

Abstract
FKBP ligand homodimers can be used to activate signaling events inside cells and animals that have been engineered to express fusions between appropriate signaling domains and FKBP. However, use of these dimerizers in vivo is potentially limited by ligand binding to endogenous FKBP. We have designed ligands that bind specifically to a mutated FKBP over the wild-type protein by remodeling an FKBP-ligand interface to introduce a specificity binding pocket. A compound bearing an ethyl substituent in place of a carbonyl group exhibited sub-nanomolar affinity and 1,000-fold selectivity for a mutant FKBP with a compensating truncation of a phenylalanine residue. Structural and functional analysis of the new pocket showed that recognition is surprisingly relaxed, with the modified ligand only partially filling the engineered cavity. We incorporated the specificity pocket into a fusion protein containing FKBP and the intracellular domain of the Fas receptor. Cells expressing this modified chimeric protein potently underwent apoptosis in response to AP1903, a homodimer of the modified ligand, both in culture and when implanted into mice. Remodeled dimerizers such as AP1903 are ideal reagents for controlling the activities of cells that have been modified by gene therapy procedures, without interference from endogenous FKBP.
A number of natural and synthetic compounds are known that bind with high affinity to the human protein FKBP12 (1–5). Bivalent versions of these compounds—chemical “dimerizers”—recently have been developed as cell-permeant reagents to control intracellular signaling events that are naturally regulated by protein–protein interactions (6, 7). In these applications, cells are engineered to express a chimeric protein comprising a signaling domain fused to one or more FKBPs; treatment with dimerizer crosslinks the proteins and initiates signaling. Because genes for such fusion proteins can be delivered via gene therapy strategies, dimerizers might be used to control a wide range of proliferation and differentiation events for therapeutic purposes (8, 9).
Use of FKBP dimerizers in vivo is complicated by their potential interactions with endogenous FKBPs, which are ubiquitous and highly expressed in mammals. These interactions could blunt potency by allowing nonproductive dimerization events and compound sequestration. High concentrations of ligand also might interfere with the physiological functions of FKBP12, such as calcium channel modulation and regulation of cardiac development (10–12). The ideal dimerizer therefore would interact minimally with endogenous FKBP; but any modifications that reduce binding would have equivalent effects on the affinity for the chimeric target protein.
We set out to solve this specificity problem by engineering a unique pocket into the target FKBP that differentiates it from the endogenous protein. This approach is feasible because dimerizer applications involve the delivery of the target as well as the drug. Our aim was to add “bumps” to ligands to block sterically binding to wild-type FKBP and then generate compensating “holes” by mutagenesis that could be installed into target fusion proteins. We report here the design of such a remodeled ligand–FKBP pair and evaluation of the resulting bumped dimerizer as a signaling reagent in vivo.
MATERIALS AND METHODS
Synthetic Chemistry.
The synthesis of compound 1 has been described (13). Isomers of compound 5 were prepared as shown in Fig. 3. Compounds 2–4 were prepared analogously by coupling amine 7 (Fig. 3) with 3,3-dimethyl-2-methoxy-pentanoic acid, 3,3-dimethyl-2-ethyl-pentanoic acid, or 2-phenyl-butanoic acid, respectively. FK506 was coupled to 4′-(aminomethyl)-fluorescein (4′-AF; Molecular Probes) analogously to the synthesis of FK1012 (6). Fluoro-5S was prepared by coupling 5S to 4′-AF by using 1,3-dicyclohexylcarbodiimide and 4-dimethylaminopyridine in dichloromethane.
Figure 3.

Chemical structure of the C9-bumped dimerizer AP1903 and scheme for its synthesis via compound 5S. Preparation of alcohol 6 has been described (13). R and S isomers of compound 5 were chromatographically separated as tert-butyl esters before final TFA deprotection. Fmoc, N-(9-fluorenylmethoxycarbonyl); DCC, 1,3-dicyclohexylcarbodiimide; DMAP, 4-dimethylaminopyridine; ClMePyrI, 2-chloro-1-methylpyridinium iodide; TFA, trifluoroacetic acid; BOP, benzotriazol-1-yloxy-tris(dimethylamino)phosphonium hexafluorophosphate; Me, methyl; Et, ethyl; iPr, isopropyl; tBu, tert-butyl.
