

Abstract
Progenitor mast cells (prMCs), derived from CD34+ precursors are CD4+/CCR5+/CXCR4+ and susceptible to CCR5(R5)-tropic virus but only marginally susceptible to CXCR4(X4)-tropic HIV. As infected prMCs mature within extravascular compartments, they become both latently infected and HIV-infection resistant, and thus capable of establishing an inducible reservoir of CCR5-tropic infectious clones. In this report we provide the first evidence that IgE-FcεRI interactions, occurring during a unique period of mast cell (MC) ontogeny, enhance prMC susceptibility to X4 and R5X4 virus. IgE-FcεRI interactions significantly increased expression of CXCR4 mRNA (∼400- to 1800-fold), enhanced prMC susceptibility to X4 and R5X4 virus (∼3000- to 16,000-fold), but had no significant effect on CD4, CCR3, or CCR5 expression, susceptibility to R5 virus, or degranulation. Enhanced susceptibility to infection with X4 virus occurred during the first 3–5 wk of MC ontogeny and was completely inhibited by CXCR4-specific peptide antagonists and omalizumab, a drug that inhibits IgE-FcεRI interactions. IgE-FcεRI coaggregation mediated by HIVgp120 or Schistosoma mansoni soluble egg Ag accelerated maximal CXCR4 expression and susceptibility to X4 virus by prMCs. Our findings suggest that for HIV-positive individuals with atopic or helminthic diseases, elevated IgE levels could potentially influence the composition of CXCR4-tropic and R5X4-tropic variants archived within the long-lived tissue MC reservoir created during infection.
Studies show HIV can be phenotypically classified by which specific chemokine coreceptors are engaged by its viral envelop glycoproteins during entry and infection of the host cell. The CCR5-tropic variants (R5 viruses) and CXCR4-tropic variants (X4 viruses) use target cell-expressed CCR5 and CXCR4, respectively, whereas R5X4 viruses can use either CCR5 or CXCR4 (1). During HIV disease, R5, X4, or R5X4 virus infection can become established within different habitats inside tissue compartments. These habitats consist of pools of various susceptible target cells that support productive or latent virus infection, and hence persistence. Collectively, these viral habitats constitute a dynamic “ecosystem” of virus infection. The composition of the virus populations and the various constituent target cell pools within the ecosystem strongly influence the predominant HIV coreceptor-using variants that emerge during the course of infection. A complex and dynamic balance of multiple immunological, virological, and physiological conditions may thus influence the range of different target cells, including both T cell and non-T cell lineages that express both CD4 and relevant chemokine coreceptors. In many individuals with HIV infection, predominance of virus with the CXCR4-tropic phenotype is associated with a more rapid progression to AIDS (1).
Among the non-T cell lineages that are susceptible to HIV infection, progenitor mast cells (prMCs)3 are unique in that they can develop into mature, latently infected tissue mast cells (MCs) (2– 4). MCs are long-lived and are also widely distributed among diverse tissue compartments through the body, including gastrointestinal tissues, which are now recognized as an important site of viral replication and T cell depletion during acute HIV infection (5– 8). Early studies by Irani et al. (9) showed that the number of tryptase/chymase-positive MCs in intestinal mucosal and submucosal tissues of patients with advanced AIDS and with severely reduced number of T cells remained unchanged relative to uninfected subjects. These finding are consistent with the concept that MCs are present at sites of active viral replication, are long-lived, and are resistant to cytotoxic effects of virus infection. Thus, due to their longevity, their resistance to viral cytopathicity, and their ability to harbor latent infectious virus, MCs can serve as an inducible reservoir of persistent HIV infection (4, 10).
FcεRI+ prMCs cultured in vitro from fetal or adult CD34+ progenitors express significant levels of not only CD4, but also both CXCR4 and CCR5. In addition, they are readily susceptible to R5-HIV but only marginally susceptible to X4-HIV (2, 4, 10). These infection susceptibility characteristics also hold for circulating prMCs isolated in vivo (10). In this study, we present evidence that FcεRI-bound IgE (IgE-FcεRI), or aggregated IgE-FcεRI significantly enhance prMC expression of CXCR4 and their susceptibility to X4 and R5X4 virus within a unique developmental period during MC ontogeny. Furthermore, IgE-FcεRI aggregation mediated by superallergens, such as HIVgp120 or soluble Schistosoma mansoni egg Ag (SmEA), also enhances susceptibility of prMCs to X4 and R5X4 virus. These findings indicate that IgE may significantly influence the overall fitness advantage for X4 and R5X4 virus in the MC reservoir of persistent HIV infection. The clinical implications of these findings are that HIV-positive individuals with pre-existing comorbid conditions associated with elevated IgE levels, such as atopic disease or helminthic infections, may have increased risk for infection with X4- and R5X4-HIV.
Materials and Methods
Materials and reagents
Human IgE myeloma protein from three different sources, supplied by Dr. A. A. Ansari and Dr. A. S. Kirshenbaum (National Institutes of Health, Bethesda, MD) and EMD Biosciences, were tested in preliminary pilot experiments as described below. IgE myeloma protein, supplied by Dr. A. A. Ansari (National Institutes of Health, Bethesda, MD), was then used in all subsequent experiments. Preparation of biotinylated human IgE myeloma protein (IgE-biotin; EMD Biosciences) was performed using the Pierce EZ-Link Sulfo-NHS-Biotinylation kit according to the manufacturer's instructions. These IgE-biotin preparations contained an average of 2.4 biotin molecules per molecule of IgE. Streptavidin (125 ng/ml; Sigma-Aldrich) was used in IgE-biotin cross-linking studies. Omalizumab (Genentech) was used to block IgE binding to prMCs. The CCR5 peptide antagonist RCP188 and the CXCR4 antagonist RCP138 were obtained from Raylight. Soluble SmEA was produced as described (11) and HIV-1Ba-L gp120 recombinant protein (catalog no. 4961) was obtained from the National Institutes of Health AIDS Research & Reference Reagent Program (Germantown, MD).
In vitro culture and infection of prMCs
Human prMCs were cultured from adult CD34+ pluripotent progenitors isolated from circulating PBMC under Institutional Review Board-approved protocols as described (10). Rhesus macaque prMCs were derived from CD34+ pluripotent progenitors isolated from freshly prepared aspirates of rhesus macaque bone marrow collected under protocols approved by the Institutional Review Board at Emory University and Yerkes National Primate Research Center. To isolate rhesus macaque pluripotent progenitors, the protocol for immunomagnetic positive selection of human CD34+ cells was modified by substituting an anti-rhesus CD34+ mAb-specific clone 563 for the positive selection of rhesus CD34+ progenitors (Stem Cell Technologies). Rhesus macaque CD34+ pluripotent progenitor-derived prMCs were then developed in vitro exactly as previously described for human prMCs using human recombinant IL-3, IL-6, and stem cell factor (PeproTech) (10). LAD2 cells were maintained in serum-free medium with stem cell factor (100 ng/ml) (12). HIV/SHIV-susceptible human or rhesus macaque prMCs were routinely used at 3–5 wk of culture for infection studies except where otherwise indicated. HIVTybe is an HIV-1/group M, subtype B, X4 virus strain (X4-HIV or virus) that replicates efficiently in macrophages exclusively through use of CXCR4 as a coreceptor. HIVTybe was obtained from the National Institutes of Health Research and Reference Reagent Program (catalog no. 10454). The well-characterized R5 HIV-1 strain (R5-HIV or virus) HIV-1Ba-L was obtained from Dr. S. Gartner, Dr. M. Popovic, and Dr. R. Gallo (Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD (13)) through the National Institutes of Health Research and Reference Reagent Program (catalog no. 510). The CXCR4-tropic strain SHIV33a (14) and the CCR5-CXCR4-tropic strain SHIV89.6P (15, 16) (R5X4-HIV or virus), which are able to infect hematopoietic cells of both rhesus macaque and human origin, were a gift from Dr. F. Villinger (Emory University, Atlanta, GA). All virus infection experiments were conducted by challenging treated target cells with virus (multiplicity of infection ∼0.1), then performing two washes in sterile PBS (pH 7.2) that did not contain Ca2+/Mg2+. Mock-infected target cell controls were prepared the same way in the absence of virus. To test the relative effects of IgE and IgG on susceptibility to virus infection, prMCs were pretreated overnight with IFN-γ (20 ng/ml; PeproTech) to up-regulate functional expression of FcγRI (17).
