are potent activators of B7-H1 protein expression in T cells, B cells, endothelial and epithelial cells [15]. We favor the hypothesis that parasite-derived molecules are responsible for the B7-H1 expression because we failed to detect an increase of either IFN-γ or TNF-α in the cell cultures after exposure to C. parvum oocysts or lysate. This not only supports the notion that these pro-inflammatory cytokines are not involved in C. parvum-induced B7-H1 expression but also excludes the possibility of LPS contamination of our parasite preparations. An increase of TNF-α mRNA expression should be detected if LPS contamination occurs. Further studies should address key questions regarding the characterization of parasite-derived molecules which may account for the induction of B7-H1 expression in host epithelial cells, such as whether their effects are heat labile, protease susceptible or dialyzable.
A key observation of our study is that C. parvum-induced B7-H1 expression in cholangiocytes may involve relief of miR-513-mediated translational suppression of B7-H1. We previously identified that miR-513 targets 3′-UTR of B7-H1 resulting in translational suppression of the protein in human cholangiocytes [18]. Whereas C. parvum induces B7-H1 protein in cholangiocytes, we detected a significant decrease of miR-513 expression in cells after exposure to viable C. parvum or C. parvum lysate. Following C. parvum infection, cells showed relief of translational suppression associated with B7-H1 3′-UTR containing the miR-513 binding site. Moreover, treatment of cells with miR-513 precursor inhibited C. parvum-induced B7-H1 protein expression. Since a control precursor showed no inhibitory effect, we speculate that inhibition of B7-H1 protein induction by miR-513 precursor is due to enhanced translational repression via targeting of miR-513 to the B7-H1 3′-UTR. Coupled with our observation of increased expression of B7-H1 mRNA following C. parvum infection, our results suggest that C. parvum induces B7-H1 protein expression in cholangiocytes through both increased transcription and relief of miR-513-mediated translational repression.
Expression of B7-H1 in cholangiocytes may represent an important element of biliary epithelial reactions in response to C. parvum infection. B7-H1 possesses dual functions in regulating T cell homeostasis: B7-H1 activates a set of positive signals in naïve T cells to stimulate early T cell priming and differentiation but also initiates a negative response in activated T cells to inhibit T cell function and survival [15,26,31,32]. Induction of apoptotic cell death in activated T cells has been implicated as one of the molecular mechanisms by which B7-H1 negatively regulates T cell functions [15,35]. Emerging clinical evidence implies that the dual functions of B7-H1 to T cells may be key to regulating host anti-microbial immune responses. In this study, we confirmed that B7-H1 expressed on the surface of human cholangiocytes following C. parvum infection induced apoptotic cell death in co-cultured activated Jurkat cells. An antibody to B7-H1 blocked apoptosis in co-cultured activated Jurkat cells (Fig. 6). Functional manipulation of miR-513 in cholangiocytes during C. parvum infection could also influence associated apoptotic cell death in co-cultured Jurkat cells. Thus, B7-H1 may have important regulatory functions to ensure a controlled and balanced inflammatory reaction in the biliary mucosa during C. parvum infection. On one hand, B7-H1 expressed on cholangiocytes may facilitate the activation of naive T cells to trigger cellular immune responses against C. parvum infection. On the other hand, B7-H1 can negatively regulate the functions of activated T cells to prevent immunopathology due to an unregulated immune response. It can be speculated that miR-513-mediated expression of B7-H1 on cholangiocytes will influence cholangiocyte-T cell interactions during biliary C. parvum infection in vivo. Infiltration of inflammatory lymphoid cells to the portal region, as well as associated apoptotic cell death of lymphocytes, has been demonstrated in C. parvum infection in the biliary tract in vivo [36].
In conclusion, our data indicate that C. parvum induces B7-H1 expression in human cholangiocytes via relief of miR-513-mediated translational suppression of B7-H1. Moreover, expression of B7-H1 by cholangiocytes is involved in cholangiocyte-T cell interactions during C. parvum infection. Thus, miR-513 plays a role in regulating B7-H1 expression by cholangiocytes in response to C. parvum infection. It will be of interest to extend these studies to C. parvum infection of intestinal epithelial cells. Future studies should also determine the mechanisms by which C. parvum infection decreases miR-513 expression and the role of miRNAs in host antiC. parvum immunity in vivo.
Acknowledgments
This work was supported by National Institutes of Health Grant R01 AI071321 and by the Nebraska Tobacco Settlement Biomedical Research Program (LB692) (to X-M.C).