Protein Engineering and Expression.
FKBP and its mutants were expressed with an N-terminal hexa-histidine purification sequence and an influenza hemaglutinin (HA) epitope tag. Expression vector pET-H6HA-FKBP was assembled by cloning an NdeI–NdeI oligonucleotide pair encoding the amino acid sequence MHHHHHHYPYDVPDYAAMAHM and an NdeI–BamHI PCR product encoding human FKBP12 into NdeI/BamHI-digested pET20b(+) (Novagen). Mutants were engineered as described (14). Proteins were expressed in Escherichia coli BL21(DE3) and released in ≥50% purity by a single freeze-thaw step as described (15) and were purified further on Ni-NTA agarose (Qiagen) under native conditions. For structural studies, an expression vector for untagged F36V-FKBP was generated by excising the NdeI fragment from pET-H6HA-F36V-FKBP followed by recircularization. Protein for crystallography was expressed and extracted as before and purified to homogeneity by DEAE ion-exchange chromatography followed by gel filtration on S-100 Sepharose.
FKBP Binding Assay.
Fluorescence polarization competition assays were performed as described (C.T.R., E. Laborde, D.A.H., and T.C., unpublished work). In brief, subsaturating concentrations of protein were incubated for 30 min with 2.5 nM fluoro-FK506 and serial dilutions of competitor ligand in the wells of a Dynatech microfluor plate. Polarization was read on a Jolley FPM-2 (Jolley Consulting and Research, Grayslake, IL). The increase in polarization on binding was used as a direct readout of percentage of bound probe, compared with controls containing no competitor (100%) or no protein (0%). The concentration of competitor resulting in a 50% probe displacement (IC50) was determined by a nonlinear least square fit by using the four-parameter algorithm
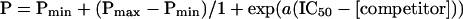 |
where Pmin, Pmax, and P represent the minimal, maximal, and measured polarization values, respectively. Direct binding experiments with fluoro-5S used 10 pM ligand, and polarization was read on a Jolley FPM-1 reader.
X-Ray Crystallography.
Crystals of the complex of F36V-FKBP with 5S were obtained by vapor diffusion in hanging drops containing 40 mg/ml complex in 1.2 M ammonium sulfate, 0.1 M sodium phosphate (pH 6.0), and 20 mM DTT over reservoirs of 2.4 M ammonium sulfate and were triclinic (a = 32.73, b = 41.85, c = 43.59 Å, α = 62.61, β = 82.75, γ = 85.29°) with two complexes in the asymmetric unit. Data were collected at room temperature with a Rigaku R-AXIS II area detector with graphite monochromated Cu Kα x-rays. Data were collected as 2° oscillation images, reduced to integrated intensities with the program denzo (16), and scaled with rotavata, and agrovata (17). The structure was solved by molecular replacement with amore (17) and refined by using x-plor (18). The current R value for reflections with F > 2σ from 10.0 to 1.9 Å is 0.21 and Rfree is 0.26; rms deviations are 0.009 Å for bonds and 1.78° for angles. Coordinates have been deposited with the Brookhaven Protein Data Bank (PDB ID code 1bl4).
Cell Line for in Vitro Fas Studies.
Construction of the retroviral plasmid pSRα-myristoylation-2xFKBP-Fas-E has been described (13). An equivalent vector incorporating the F36V mutation in both FKBPs was prepared analogously. Cloned HT1080 cell lines (ATCC CCL-121) retrovirally transduced with Fas constructs were prepared as described (13). Cell viability after overnight incubation with AP1903 was measured by Alamar Blue assay (13).