Determining relative expression of chemokine receptor gene expression by real-time quantitative PCR
Total RNA was prepared from 1–2 × 106 MCs using the QIAamp RNAe-asy Mini kit protocol (Qiagen). Real-time quantitative PCR was performed in triplicate in 50-μl volumes with iScript One-step RT-PCR Mastermix with SYBR Green (Bio-Rad), 200–400 of ng RNA, and the chemokine coreceptor gene-specific validated primers (SuperArray) for CCR3 (catalog no. PPH00612A, band size 170 bp), CCR5 (catalog no. PPH00615A, band size 188 bp), and CXCR4 (catalog no. PPH00621A, band size 150 bp). PCR was performed using a Bio-Rad Icycler real-time PCR system with the following program: 95°C, 10 min; 50 cycles of 95°C, 30 s; 55°C, 30 s; and 72°C, 30 s.
Determining viral entry and infection of prMCs by detection of viral strong stop (SS) cDNA
Susceptibility of MCs to infection with HIV at the level of viral entry was determined by measuring relative levels of R5 and X4 virus SS DNA by real-time quantitative PCR using the common SS primers M667 (sense) GGCTAACTAGGGAACCCACTG and AA55 (antisense) CTGCTA GAGATTTTCCACACTGAC, used at a final concentration of 200 nM. The viral SS DNA, amplified from 400 ng of sample DNA, consisted of an 89-bp DNA product reverse-transcribed from the U5 and R sequences from the 5′ untranslated region as previously described (10).
Immunophenotyping by flow microfluorometry
Analysis of the surface expression of CXCR4 and FcεRI-bound IgE on paraformaldehyde-treated prMC was performed by immunophenotyping and flow microfluorometry with a BD Biosciences FACSCalibur using CellQuest software as reported previously (10).
β-Hexosaminidase (β-hex) release assay
The prMCs were sensitized overnight with IgE-biotin (100 ng/ml). Cells were then washed to remove excess IgE-biotin and cross-linked using streptavidin as reported (17). Levels of β-hex were reported as a percentage of total cell contents released.
Statistical analyses
Results from real-time quantitative PCR and β-hex release assays are reported as mean ± SEM. Data presented are representative of results obtained from experiments using prMCs from three different human and rhesus macaque donors. The fold increase expression of chemokine coreceptor gene mRNA and viral SS cDNA expression relative to reference control groups as determined by real-time quantitative PCR was computed by the 2−ΔΔCt method (18). Where indicated, p values for statistically significant differences in gene expression between experimental groups or between experimental groups and reference controls were computed by the pairwise fixed reallocation randomization test using the relative expression software tool (REST software) (19). Significant differences reported for β-hex release were determined by one-way ANOVA. A value for p < 0.05 was considered significant.
Results
IgE enhances CXCR4 expression and X4-HIV susceptibility by prMCs
To determine the effect of IgE-FcεRI interactions on CXCR4 expression and X4-HIV susceptibility, prMCs derived from human CD34+ cells (3–5 wk of culture) were incubated with varying concentrations of IgE, and the kinetics of viral chemokine coreceptor expression were determined. A significant concentration-dependent increase (∼1600-fold) over the constitutive expression of CXCR4 mRNA was observed in prMCs after treatment with IgE (n = 3 experiments) (Fig. 1A). Induced CXCR4 mRNA expression, which appeared to peak at 48 h, was IgE concentration-dependent over a broad range from 10 to 1000 ng/ml. The cell surface expression of IgE-induced CXCR4 was slightly delayed, peaking after 72 h, but subsequently followed a similar pattern of kinetics (Fig. 1B). The mean fluorescent intensity in the expression of CXCR4 increased by an average of 7.7- and 9.7-fold after 72 h treatment (n = 2 experiments) with IgE concentrations of 100 and 1000 ng/ml, respectively (Fig. 1C). In contrast, IgE treatment showed no measurable inductive effect above the constitutive expression for either CCR3 or CCR5 (Fig. 1, A and B).
FIGURE 1.
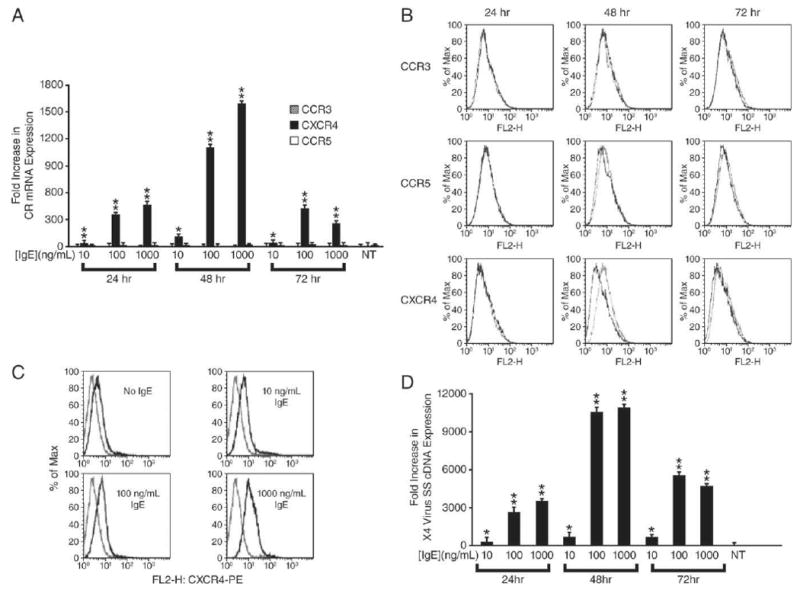
IgE-induced expression of CXCR4 and enhanced susceptibility to infection with X4-HIV by prMCs from 3–5 wk cultures is dose- and time-dependent. A, Relative fold increase in real-time quantitative PCR measurement of chemokine coreceptor mRNA expression by prMCs pulsed for 24, 48, or 72 h with 10, 100, or 1000 ng/ml IgE. **, p ≤ 0.017 significant difference between experimental groups; *, p ≤ 0.017 significant difference between untreated (NT) reference controls. B, Flow microfluorometry of CXCR4 expression (x-axis) of gated populations of CD117+ prMCs pulsed for 48, 72, or 96 h in the absence or presence of 100 ng/ml IgE and immunostained with PE-labeled human CXCR4-specific mAb. The maximum percentage (% Max) of total gated cells (y-axis) at a given fluorescent intensity (represented on the x-axis) is shown. C, Flow microfluorometry of CXCR4 expression (x-axis) of gated populations of CD117+ prMCs pulsed for 72 h in the absence or presence of 10, 100, or 1000 ng/ml IgE and immunostained with PE-labeled human CXCR4-specific mAb. The maximum percentage (% Max) of total gated cells (y-axis) at a given fluorescent intensity (represented on the x-axis) is shown. D, Relative fold increase in real-time quantitative PCR measurements of X4-HIV SS cDNA expression in prMCs pulsed for 24, 48, or 72 h with 10, 100, or 1000 ng/ml IgE and then challenged overnight (18 h) with X4-HIV before quantitative PCR measurements of SS cDNA. **, p ≤ 0.017 significant difference between experimental groups; *, p ≤ 0.017 significant difference between untreated (NT) reference controls.