We thank Mr. J Mitchell and Dr. R. G. Townley for technical assistance with the bead-based multiplex sandwich immunoassay.
Footnotes
The authors have no financial conflict of interest.
References
- 1.Viswanathan VK, Hecht G. Innate immunity and the gut. Curr Opin Gastroenterol. 2000;16:546–51. doi: 10.1097/00001574-200011000-00015. [DOI] [PubMed] [Google Scholar]
- 2.Han J, Ulevitch RJ. Limiting inflammatory responses during activation of innate immunity. Nat Immunol. 2005;6:1198–205. doi: 10.1038/ni1274. [DOI] [PubMed] [Google Scholar]
- 3.Alpini G, McGill JM, LaRusso NF. The pathobiology of biliary epithelia. Hepatology. 2002;35:1256–68. doi: 10.1053/jhep.2002.33541. [DOI] [PubMed] [Google Scholar]
- 4.Strazzabosco M, Fabris L, Spirli C. Pathophysiology of cholangiopathies. J Clin Gastroenterol. 2005;39:S90–102. doi: 10.1097/01.mcg.0000155549.29643.ad. [DOI] [PubMed] [Google Scholar]
- 5.Chen XM, Keithly JS, Paya CV, LaRusso NF. Cryptosporidiosis. N Engl J Med. 2002;346:1723–31. doi: 10.1056/NEJMra013170. [DOI] [PubMed] [Google Scholar]
- 6.Rogers KA, Rogers AB, Leav BA, et al. MyD88-dependent pathways mediate resistance to Cryptosporidium parvum infection in mice. Infect Immun. 2006;74:549–56. doi: 10.1128/IAI.74.1.549-556.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen XM, Nelson JB, O’Hara SP, et al. Multiple Toll-like Receptors are expressed in human cholangiocytes and mediate host epithelial responses to Cryptoaporidium parvum via activation of NF-kappaB. J Immunol. 2005;175:7447–56. doi: 10.4049/jimmunol.175.11.7447. [DOI] [PubMed] [Google Scholar]
- 8.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 9.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–5. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 10.Asirvatham AJ, Gregorie CJ, Hu Z, Magner WJ, Tomasi TB. MicroRNA targets in immune genes and the Dicer/Argonaute and ARE machinery components. Mol Immunol. 2008;45:1995–2006. doi: 10.1016/j.molimm.2007.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pedersen IM, Cheng G, Wieland S, et al. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature. 2007;449:919–22. doi: 10.1038/nature06205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A. 2007;104:1604–9. doi: 10.1073/pnas.0610731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bezerra JA. Biliary atresia--translational research on key molecular processes regulating biliary injury and obstruction. Chang Gung Med J. 2006;29:222–30. [PubMed] [Google Scholar]
- 14.Kamihira T, Shimoda S, Nakamura M, et al. Biliary epithelial cells regulate autoreactive T cells: implications for biliary-specific diseases. Hepatology. 2005;41:151–9. doi: 10.1002/hep.20494. [DOI] [PubMed] [Google Scholar]
- 15.Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–9. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 16.Dong H, Zhu G, Tamada K, et al. B7-H1 determines accumulation and deletion of intrahepatic CD8(+) T lymphocytes. Immunity. 2004;20:327–36. doi: 10.1016/s1074-7613(04)00050-0. [DOI] [PubMed] [Google Scholar]
- 17.Mataki N, Kikuchi K, Kawai T, et al. Expression of PD-1, PD-L1, and PD-L2 in the liver in autoimmune liver diseases. Am J Gastroenterol. 2007;102:302–12. doi: 10.1111/j.1572-0241.2006.00948.x. [DOI] [PubMed] [Google Scholar]
- 18.Gong A, Zhou R, Hu G, et al. MicroRNA-513 regulates B7-H1 translation and is involved in interferon-gamma-induced B7-H1 expression in cholangiocytes. J Immunol. 2009;182:1325–1333. doi: 10.4049/jimmunol.182.3.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grubman SA, Perrone RD, Lee DW, et al. Regulation of intracellular pH by immortalized human intrahepatic biliary epithelial cell lines. Am J Physiol. 1994;266:G1060–70. doi: 10.1152/ajpgi.1994.266.6.G1060. [DOI] [PubMed] [Google Scholar]
- 20.Thea DM, Pereira ME, Kotler D, Sterling CR, Keusch GT. Identification and partial purification of a lectin on the surface of the sporozoite of Cryptosporidium parvum. J Parasitol. 1992;78:886–93. [PubMed] [Google Scholar]