Cell Line for in Vivo Fas Studies.
pSRα-LNGFR-2x(F36V-FKBP)-Fas-E encodes the extracellular and transmembrane portions (amino acids 1–274) of the human low affinity nerve growth factor receptor (19), derived by PCR from a human cerebellum cDNA library, in place of a myristoylation sequence. The constitutive human growth hormone (hGH) expression plasmid pCEP4-hGH was made by subcloning a genomic hGH clone (20) into the episomal plasmid pCEP4 (Invitrogen). To prepare cells that constitutively express both F36V-FKBP-Fas and hGH, a G418-selected population of HT1080 cells retrovirally transduced with pSRα-LNGFR-2FKBP F36V-Fas-E was transfected with pCEP4-hGH by lipofection, and clones were selected in hygromycin B. Clone HTFasGH-3 was selected for its high response to AP1903 in the absence of actinomycin D, and high hGH production.
Animal Experiments.
Male nu/nu mice were obtained from Charles River Laboratories (Wilmington, MA). For injection, HTFasGH-3 cells were harvested from tissue culture dishes in PBS/0.1% glucose/10 mM EDTA, washed, and resuspended in PBS/0.1% BSA/0.1% glucose at a concentration of 2 × 107 cells/ml. Between 2 and 4 × 106 cells were implanted into two i.m. sites. After 24 h, mice were administered i.v. AP1903 formulated in [50% N,N-dimethylacetamide/50% (90% PEG-400/10% Tween 80)] at 2 ml/kg. After a further 24 h mice were killed and serum hGH concentrations were determined by ELISA (Boehringer Mannheim 1-585-878).
RESULTS
Molecular Design.
The natural product FK506 and synthetic derivatives exemplified by compound 1 (Fig. 1A) share a conserved α-keto-pipecolylamide core that binds in a complementary hydrophobic pocket on FKBP, as revealed by x-ray crystallography (Fig. 1B) (2, 5, 21). We focused on the C9 carbonyl oxygen, which packs tightly against Tyr26, Phe36, and Phe99, all making unusual edge-on ɛ-hydrogen contacts. Inspection of crystal structures suggested that replacing this carbonyl with larger substituents would dramatically reduce FKBP binding through steric clashes with either Phe36 or Tyr26 (depending on the stereochemistry at a tetrahedral C9 carbon; Fig. 1A). This in turn suggested that mutations that truncate these side chains might provide compensatory holes that would restore binding.
Figure 1.

(A) Chemical structure of synthetic FKBP ligand 1, with the α-keto-pipecolylamide core region colored red. The arrow indicates the C9 carbonyl position modified in this study. A carboxylate group (Right) was included in all ligands to facilitate subsequent engineering of dimers. (B) Portion of the x-ray crystal structure of the complex between human FKBP12 and a synthetic compound related to 1 (from ref. 2). Only the α-keto-pipecolylamide core of the compound is shown, corresponding to the region colored red in Fig. 1A.
Interface Remodelling.
We engineered a set of FKBP mutants in which Phe36 was replaced with smaller hydrophobic residues and measured their affinities for C9-alkylated analogs of compound 1 by using a competitive fluorescence polarization assay (Table 1). This assay reports IC50 values that are proportional to dissociation constants (Kd). Compound 1 bound to wild-type FKBP with an IC50 of 86 nM [corresponding to a Kd of ≈20 nM (2)], and binding was largely unchanged for the Phe36 mutants. Even modest enlargements of the C9 substituent to methoxy (compound 2) or ethyl (compound 3) reduced affinity to undetectable levels (IC50 > 10,000 nM), as expected. However, binding could be rescued by any of the Phe36 mutations, and although affinities were fairly low, the compounds showed apparent selectivity of up to 50-fold for the mutants over wild type. Two further trends were apparent. First, compound 3 bound more tightly than 2, indicating that nonpolar substituents may be preferred. Second, the mutants clearly favored the S-isomer of compounds over the R. This is the preference predicted by modeling, so it suggested that the modifications were acting as specificity determinants as envisaged by our design.