IgE-induced CXCR4 mRNA and cell surface expression also correlated with an increased susceptibility of prMCs to infection with X4-HIV. The critical determinant for HIV replication and productive infection in MCs is at the level of viral entry and not a function of viral reverse transcription, proviral integration and transcription, or posttranscriptional processing and virion assembly (2). Similarly, our laboratory has shown that virus entry, reverse transcription, proviral integration, and productive or inducible latent infection occurs in circulating prMCs and placental tissue MCs obtained from an HIV-infected pregnant women on highly active antiretroviral therapy (HAART) (10). These studies therefore suggest that measurement of viral SS DNA, one of the earliest indicators of viral entry, is an effective measure to determine infection of prMCs. At each IgE treatment time point (24, 48, or 72 h), prMCs were challenged overnight with X4-HIV. Measurements of relative levels of viral SS cDNA indicated that prMCs were most susceptible to overnight infection with X4 virus 48 h after IgE exposure, paralleling the IgE-induced increase in CXCR4 expression (n = 3 experiments) (Fig. 1, compare D with A).
In contrast, no enhancement above constitutive levels of susceptibility to infection with R5-HIV was observed in IgE-treated prMCs (n = 3 experiments) (Fig. 2A). Thus, IgE-induced up-regulation of CXCR4 expression uniquely correlated with IgE-induced susceptibility to infection with X4-HIV by prMCs. To confirm that exposure to X4-HIV leads to both viral entry and integration and productive infection of MCs, the human MC line LAD2 (12), which expresses high levels of CD4, CCR5, and CXCR4, was challenged overnight (18 h) with virus. Increased levels of viral SS DNA measured in LAD2 cells corresponded with high levels of HIV p24 at day 35 postinfection in parallel cultures of virus-infected LAD2 cells, thus confirming that measurement of HIV viral entry predicts productive infection in MCs (Fig. 2B).
FIGURE 2.
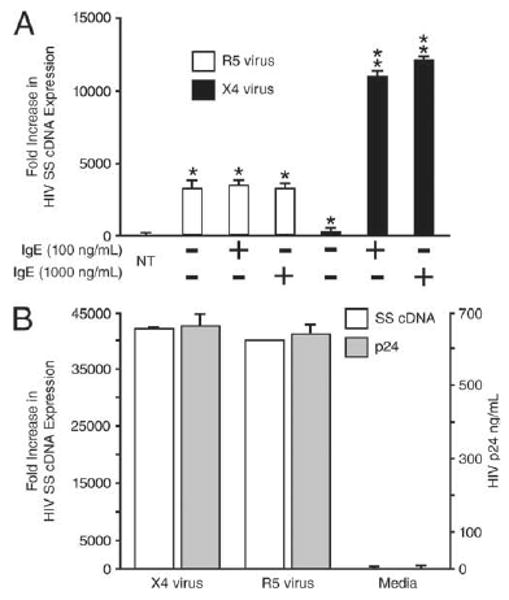
IgE-FceRI interactions do not influence susceptibility to R5 HIV infection. A, Relative fold increase in real-time quantitative PCR measurements of expression of HIV SS cDNA expression in 4–5 wk prMCs (R5 virus) and 3–4 wk prMCs (X4 virus) pulsed for 48 h with 100 or 1000 ng/ml IgE and then challenged overnight with R5 or X4-HIV. **, p ≤ 0.017 significant difference between experimental groups; *, p ≤ 0.017 significant difference between untreated (NT) reference controls. B, Relative fold increase in real-time quantitative PCR measurements (histograms and primary y-axis on the left) of HIV SS cDNA expression in LAD2 MCs challenged overnight (18 h) with R5- or X4-HIV before quantitative PCR measurement of SS cDNA. Levels of HIV p24 in culture supernatant fluids from parallel cultures of LAD2 cells infected with either R5 or X4 virus or left uninfected (Media) were determined by ELISA after day 35 in culture (line graph and secondary y-axis on the right).
Specificity of IgE-enhanced susceptibility for X4 and R5X4 virus and CXCR4 expression
Next, we characterized the specificity of IgE treatment for enhanced susceptibility to X4 virus by prMCs. The prMCs (3–5 wk) were stimulated with IgE (100 ng/ml) for 48 h and challenged overnight with X4 (HIV-1Tybe), R5 (HIV-1Ba-L), or R5X4 (SHIV89.6P) virus in the presence or absence of CXCR4 or CCR5 peptide antagonists. The level of viral SS cDNA was then determined by real-time quantitative PCR. The CXCR4 peptide antagonist RCP138 completely blocked infection of prMCs challenged with X4 virus but had no effect on SS cDNA expression in R5 virus-challenged prMCs (n = 3 experiments) (Fig. 3A). Similarly, the CCR5 peptide antagonist RCP188 completely inhibited infection of prMCs challenged with R5 but did not alter X4 virus infection of IgE-stimulated prMCs (Fig. 3A).
FIGURE 3.
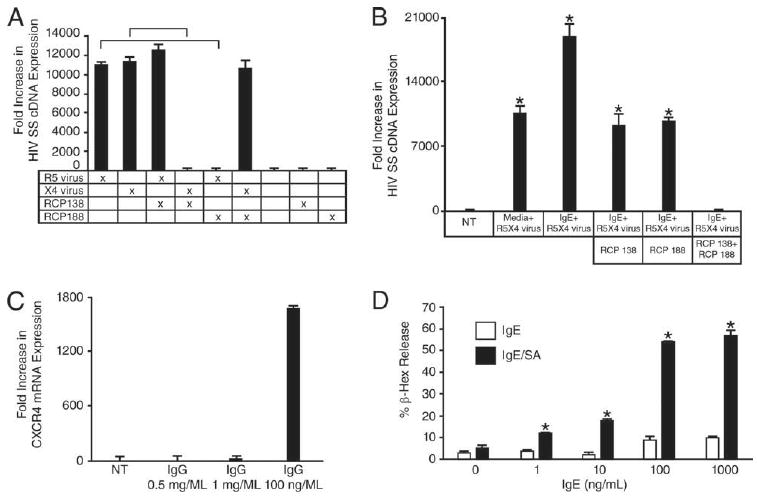
IgE-enhanced susceptibility for X4- and R5X4-HIV is mediated specifically through up-regulation of CXCR4 with minimal MC activation. A, Relative fold increase in real-time quantitative PCR measurements of X4- and R5-HIV SS cDNA expression in 3–4 wk prMCs pulsed with IgE (100 ng/ml, 48 h) and then challenged overnight with R5- or X4-HIV in the presence or absence of the CXCR4 peptide antagonist RCP138 or the CCR5 peptide antagonist RCP188. Significant difference (p ≤ 0.017) in the levels of HIV SS cDNA between prMCs infected with either R5 or X4 virus in the absence or presence of the specific chemokine coreceptor peptide antagonists RCP188 or RCP138, respectively, is indicated with horizontal bar. B, Relative fold increase in real-time quantitative PCR measurements of R5X4-HIV SS cDNA expression in 3–4 wk prMCs pulsed with IgE (100 ng/ml, 48 h) and then challenged overnight with R5X4 virus in the absence or presence of peptide antagonists RCP138, RCP188 or a combination of both. *, p ≤ 0.03 significant difference between prMCs infected with R5X4 virus in the presence of either RCP188 or RCP138, and prMCs infected with R5X4 virus in the presence of both RCP188 and RCP138 and untreated (NT) reference controls. C, Relative fold increase in CXCR4 mRNA expression in prMCs pulsed with IgE-(100 ng/ml) or IFN-γ–pretreated prMCs pulsed with IgG (500 and 1000 μg/ml) for 48 h. D, Percentage of release of β-hex in prMCs pulsed with IgE (1, 10, 100, or 1000 ng/ml) for 48 h or treated with IgE (1, 10, 100, or 1000 ng/ml) for 2 h followed by aggregation with streptavidin (SA, 125 ng/ml) for 2 h. *, p ≤ 0.05 significant difference determined by ANOVA between experimental groups.