- 21.Das S, Suarez G, Beswick EJ, Sierra JC, Graham DY, Reyes VE. Expression of B7-H1 on gastric epithelial cells: its potential role in regulating T cells during Helicobacter pylori infection. J Immunol. 2006;176:3000–9. doi: 10.4049/jimmunol.176.5.3000. [DOI] [PubMed] [Google Scholar]
- 22.Wu H, Neilson JR, Kumar P, et al. miRNA profiling of naive, effector and memory CD8 T cells. PLoS ONE. 2007;2:e1020. doi: 10.1371/journal.pone.0001020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller JE, Ghoshal K, Ramaswamy B, et al. MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27 (Kip1) J Biol Chem. 2008;283:29897–903. doi: 10.1074/jbc.M804612200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lédée N, Lombroso R, Lombardelli L, et al. Cytokines and chemokines in follicular fluids and potential of the corresponding embryo: the role of granulocyte colony-stimulating factor. Hum Reprod. 2008;23:2001–9. doi: 10.1093/humrep/den192. [DOI] [PubMed] [Google Scholar]
- 25.Chen XM, Splinter PL, O’Hara SP, LaRusso NF. A cellular miRNA, let-7i, regulates toll-like receptor 4 expression and contributes to cholangiocyte immune responses against Cryptosporidium parvum infection. J Biol Chem. 2007;282:28929–38. doi: 10.1074/jbc.M702633200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dong H, Strome SE, Salomao DR, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 27.Filippatos G, Ang E, Gidea C, et al. Fas induces apoptosis in human coronary artery endothelial cells in vitro. BMC Cell Biol. 2004;5:6. doi: 10.1186/1471-2121-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen XM, Gores GJ, Paya CV, LaRusso NF. Cryptosporidium parvum induces apoptosis in biliary epithelia by a Fas/Fas ligand-dependent mechanism. Am J Physiol. 1999;277:G599–08. doi: 10.1152/ajpgi.1999.277.3.G599. [DOI] [PubMed] [Google Scholar]
- 29.McDonald V. Host cell-mediated responses to infection with Cryptosporidium. Parasite Immunol. 2000;22:597–604. doi: 10.1046/j.1365-3024.2000.00343.x. [DOI] [PubMed] [Google Scholar]
- 30.Park J, Gores GJ, Patel T. Lipopolysaccharide induces cholangiocyte proliferation via an interleukin-6-mediated activation of p44/p42 mitogen-activated protein kinase. Hepatology. 1999;29:1037–43. doi: 10.1002/hep.510290423. [DOI] [PubMed] [Google Scholar]
- 31.Yamazaki T, Akiba H, Iwai H, et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol. 2002;169:5538–45. doi: 10.4049/jimmunol.169.10.5538. [DOI] [PubMed] [Google Scholar]
- 32.Ding H, Wu X, Gao W. PD-L1 is expressed by human renal tubular epithelial cells and suppresses T cell cytokine synthesis. Clin Immunol. 2005;115:184–91. doi: 10.1016/j.clim.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 33.O’Hara SP, Small AJ, Nelson JB, et al. The human immunodeficiency virus type 1 tat protein enhances Cryptosporidium parvum-induced apoptosis in cholangiocytes via a Fas ligand-dependent mechanism. Infect Immun. 2007;75:684–96. doi: 10.1128/IAI.01348-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heinecke L, Proud D, Sanders S, Schleimer RP, Kim J. Induction of B7-H1 and B7-DC expression on airway epithelial cells by the Toll-like receptor 3 agonist double-stranded RNA and human rhinovirus infection: In vivo and in vitro studies. J Allergy Clin Immunol. 2008;121:1155–60. doi: 10.1016/j.jaci.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dong H, Chen XM. Immunoregulatory role of B7-H1 in chronicity of inflammatory responses. Cell Mol Immunol. 2006;3:179–87. [PMC free article] [PubMed] [Google Scholar]
- 36.Lacroix S, Mancassola R, Naciri M, Laurent F. Cryptosporidium parvum-specific mucosal immune response in C57BL/6 neonatal and gamma interferon-deficient mice: role of tumor necrosis factor alpha in protection. Infect Immun. 2001;69:1635–42. doi: 10.1128/IAI.69.3.1635-1642.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]