Table 1.
Binding affinities (IC50s in nM) of unmodified and C9-bumped ligands 1–5 for a panel of FKBP Phe36 mutants

For compounds 2–5, the affinities of the separated R and S isomers were determined. Only the lower portion of each compound is shown (see Fig. 1A), with the C9 carbonyl or bump substituents colored red. The high affinity interaction between compound 5S and the Phe36Val mutant is boxed.
To try to improve the affinities of these specific pairings, we retained the ethyl bump of 3 and varied the tert-pentyl “bottom” C9 substituent (2, 4, 5). Installing a phenyl group (compound 4) improved affinities for each of the mutants by 3- to 5-fold while maintaining the lack of detectable binding to wild-type protein, and additional 5- to 20-fold increases were observed when a trimethoxyphenyl group was incorporated (compound 5). As the affinities improved, so did the preference for the S-isomer over R, suggesting that the changes were not altering the binding of the engineered bump. Strikingly, none of the bumped compounds showed any major preference for a particular mutant; instead, affinities were largely insensitive to the size of the new side chain. This implied that, rather than a precise “lock-and-key” fit, molecular recognition in the new pocket was rather loose and flexible.
The best combination was the pairing of 5S with the Phe->Val mutant (F36V-FKBP), with an IC50 (1.8 nM) close to the resolution limit of the assay. To assess the true affinity of the compound, we coupled it to fluorescein and performed direct fluorescence polarization titration. The labeled compound, fluoro-5S, had a Kd of 0.094 nM for the F36V mutant, compared with 67 nM for the wild-type protein (data not shown). Thus, although binding to the wild-type protein was still detectable, specificity for F36V-FKBP was almost three orders of magnitude. Moreover, the affinity of fluoro-5S for F36V-FKBP exceeded that of any known ligand for the wild-type protein (1, 5).
Crystallographic Analysis of Remodeled Interface.
To determine the structural basis for these binding characteristics, we crystallized the complex between 5S and F36V-FKBP and determined its structure to 1.9-Å resolution (Fig. 2). Overall, except for the mutation, the structure of the protein is essentially superimposable with wild-type FKBPs from unmodified complexes (rms deviation for main chain atoms 0.45 Å). The F36V substitution opens up a cavity of ≈90 Å3 (Fig. 2A), and surprisingly—apart from a 0.2-Å movement of the Val36 main chain—the protein does not relax to fill it. As a result, the pocket is lined by the predominantly hydrophobic side chains that usually pack against Phe36, and the Gly28 mainchain is exposed at the base of the pocket (Fig. 2B). The S-ethyl bump of the ligand inserts into this cavity exactly as predicted, penetrating what would be the surface of Phe36 in the wild-type protein and making multiple van der Waals contacts (Fig. 2 C and D). Compared with a complex between wild-type FKBP and a conventional ligand (2), the atoms of the core motif move by only 0.2–0.4 Å (Fig. 2D). Thus the modifications lead to a largely “surgical” deletion and insertion of contacts.
Figure 2.

X-ray crystal structure of the 5S-F36V-FKBP complex. (A) Overview of the complex. (B) Sidechains contacting the ethyl bump of 5S. Side chains are shown in gray, and main chain is shown in brown. The van der Waals surface of the ethyl bump of 5S is shown (yellow dots). (C) A section through the ligand binding site of the complex showing the large cavity created by the F36V mutation. (D) Equivalent section through the complex between the wild-type protein and an unbumped compound related to 1, shown in white (2). For comparison, the Val36 side chain and the 5S core region from C are overlaid (yellow), based on a superposition of the main chain atoms in the two structures.