This same approach was used to assess the specific effects of IgE stimulation on the susceptibility of prMCs to R5X4 virus. As shown in Fig. 3B, in the absence of chemokine coreceptor peptide antagonists, IgE stimulation of prMCs nearly doubled the levels of viral SS cDNA over unstimulated prMCs after challenge with R5X4 virus. Conversely, challenge with R5X4 virus in the presence of both RCP138 and RCP188 completely inhibited infection of IgE-treated prMCs. Furthermore, levels of SS cDNA in IgE-treated prMCs after R5X4 virus infection in the presence of RCP138 or RCP188 alone were comparable to those present in virus-infected IgE-naive prMCs in the absence of either chemokine coreceptor peptide antagonist (Fig. 3B).
IgE uniquely enhances CXCR4 expression with minimal MC activation
Induction of CXCR4 expression by IgE was compared with induction by IgG to measure the specificity of the response by prMCs. IgE is the least abundant serum Ig with a concentration ranging from 100 to 150 ng/ml in normal (nonatopic) individuals and 1–2 μg/ml in atopic individuals. In contrast, the normal range for plasma levels of IgG is from 1 to 10 mg/ml (20). Therefore, to compare how IgG-FcγRI interactions affected CXCR4 expression relative to IgE, experimental groups of 3- to 5-wk-cultured prMCs were pretreated overnight with or without IFN-γ (20 ng/ml), which up-regulates FcγRI receptors on human MCs (17). The cultured prMCs were then incubated with human IgG or with IgE for 48 h. The functional expression of CXCR4 on prMCs was induced only by IgE and not IgG, even when prMCs were treated with IgG at levels 5000- to 10,000-fold greater than IgE (n = 3 experiments) (Fig. 3C). We also verified that IgE treatment-enhanced CXCR4 expression of prMCs was not simply the result of nonspecific activation as IgE treatment induced minimal MC degranulation. IgE levels that stimulated a 1500-fold increase in CXCR4 expression caused β-hex release of less than 10% compared with ∼60% release from prMCs incubated with cross-linked IgE (IgE-streptavidin) (Fig. 3D).
Cross-linked IgE enhances CXCR4 expression and susceptibility to X4 virus
MC may be activated either by IgE (monomeric IgE) or by IgE-streptavidin (cross-linked IgE) through interactions with MC-bound FcεRI. To account for the possible interaction between monomeric IgE and undetected serum proteins, IgE purified from three different myeloma sources were tested under serum-free culture conditions. Comparable levels of CXCR4 mRNA expression (1200- to 1800-fold increase) were induced on 3- to 5-wk-cultured prMCs after 48 h with all three IgE preparations (data not shown). All subsequent experiments were conducted using a single source of myeloma IgE, as described in Materials and Methods. However, another possibility is that even in the absence of cross-linking, monomeric IgE interactions with FcεRI may still result in FcεRI aggregation and as a result low level MC activation over time (21). Considering this possibility, we tested the hypothesis that IgE-FcεRI cross-linking would accelerate prMC expression of CXCR4 and enhanced susceptibility to X4-HIV. Streptavidin-mediated cross-linking of IgE-biotin bound to FcεRI on prMCs for 2 h induced levels of CXCR4 mRNA expression (n = 3 experiments) (Fig. 4A) and X4-HIV susceptibility (n = 3 experiments) (Fig. 4B) that were similar to prMC treated with IgE alone for 48 h. Thus, maximum levels of induced CXCR4 expression occurred much more rapidly on prMCs following FcεRI aggregation.
FIGURE 4.
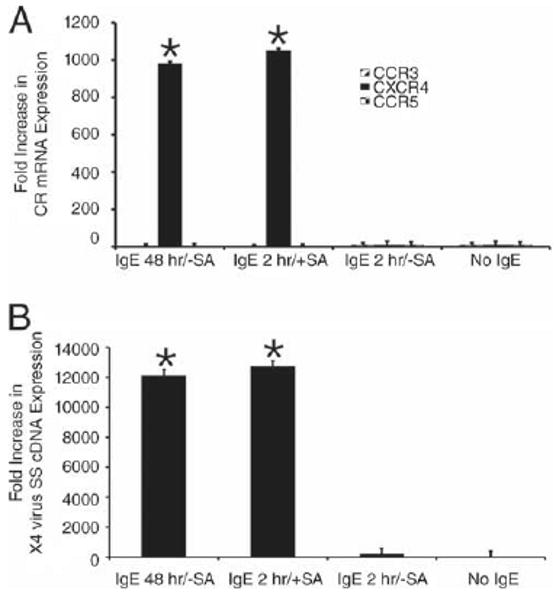
Cross-linked IgE enhances CXCR4 mRNA expression and susceptibility to X4-HIV. A, Relative fold increases in CCR3, CXCR4, and CCR5 mRNA expression in 3- to 5-wk-cultured prMCs that were treated with IgE (100 ng/ml) for 48 h without streptavidin (IgE 48 h/−SA), IgE for 2 h followed by aggregation with streptavidin for 2 h (IgE 2 h/+SA), IgE for 2 h without streptavidin (IgE 2 h/−SA), and in control (No IgE) prMCs. *, p ≤ 0.001 significant difference between experimental groups and medium control groups (No IgE). B, Relative fold increase in X4-HIV SS cDNA expression in prMCs treated with IgE (100 ng/ml) for 48 h without streptavidin (IgE 48 h/−SA), IgE for 2 h followed by aggregation with streptavidin for 2 h (IgE 2 h/+SA), IgE for 2 h without streptavidin (IgE 2 h/−SA), and untreated (No IgE), and then challenged overnight with X4-HIV. *, p ≤ 0.001 significant difference between experimental groups and medium controls (No IgE).
Superallergen-mediated enhancement of CXCR4 expression and susceptibility to X4-HIV is IgE-dependent
Certain viral, bacterial, and helminthic proteins cross-link cell-bound IgE nonspecifically and activate MCs and basophils by acting as “superallergens” that directly induce IgE-FcεRI complex clustering or aggregation (22–24). We thus chose to examine the ability of two well-characterized superallergens, SmEA and HIV-1gp120, to mediate IgE-FcεRI aggregation-dependent enhancement of the functional expression of CXCR4 and the susceptibility of prMCs to X4 virus. Schistosoma mansoni infection increases SHIV replication and susceptibility to viral infection (25) in rhesus macaques. The effects of SmEA-mediated IgE-FcεRI aggregation on enhancement of CXCR4 expression and X4 virus susceptibility were therefore evaluated for both human (HIV-1Tybe strain) and rhesus macaque (SHIV33a strain) prMCs because S. mansoni infection models in rhesus macaques have been well established (26) and the results of these in vitro studies could thus be readily extended in this animal model.