One remarkable consequence of this is that the bump only occupies ≈40% of the engineered hole, so that a large, fully enclosed cavity of ≈60 Å3 remains within which no ordered water molecules are visible. Improving the fit by extending the ethyl bump of 5S to an allyl group did increase affinity but only ≈3-fold (data not shown). Thus, affinity is largely insensitive to the quality of packing in the engineered pocket, possibly because recognition occurs mainly through nondirectional hydrophobic interactions (5). These structural observations correlate with our binding data showing that many Phe36 substitutions confer good binding affinity and that nonpolar bumps are preferred (Table 1).
The remaining peripheral substituents of 5S pack into hydrophobic channels, essentially as observed for unbumped compounds (2). The conformations of these groups can vary widely between ligands (2, 4, 5), and because we do not have the structure of unbumped 5S for direct comparison, we cannot assess effects of the modification—although the C9-trimethoxyphenyl group would have to shift because of the altered C9 stereochemistry (Fig. 2D). The substantial binding energy this group confers to 5S (Table 1) probably stems from two good hydrogen bonds to Arg41 and His87 and also extensive intramolecular contacts (Fig. 2 A and B) that may preorganize the ligand and decrease the entropic cost of binding (2, 5).
Activation of Fas Signaling with a Remodeled Dimerizer.
Having identified 5S as an appropriately specific ligand, we coupled together two molecules through a previously identified linker (13) to create the bumped dimerizer AP1903 (Fig. 3). The binding affinity and specificity characteristics of AP1903 mirrored those of its monomeric precursor (Fig. 4), confirming that AP1903 should interact minimally with endogenous FKBP inside cells. As a first test of the potency of AP1903, we determined its ability to activate signaling through a chimeric Fas receptor. Previous work showed that cells expressing membrane-tethered fusions between FKBP and the intracellular domain of the Fas receptor undergo apoptosis in response to appropriate dimeric FKBP ligands (8, 13). This approach is a promising general method for eliminating engineered cells following gene therapy procedures, but clinical application may require ligands that can operate independently of endogenous FKBP.
Figure 4.

Binding affinity and specificity of AP1903 determined by competition fluorescence polarization assay. Fluoresceinated FK506 probe was bound to wild-type FKBP (open circles) or F36V-FKBP (closed circles), and serial dilutions of AP1903 were added. AP1903 displaced the probe from F36V-FKBP with an IC50 of 5 nM, but binding to the wild-type protein was negligible.
The human fibrosarcoma line HT1080 was engineered to express stably a fusion protein comprising a myristoylation sequence, two copies of F36V-FKBP, and the human Fas intracellular domain. AP1903 elicited potent and dose-dependent apoptotic death of these engineered cells in culture, with an EC50 of ≈0.1 nM (Fig. 5A). This potency corresponds directly to the binding affinity and represents a 60-fold improvement over the previous best unbumped compound (AP1510, a synthetic dimer of 1; Fig. 1B) (13). By contrast, activity was reduced by over two orders of magnitude on an analogous cell line expressing a wild-type FKBP Fas fusion protein. Of interest, the level of discrimination (200-fold) was lower than that observed for binding in vitro (1,000-fold). We speculate that this reflects stabilization of the weak interactions between AP1903 and membrane-tethered tandem wild-type FKBP fusion proteins (the avidity effect). No effect was observed on unmodified HT1080 cells at concentrations up to 1 μM (data not shown). Thus, the affinity and specificity of 5S were reflected in dimerizer potency in cells.
Figure 5.

Activation of Fas signaling by AP1903 in vitro and in vivo. (A) AP1903-induced killing of cells in culture expressing dimerizer-dependent Fas constructs. HT1080 cells stably transduced with retrovirus pSRα-myr-2FKBP-Fas-E (open circles) or pSRα-myr-2(F36V-FKBP)-Fas-E (closed circles) were treated overnight with the concentrations of AP1903 shown, and viability then was measured by Alamar Blue assay. Values shown are the means of triplicate wells. (B) AP1903-dependent elimination of hGH-secreting HTFasGH-3 cells implanted into nude mice. Serum hGH levels directly reflect the number of viable cells (see text). Values (mean ± 1 SEM) are from three to six separate experiments (at least three mice per point per experiment).