In a series of parallel experiments, 3- to 5-wk-cultured human and rhesus macaque prMCs were pulsed with IgE for 2 h in the absence or presence of different concentrations of omalizumab (10, 100, and 1000 ng/ml), a drug that blocks IgE-FcεRI interactions. IgE-untreated prMCs were included as negative (medium) controls. The prMCs were then cocultured with different concentrations of SmEA (10–1000 ng/ml) for an additional 2 h. Results from real-time quantitative PCR revealed a significant SmEA dose-dependent enhancement of CXCR4 mRNA expression in the presence of IgE that was completely inhibited by omalizumab at higher concentrations (100 or 1000 ng/ml) in both human and rhesus macaque prMCs (n = 3 experiments) (Fig. 5A). The increased levels of SmEA-IgE-induced CXCR4 expression also significantly enhanced susceptibility of both human and rhesus prMCs to X4 virus (n = 3 experiments) (Fig. 5B). Results from real-time quantitative PCR revealed a dose-dependent increase in X4 virus SS cDNA expression relative to medium controls (no IgE treatment) that correlated with the degree of SmEA-induced CXCR4 expression (Fig. 5, compare B and A). Although the level of CXCR4 mRNA expression induced by SmEA-IgE interactions was less than half of the level of expression seen in prMCs after IgE-streptavidin biotin-treated activation (compare Figs. 5A and 4A), comparable levels of X4 virus SS cDNA and virus susceptibility were observed with both treatments (compare Figs. 5B and 4B). Similarly, 2 h of pretreatment of prMCs with IgE followed by aggregation with recombinant HIVgp120 (0.1, 1.0, 10, or 20 μg/ml) resulted in a significant dose-dependent increase in susceptibility to X4-HIV with viral SS cDNA expression comparable to levels of SS cDNA measured in SmEA-IgE and IgE-streptavidin biotin-treated prMCs (Fig. 5, compare C and B).
FIGURE 5.
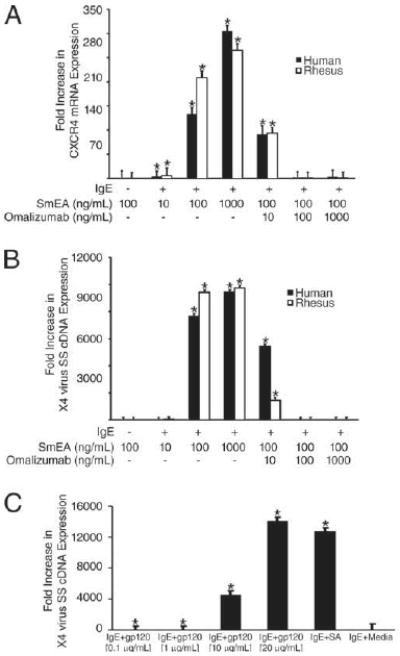
Superallergens (SmEA or HIVgp120) mediate IgE cross-linking-dependent enhancement of CXCR4 mRNA expression and susceptibility to X4-HIV. A, Relative fold increase in CXCR4 mRNA expression in human and rhesus prMCs (3–5 wk cultures) untreated or pulsed with IgE (100 ng/ml) in the absence or presence of omalizumab (10, 100, and 1000 ng/ml) for 2 h and then treated with SmEA (10, 100, or 1000 ng/ml) for 2 h. *, p ≤ 0.03 significant difference between experimental groups and medium control groups (No IgE). B, Relative fold increase in X4 virus SS cDNA expression in human and rhesus prMCs untreated or pulsed with IgE (100 ng/ml) in the absence or presence of omalizumab (10, 100, and 1000 ng/ml) for 2 h, treated with SmEA (10, 100, or 1000 ng/ml) for 2 h and then challenged overnight with CXCR4-tropic HIVTybe (human prMCs) or SHIV33a (rhesus prMCs). *, p ≤ 0.03 significant difference between experimental groups and medium control groups (No IgE). C, Relative fold increase in X4-HIV SS cDNA expression in human prMCs treated with IgE-biotin (100 ng/ml) alone for 2 h, IgE (100 ng/ml) plus medium (IgE + Media), or IgE-biotin (100 ng/ml) for 2 h followed by aggregation with different doses (0.1, 1.0, 10, or 20 μg/ml) of soluble recombinant HIVgp120 or streptavidin (SA, 125 ng/ml) for 2 h. The prMCs were then incubated overnight with X4-HIV. *, p ≤ 0.03 significant difference between experimental groups and IgE plus medium control groups.
IgE-enhanced CXCR4 expression and susceptibility to X4-HIV is determined by prMC maturational stage
MC susceptibility to infection with HIV is uniquely limited to discreet maturational stages during which they express CD4 and relevant chemokine coreceptors to permit viral entry and infection (2, 4). We thus investigated whether prMCs have a unique developmental stage during which treatment with IgE induces an up-regulation of CXCR4 expression and a corresponding enhanced susceptibility to X4 HIV. As observed in previous experiments (Fig. 1A), treatment with IgE (48 h) had no effect on the constitutive expression of CCR3 or CCR5 in prMCs (Fig. 6, A and B). However, a striking pattern of IgE-induced enhancement of CXCR4 mRNA expression was observed. As shown in Fig. 6C, IgE-induced CXCR4 mRNA expression was detectable in cells cultured for 2 wk, peaked in prMCs cultured for 4 wk, and decreased in older cultures. By wk 8–9, MCs had become refractory to IgE-induced expression of CXCR4 (n = 3 experiments).
FIGURE 6.
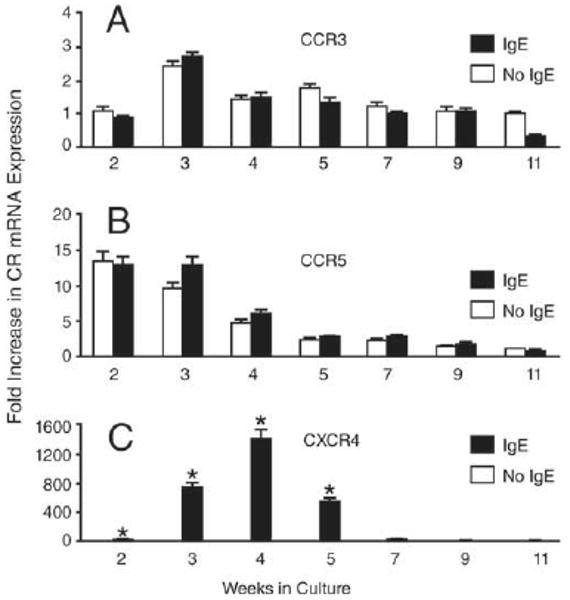
IgE-dependent enhancement of CXCR4 mRNA expression is determined by the maturational stage of prMCs. Relative fold increase in real-time quantitative PCR measurements of CCR3 (A), CCR5 (B), and CXCR4 (C) mRNA expression in human prMCs pulsed with IgE (100 ng/ml, 48 h) at weekly intervals (2–11 wk in culture). Fold increase in chemokine coreceptor mRNA expression was normalized against the mean value of triplicate measurements of the lowest specific mRNA expression coinciding with prMCs at 11 wk in culture. *, p ≤ 0.03 significant difference between IgE-treated and untreated groups at each weekly interval.
IgE-FcεRI interactions exclusively increased susceptibility to X4 virus in prMC. Measurements of viral SS cDNA indicated that stimulation with IgE had no effect on the susceptibility of prMCs to R5 HIV at any point during development (Fig. 7A). However, stimulation with IgE (48 h) resulted in a dramatic enhancement of prMC susceptibility to X4-HIV, which peaked in 4-wk cultures, parallel with IgE enhancement of CXCR4 expression during the same period (compare Figs. 7B and 6C). Again, mature (7 wk or more) MCs were refractory to IgE-induced susceptibility to X4-HIV infection (n = 3 experiments).
FIGURE 7.
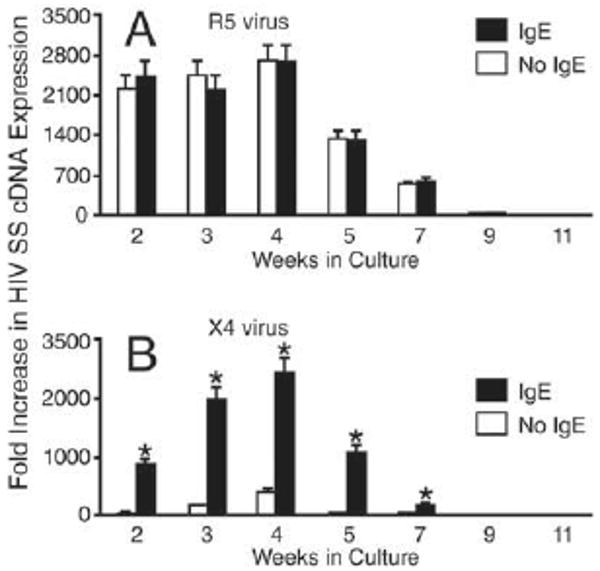
IgE-dependent enhancement of susceptibility to infection with X4-HIV is determined by the maturational stage of the prMCs. Relative fold increase in real-time quantitative PCR measurements of R5-HIV (A) and X4-HIV (B) SS cDNA expression in human prMCs pulsed with IgE (100 ng/ml, 48 h) at weekly intervals (2–11 wk in culture) and then incubated overnight with R5- or X4-HIV. Fold increase in SS cDNA expression was normalized against the mean value of triplicate reproducible measurements of the lowest specific cDNA expression coinciding with prMCs at 11 wk in culture. *, p ≤ 0.03 significant difference between IgE-treated and untreated groups at each weekly interval.
Discussion
In ∼50% of HIV patients, a “switch” to a dominant CXCR4-tropic phenotype occurs, corresponding to an accelerated rate of CD4+ T cell loss and a shift to TH2 immunity (1). Although the precise mechanisms have not been elucidated, the CCR5-to-CXCR4 tropic shift in coreceptor usage among emerging variants is influenced by HAART, antiviral immune responses and the dynamic interactions between viral quasispecies and their various target cell populations within viral habitats and reservoirs (27–30). Our findings that IgE-FcεRI interactions up-regulate functional expression of CXCR4 and thus expand the window of susceptibility of prMCs to both X4 and R5X4 viruses (1), and that this expanded range of virus susceptibility occurs during a well-defined period of early MC development (2), are significant because they provide new insights into how IgE-mediated allergy or helminthic coinfection may facilitate the establishment of a MC reservoir of persistent X4 virus infection. This mechanism may influence a movement to a predominant X4 virus phenotype and the progression of HIV/AIDS in infected individuals.
We have reported that mature tissue MCs can archive latent, inducible infectious HIV in vivo (10). Data presented in this study now show that IgE-FcεRI binding, even in the absence of cross-linking Ag, and at IgE concentrations as low as 100 ng/ml, can expand the presence of both X4- and R5X4-HIV in populations of prMCs that supply the developing MC reservoir of persistent infection (Figs. 1C and 3, A and B). Although the data obtained with IgE in the absence of Ag would support a role for monomeric IgE in the regulation of specific MC responses as has been previously reported (31), we cannot rule out the possibility that the responses were due to dimers or small oligomers of IgE forming over the duration of the experiments. Regardless, these responses were observed under conditions, which were not sufficient to induce significant MC degranulation (Fig. 3D).
Our data also revealed that, in the presence of streptavidin- or superallergen-induced coaggregation, up-regulation of CXCR4 expression occurs much more rapidly and suggest that maximum levels of virus susceptibility can be achieved with submaximal induction of CXCR4 mRNA expression (compare Figs. 1, 4, and 5, A and B). These findings indicate that IgE-FcεRI aggregation can promote the inductive signaling pathway leading to functional expression of CXCR4 on prMCs. Furthermore, the IgE effect is specifically mediated through an FcεRI-dependent pathway. Omalizumab, which effectively inhibits binding of IgE to FcεRI (the predominant IgE receptor expressed throughout MC ontogeny) (32), specifically uncoupled and completely abolished IgE-dependent enhancement of prMC CXCR4 expression and susceptibility to X4-HIV (Fig. 5, A and B). The broader implications of these findings are that pre-existing comorbid conditions such as atopic disease or helminthic infections with elevated IgE levels may significantly contribute to the establishment of X4 and R5X4 virus within the MC reservoir during HIV infection.
Our observation that 3- to 5-wk-old prMCs, but not mature (8-wk-old or more) MCs (Figs. 6 and 7), were susceptible to X4 virus infection in the presence of IgE or cross-linked IgE has at least two important consequences. First, transmission of X4 virus to prMCs occurs not only in the presence of receptor-bound IgE but also predominates in those tissue compartments that support the growth and maintenance of the HIV-susceptible FcεRI+/CD4+/CCR5+/CXCR4+ prMC phenotype. MCs with this progenitor phenotype are found in the circulation (33). However, whether tissue MCs with a similar IgE-sensitive progenitor phenotype are present within specialized tissue compartments (e.g., the gastrointestinal tract) remains to be determined. Secondly, up-regulation of the functional cell surface expression CXCR4 in CD34+ progenitor cells is mediated by a cAMP-dependent pathway (34). Up-regulation of cell surface expression of CXCR4 on lymphocytes by cAMP (35, 36) has not only been shown to enhance chemotaxis in response to SDF-1, but also susceptibility to infection with HIV (37). Ongoing studies in our laboratory with LAD cells (12), which emulate in many ways the IgE inducible expression of CXCR4 on primary 3- to 5-wk-old prMCs, support the involvement of cAMP in the inducible expression of CXCR4 on these prMCs (our unpublished observation). Therefore, our findings that IgE-mediated regulation of CXCR4 on MCs recedes as they mature (Figs. 6 and 7) suggest that signaling pathways mediated by IgE-FcεRI interactions may also coordinately change and evolve as prMCs mature. A broader implication of these findings is that progenitors of potentially other, yet to be described, cell lineages may also become transiently susceptible to HIV infection during unique stages of development and then covertly maintain latent persistent infection in a similar fashion as they mature into otherwise HIV infection-resistant cell phenotypes.
These results also underscore the general theme and overall significance of the relationship between MC maturational or developmental stages and susceptibility to both R5- and (now) X4-HIV infection. The dynamic constitutive (IgE-independent) expression of CXCR5 and corresponding relative susceptibility to infection with R5 virus varies with the developmental stage of prMCs derived from either fetal (2, 4) or adult CD34+-derived committed progenitors (Figs. 6 and 7). As shown in Fig. 6, constitutive expression of CXCR5 is maximal at 2–3 wk of culture and then begins a dramatic decline. These observations may explain differences in maximum levels of R5 SS cDNA obtained from prMCs exposed to virus at wk 4–5 (Fig. 2A) and wk 3–4 (Fig. 3, A and B). MCs appear unique among non-T cell lineages in that their transient susceptibility to infection to both R5 and X4 viruses is related to biological and IgE-independent and – dependent physiologic characteristics at different stages of development. Thus, during ongoing HIV infection, conditions that are associated with MC mobilization, recruitment and survival, such as IgE-mediated allergy, schistosomiasis, or other helminth infections (31, 38), may establish a reservoir of latently infected “tissue” MCs harboring infectious R5-, X4-, and R5X4-HIV within diverse tissue compartments.