Activation of Fas Signaling in Vivo by AP1903.
To test whether AP1903 can function in vivo, we determined its efficacy in an mouse model of conditional cell ablation. We engineered a second HT1080 cell line expressing a Fas-F36V-FKBP construct that also constitutively secretes hGH. Constitutive hGH secretion provides a convenient and accurate way to monitor the number of viable cells in vivo because hGH has a serum half-life of only ≈3 min in mice (22, 23). Over 3 consecutive days, we implanted these cells i.m. into nude mice, treated the animals i.v. with various doses of AP1903, and then determined serum hGH levels as a measure of the number of surviving cells. Fig. 5B shows that AP1903 elicited a dose-dependent decrease in serum hGH levels, with a half-maximal effective dose of 0.4 ± 0.1 mg/kg. Thus, the bumped dimerizer retains potent activity in vivo, indicating that the remodeled interface is functional in a whole animal context.
DISCUSSION
General Implications for Drug Design.
In this study, we redesigned a molecular interface to generate compounds that can differentiate 1,000-fold between two proteins differing by a single point mutation. This problem resembles a common challenge in conventional drug discovery, where efficacy can depend on discrimination between closely related homologs. For example, anti-inflammatory drugs that specifically inhibit human cyclooxygenase-2 (COX-2) while sparing COX-1 are expected to show significantly reduced toxicity (24). A cavity caused by the amino acid substitution Ile523Val is the major differentiating feature of the COX-2 binding site (25). Therefore, lessons learned in our work may be generally relevant to drug design.
In particular, we found that our designed ligand has subnanomolar affinity for its target despite the presence of an enclosed hole in the binding interface. Cavities at interfaces previously have been considered incompatible with high affinity ligand binding, which is invariably characterized by excellent shape complementarity (5). Furthermore, holes engineered into protein cores are destabilizing (26). Our observations suggest surprisingly that such holes need not be detrimental to affinity. Selectivity of conventional drugs might therefore be attained by relatively imprecise recognition of surface cavities in target proteins—particularly when those cavities are largely nonpolar—and binding affinity then improved by secondary modifications, as in this work.
Comparison to Other Remodeled Interfaces.
We found that our interface modifications caused only local structural changes. Redesigned interfaces also have been engineered between cyclophilin and cyclosporin (27, 28) and between Src tyrosine kinase and ATP (29). Of interest, molecular modelling generally suggests that, in these cases, holes would remain unless rearrangements occurred (30), but no direct structural data are yet available. However, our results are in striking contrast to a recent report of a remodeled complex between two proteins, where major structural adjustments were propagated across the entire interface (31). Additional studies will be required to determine whether this reflects an inherent difference in “plasticity” between the two types of complex.
Future Applications of AP1903.
By engineering out binding to endogenous FKBP, we have eliminated an important potential obstacle to the therapeutic use of chemical dimerizers. Unexpectedly, our design efforts also produced a ligand with higher affinity than any previous dimerizer. AP1903 should therefore be effective for controlling a wide range of cellular events inside cells and whole organisms, at low concentrations and independently of endogenous FKBP.
Inducible apoptosis may be an important early clinical application because the ability to eliminate engineered cells could be broadly useful—for example, to abort a graft-versus-host response after allogeneic bone marrow transplantation (32) or as a general failsafe for gene therapy procedures. Current approaches involve expressing nonhuman enzymes that can metabolize pro-drugs to lethal products (for example, the combination of herpesvirus thymidine kinase and gancyclovir), but immune recognition of the foreign protein is a serious problem (32, 33). AP1903-inducible Fas activation is an attractive alternative because apoptosis is a cell-autonomous and noninflammatory process that occurs rapidly and in the absence of cell division, and Fas-FKBP-F36V is built from human proteins and should be minimally immunogenic.