Many questions remain regarding the clinical significance and the role of IgE in the pathogenesis of HIV infection in the presence of coinfection, especially in the gut. Adult female S. mansoni residing in the intestinal vasculature release eggs that potently induce IgE and Th2 immune responses (39). The shift toward a Th2 immune environment limits host antiviral immune responses and promotes survival of MCs (40). One prediction based on our findings that IgE-FcεRI interactions enhance expression of CXCR4 and susceptibility to X4 and R5/X4-tropic HIV/SIV (Figs. 1 and 3) is that increased levels of IgE and prMCs during allergy or a helminth infection would expand the presence of archived X4 and R5X4 viruses in the MC reservoir and thus proportionately increase the opportunity for X4 and R5X4 viruses to persist and eventually emerge as viral fitness landscapes change during disease progression. To our knowledge, no formal studies correlating IgE levels and CCR5-to-CXCR4 tropism shifts during HIV or SIV infection have been conducted. However, several new reports of clinical findings indicate that effective HAART, which suppresses viral replication and evolution, creates an environment necessary for the emergence of CXCR4-tropic variants archived in cellular reservoirs, and are thus consistent with this prediction from our model (27–29). Acute S. mansoni infection also increases susceptibility to systemic SHIV Clade C infection in rhesus macaques after mucosal virus exposure (25). The IgE-SmEA-mediated enhancement of rhesus macaque prMC susceptibility to X4-tropic SHIV33a (Fig. 5) makes it feasible to study the consequences of schistosomiasis coinfection and SHIV disease progression in this nonhuman primate model of AIDS.
Many questions also remain regarding the relative clinical and pathological significance of the MC lineage as a target of HIV infection. Knowledge of the scope and diversity of susceptible target cell populations that comprise different reservoirs of persistent HIV infection during HAART now includes many non-T cell lineages, such as macrophages, dendritic cells (DCs), and others (41, 42). MCs represent a new lineage of target cells that share many important similarities with macrophages and DCs in terms of their ability to serve as agents of disseminated HIV infection (4, 10, 41, 42). All three of these cell lineages are primarily susceptible to R5-HIV and only to a lesser extent X4-HIV (4, 10, 43, 44). HIV-infected MCs, macrophages, and DCs are all capable of interacting with and spreading HIV infection to susceptible populations of T cells (4, 10, 44, 45). Like macrophages, MCs are widely distributed among various tissues, including the intestinal lamina propria and gut mucosal tissues, which are now recognized as the major anatomical sites for HIV transmission and replication during acute infection (5, 7). MCs, like DCs, can be found in regional lymph nodes and secondary lymphoid organs and can interact with and induce clonal expansion of Ag-specific susceptible T cells (46, 47). However, although macrophages and DCs, as well as many progenitor cells, constitutively express CXCR4, they remain refractory or only marginally susceptible to infection with X4-HIV (43, 44). Therefore, unlike MCs, their potential role in X4 virus infection, expansion, and viral persistence is most likely limited.
Human MCs also differ from macrophages and DCs in their ability to harbor latent inducible infectious HIV proviral DNA (4, 10). Reactivation of viral replication in latently infected mature MCs does not affect MC viability and can be achieved through multiple activation pathways, including signaling through TLR2, TLR4, TLR9, and IgE cross-linking (4, 10). Also, because latency is only established after infected prMCs differentiate into HIV-infection resistant mature forms, clonally conserved virus can become archived in the MC reservoir (4, 10).
The significance of this observation is becoming increasingly apparent with reports of two important findings from studies of persistent HIV infection during antiretroviral therapy. First, in some patients on long-term effective HAART, the source of residual viremia from invariant clones not found in circulating T cells may be a monocyte-macrophage lineage progenitor capable of producing virus that can infect CD4+ T cells (48). It is unlikely that macrophages are the source because viral evolution is accelerated in infected tissue macrophages relative to T cells (49). Second, there is new evidence that X4-HIV is archived during periods of productive infection and is able to persist even after long-term administration of effective HAART (27, 50). How this action happens and the extents to which MCs contribute to X4 virus persistence during HAART have not been elucidated.
Our data suggest that MCs are the primary, if not exclusive, FcεRI-expressing cell lineage that may serve as an inducible target for X4-HIV infection. Although other cell lineages, such as eosinophils, do express mRNA and protein for FcεRI, evidence from both flow microfluorometry and functional assays indicate that FcεRI is not expressed in any significant levels on the cell surface; thus limiting IgE-FcεRI-induced responses by eosinophils (33, 51). Mature eosinophils do express low levels of CD4 and CXCR4 and are moderately susceptible to X4, but not R5, tropic HIV (52). However, unlike MCs, they succumb to viral-mediated apoptosis and necrosis and are thereby unlikely to serve as reservoirs of latent infection. Furthermore, unlike MCs, eosinophils develop and mature in the bone marrow from CD34+ progenitors and are released to the peripheral blood as mature cells. Because of their relatively short half-life (7–14 days) compared with tissue MCs (months) and the susceptibility of eosinophils to viral cytotoxicity (unlike MCs), they would only present a minor contribution, if any, to persistent retroviral infection.
Many important details regarding the clinical significance and basic pathogenic mechanisms of the role of IgE in the establishment of a MC reservoir of persistent X4-HIV infection in vivo must be elucidated. For instance, the kinetics and conditions for establishing a MC reservoir in vivo during atopy or helminthic parasitic coinfection, as well as its significance to HIV disease progression, especially during HAART, will have to be addressed with the proper animal models. Our findings, however, make the design and implementation of these studies conceivable. The results of IgE-SmEA-mediated enhancement of rhesus macaque prMC susceptibility to CXCR4-tropic SHIV33a (Fig. 5) provide the opportunity to address these questions using the established rhesus macaque models of schistosomiasis/SIV coinfection (40).
Footnotes
This work was supported in part by Grant R01AI062383 from the National Institutes of Health (to J.B.S.), in part by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases/National Institutes of Health, and in part by base Grant RR-00165 from the Animal Resources Program of the National Institutes of Health awarded to the Yerkes National Primate Research Center at Emory University.
Abbreviations used in this paper: prMC, progenitor mast cell; MC, mast cell; SS, strong stop; β-hex, β-hexosaminidase; SmEA, Schistosoma mansoni egg Ag; DC, dendritic cell; HAART, highly active antiretroviral therapy.
Disclosures
The authors have no financial conflict of interest. The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention.