Acknowledgments
We thank Terry Keenan and Edgardo Laborde for reagents, Brian Gladstone and Stuart Schreiber for early discussions and advice, and Dale Talbot and Rebecca Ward for helpful comments on the manuscript.
ABBREVIATIONS
- FKBP
human FK506-binding protein 12
- F36V-FKBP
FKBP mutant with Phe36 replaced by Val
- hGH
human growth hormone
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, Biology Department, Brookhaven National Laboratory, Upton, NY 11973 (PDB ID code 1bl4).
References
- 1.Schreiber S L. Science. 1991;251:283–287. doi: 10.1126/science.1702904. [DOI] [PubMed] [Google Scholar]
- 2.Holt D A, Luengo J I, Yamashita D S, Oh H-J, Konialian A K, Yen H-K, Rozamus L W, Brandt M, Bossard M J, Levy M A, et al. J Am Chem Soc. 1993;115:9925–9938. [Google Scholar]
- 3.Hauske J R, Dorff P, Julin S, DiBrino J, Spencer R, Williams R. J Med Chem. 1992;35:4284–4296. doi: 10.1021/jm00101a005. [DOI] [PubMed] [Google Scholar]
- 4.Armistead D M, Badia M C, Deininger D D, Duffy J P, Saunders J O, Ting R D, Thomson J A, DeCenzo M T, Futer O, Livingston D J, et al. Acta Crystallogr D. 1995;51:522–528. doi: 10.1107/S0907444994014502. [DOI] [PubMed] [Google Scholar]
- 5.Babine R E, Bender S L. Chem Rev. 1997;97:1359–1472. doi: 10.1021/cr960370z. [DOI] [PubMed] [Google Scholar]
- 6.Spencer D M, Wandless T J, Schreiber S L, Crabtree G R. Science. 1993;262:1019–1024. doi: 10.1126/science.7694365. [DOI] [PubMed] [Google Scholar]
- 7.Spencer D M. Trends Genet. 1996;12:181–187. doi: 10.1016/0168-9525(96)10013-5. [DOI] [PubMed] [Google Scholar]
- 8.Spencer D M, Belshaw P J, Chen L, Ho S N, Randazzo F, Crabtree G R, Schreiber S L. Curr Biol. 1996;6:839–847. doi: 10.1016/s0960-9822(02)00607-3. [DOI] [PubMed] [Google Scholar]
- 9.Blau C A, Peterson K R, Drachman J G, Spencer D M. Proc Natl Acad Sci USA. 1997;94:3076–3081. doi: 10.1073/pnas.94.7.3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Snyder S H, Sabatini D M. Nat Med. 1995;1:32–37. doi: 10.1038/nm0195-32. [DOI] [PubMed] [Google Scholar]
- 11.Marks A R. Physiol Rev. 1996;76:631–649. doi: 10.1152/physrev.1996.76.3.631. [DOI] [PubMed] [Google Scholar]
- 12.Shou W, Aghdasi B, Armstrong D L, Guo Q, Bao S, Charng M-J, Mathews L M, Schneider M D, Hamilton S L, Matzuk M M. Nature (London) 1998;391:489–492. doi: 10.1038/35146. [DOI] [PubMed] [Google Scholar]
- 13.Amara J F, Clackson T, Rivera V M, Guo T, Keenan T, Natesan S, Pollock R, Yang W, Courage N L, Holt D A, et al. Proc Natl Acad Sci USA. 1997;94:10618–10623. doi: 10.1073/pnas.94.20.10618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kunkel T A, Bebenek K, McClary J. Methods Enzymol. 1991;204:125–139. doi: 10.1016/0076-6879(91)04008-c. . (1991). [DOI] [PubMed] [Google Scholar]
- 15.Wiederrecht G, Hung S, Chan H K, Marcy A, Martin M, Calaycay J, Boulton D, Sigal N, Kincaid R L, Siekierka J J. J Biol Chem. 1992;267:21753–21760. [PubMed] [Google Scholar]
- 16.Otwinowski Z. In: Proceedings of the CCP4 Study Weekend. Sawyer L, Isaac L N, Borley S, editors. Daresbury, UK: SERC; 1993. pp. 56–62. [Google Scholar]
- 17.Collaborative Computational Project, Number 4. Acta Crystallogr D. 1994;50:760–763. [Google Scholar]
- 18.Brünger A T. Curr Opin Struct Biol. 1991;1:1016–1022. [Google Scholar]
- 19.Johnson D, Lanahan A, Buck C R, Sehgal A, Morgan C, Mercer E, Bothwell M, Chao M. Cell. 1986;47:545–544. doi: 10.1016/0092-8674(86)90619-7. [DOI] [PubMed] [Google Scholar]
- 20.Rivera V M, Clackson T, Natesan S, Pollock R, Amara J F, Keenan T, Magari S R, Phillips T, Courage N L, Cerasoli F, et al. Nat Med. 1996;2:1028–1032. doi: 10.1038/nm0996-1028. [DOI] [PubMed] [Google Scholar]
- 21.Van Duyne G D, Standaert R F, Karplus P A, Schreiber S L, Clardy J. J Mol Biol. 1993;229:105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- 22.Magari S R, Rivera V M, Iuliucci J D, Gilman M, Cerasoli F. J Clin Invest. 1997;100:2865–2872. doi: 10.1172/JCI119835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heartlein M W, Roman V A, Jiang J L, Sellers J W, Zuliani A M, Treco D A, Selden R F. Proc Natl Acad Sci USA. 1994;91:10967–10971. doi: 10.1073/pnas.91.23.10967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Copeland R A, Williams J M, Giannaras J, Nurnberg S, Covington M, Pinto D, Pick S, Trzaskos J M. Proc Natl Acad Sci USA. 1994;91:11202–11206. doi: 10.1073/pnas.91.23.11202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kurumbail R G, Stevens A M, Gierse J K, McDonald J J, Stegeman R A, Pak J Y, Gildehaus D, Miyashiro J M, Penning T D, Seibert K, et al. Nature (London) 1996;384:644–648. doi: 10.1038/384644a0. [DOI] [PubMed] [Google Scholar]
- 26.Eriksson A E, Baase W A, Zhang X J, Heinz D W, Blaber M, Baldwin E P, Matthews B W. Science. 1992;255:178–183. doi: 10.1126/science.1553543. [DOI] [PubMed] [Google Scholar]
- 27.Belshaw P J, Schoepfer J G, Liu K-Q, Morrison K L, Schreiber S L. Angew Chem Int Ed Engl. 1995;34:2129–2132. [Google Scholar]
- 28.Belshaw P J, Schreiber S L. J Am Chem Soc. 1997;119:1805–1806. [Google Scholar]
- 29.Shah K, Liu Y, Deirmengian C, Shokat K M. Proc Natl Acad Sci USA. 1997;94:3565–3570. doi: 10.1073/pnas.94.8.3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pierce A C, Jorgensen W L. Angew Chem, Int Ed Engl. 1997;36:1466–1469. [Google Scholar]
- 31.Atwell S, Ultsch M, de Vos A M, Wells J A. Science. 1997;278:1125–1128. doi: 10.1126/science.278.5340.1125. [DOI] [PubMed] [Google Scholar]
- 32.Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, Ruggieri L, Ponzoni M, Rossini S, Mavilio F, Traversari C, et al. Science. 1997;276:1719–1724. doi: 10.1126/science.276.5319.1719. [DOI] [PubMed] [Google Scholar]
- 33.Riddell S R, Elliott M, Lewinsohn D A, Gilbert M J, Wilson L, Manley S A, Lupton S D, Overell R W, Reynolds T C, Corey L, et al. Nat Med. 1996;2:216–223. doi: 10.1038/nm0296-216. [DOI] [PubMed] [Google Scholar]