References
- 1.Berger EA, Murphy PM, Farber JM. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu Rev Immunol. 1999;17:657–700. doi: 10.1146/annurev.immunol.17.1.657. [DOI] [PubMed] [Google Scholar]
- 2.Bannert N, Farzan M, Friend DS, Ochi H, Price KS, Sodroski J, Boyce JA. Human Mast cell progenitors can be infected by macrophagetropic human immunodeficiency virus type 1 and retain virus with maturation in vitro. J Virol. 2001;75:10808–10814. doi: 10.1128/JVI.75.22.10808-10814.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Y, Li L, Wadley R, Reddel SW, Qi JC, Archis C, Collins A, Clark E, Cooley M, Kouts S, et al. Mast cells/basophils in the peripheral blood of allergic individuals who are HIV-1 susceptible due to their surface expression of CD4 and the chemokine receptors CCR3, CCR5, and CXCR4. Blood. 2001;97:3484–3490. doi: 10.1182/blood.v97.11.3484. [DOI] [PubMed] [Google Scholar]
- 4.Sundstrom JB, Little DM, Villinger F, Ellis JE, Ansari AA. Signaling through Toll-like receptors triggers HIV-1 replication in latently infected mast cells. J Immunol. 2004;172:4391–4401. doi: 10.4049/jimmunol.172.7.4391. [DOI] [PubMed] [Google Scholar]
- 5.Brenchley JM, Schacker TW, Ruff LE, Price DA, Taylor JH, Beilman GJ, Nguyen PL, Khoruts A, Larson M, Haase AT, Douek DC. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med. 2004;200:749–759. doi: 10.1084/jem.20040874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mehandru S, Poles MA, Tenner-Racz K, Horowitz A, Hurley A, Hogan C, Boden D, Racz P, Markowitz M. Primary HIV-1 infection is associated with preferential depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal tract. J Exp Med. 2004;200:761–770. doi: 10.1084/jem.20041196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mehandru S, Poles MA, Tenner-Racz K, Manuelli V, Jean-Pierre P, Lopez P, Shet A, Low A, Mohri H, Boden D, et al. Mechanisms of gastrointestinal CD4+ T-cell depletion during acute and early human immunodeficiency virus type 1 infection. J Virol. 2007;81:599–612. doi: 10.1128/JVI.01739-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Veazey R, Lackner A. The mucosal immune system and HIV-1 infection. AIDS Rev. 2003;5:245–252. [PubMed] [Google Scholar]
- 9.Irani AM, Craig SS, DeBlois G, Elson CO, Schechter NM, Schwartz LB. Deficiency of the tryptase-positive, chymase-negative mast cell type in gastrointestinal mucosa of patients with defective T lymphocyte function. J Immunol. 1987;138:4381–4386. [PubMed] [Google Scholar]
- 10.Sundstrom JB, Ellis JE, Hair GA, Kirshenbaum AS, Metcalfe DD, Yi H, Cardona AC, Lindsay MK, Ansari AA. Human tissue mast cells are an inducible reservoir of persistent HIV infection. Blood. 2007;109:5293–5300. doi: 10.1182/blood-2006-11-058438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carter CE, Colley DG. An electrophoretic analysis of Schistosoma mansoni soluble egg antigen preparation. J Parasitol. 1978;64:285–290. [PubMed] [Google Scholar]
- 12.Kirshenbaum AS, Akin C, Wu Y, Rottem M, Goff JP, Beaven MA, Rao VK, Metcalfe DD. Characterization of novel stem cell factor responsive human mast cell lines LAD 1 and 2 established from a patient with mast cell sarcoma/leukemia; activation following aggregation of FcεRI or FcγRI. Leukocyte Res. 2003;27:677–682. doi: 10.1016/s0145-2126(02)00343-0. [DOI] [PubMed] [Google Scholar]
- 13.Gartner S, Markovits P, Markovitz DM, Kaplan MH, Gallo RC, Popovic M. The role of mononuclear phagocytes in HTLV-III/LAV infection. Science. 1986;233:215–219. doi: 10.1126/science.3014648. [DOI] [PubMed] [Google Scholar]
- 14.Harouse JM, Gettie A, Tan RC, Blanchard J, Cheng-Mayer C. Distinct pathogenic sequela in rhesus macaques infected with CCR5 or CXCR4 utilizing SHIVs. Science. 1999;284:816–819. doi: 10.1126/science.284.5415.816. [DOI] [PubMed] [Google Scholar]
- 15.Doranz BJ, Rucker J, Yi Y, Smyth RJ, Samson M, Peiper SC, Parmentier M, Collman RG, Doms RW. A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell. 1996;85:1149–1158. doi: 10.1016/s0092-8674(00)81314-8. [DOI] [PubMed] [Google Scholar]
- 16.Reimann KA, Li JT, Veazey R, Halloran M, Park IW, Karlsson GB, Sodroski J, Letvin NL. A chimeric simian/human immunodeficiency virus expressing a primary patient human immunodeficiency virus type 1 isolate env causes an AIDS-like disease after in vivo passage in rhesus monkeys. J Virol. 1996;70:6922–6928. doi: 10.1128/jvi.70.10.6922-6928.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woolhiser MR, Okayama Y, Gilfillan AM, Metcalfe DD. IgG-dependent activation of human mast cells following up-regulation of FcγRI by IFN-γ. Eur J Immunol. 2001;31:3298–3307. doi: 10.1002/1521-4141(200111)31:11<3298::aid-immu3298>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 18.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔCT) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 19.Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gould HJ, Sutton BJ, Beavil AJ, Beavil RL, McCloskey N, Coker HA, Fear D, Smurthwaite L. The biology of IGE and the basis of allergic disease. Annu Rev Immunol. 2003;21:579–628. doi: 10.1146/annurev.immunol.21.120601.141103. [DOI] [PubMed] [Google Scholar]
- 21.Schweitzer-Stenner R, Pecht I. Death of a dogma or enforcing the artificial: monomeric IgE binding may initiate mast cell response by inducing its receptor aggregation. J Immunol. 2005;174:4461–4464. doi: 10.4049/jimmunol.174.8.4461. [DOI] [PubMed] [Google Scholar]
- 22.Marone G, Rossi FW, Detoraki A, Granata F, Marone G, Genovese A, Spadaro G. Role of superallergens in allergic disorders. Chem Immunol Allergy. 2007;93:195–213. doi: 10.1159/000100896. [DOI] [PubMed] [Google Scholar]
- 23.Schramm G, Falcone FH, Gronow A, Haisch K, Mamat U, Doenhoff MJ, Oliveira G, Galle J, Dahinden CA, Haas H. Molecular characterization of an interleukin-4-inducing factor from Schistosoma mansoni eggs. J Biol Chem. 2003;278:18384–18392. doi: 10.1074/jbc.M300497200. [DOI] [PubMed] [Google Scholar]
- 24.Schramm G, Mohrs K, Wodrich M, Doenhoff MJ, Pearce EJ, Haas H, Mohrs M. Cutting edge: IPSE/α-1, a glycoprotein from Schistosoma mansoni eggs, induces IgE-dependent, antigen-independent IL-4 production by murine basophils in vivo. J Immunol. 2007;178:6023–6027. doi: 10.4049/jimmunol.178.10.6023. [DOI] [PubMed] [Google Scholar]
- 25.Chenine AL, Shai-Kobiler E, Steele LN, Ong H, Augostini P, Song R, Lee SJ, Autissier P, Ruprecht RM, Secor WE. Acute Schistosoma mansoni infection increases susceptibility to systemic SHIV Clade C infection in rhesus macaques after mucosal virus exposure. PLoS Negl Trop Dis. 2008;2:e265. doi: 10.1371/journal.pntd.0000265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ayash-Rashkovsky M, Chenine AL, Steele LN, Lee SJ, Song R, Ong H, Rasmussen RA, Hofmann-Lehmann R, Else JG, Augostini P, et al. Coinfection with Schistosoma mansoni reactivates viremia in rhesus macaques with chronic simian-human immunodeficiency virus Clade C infection. Infect Immun. 2007;75:1751–1756. doi: 10.1128/IAI.01703-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Delobel P, Sandres-Saune K, Cazabat M, Pasquier C, Marchou B, Massip P, Izopet J. R5 to X4 switch of the predominant HIV-1 population in cellular reservoirs during effective highly active antiretroviral therapy. J Acquir Immune Defic Syndr. 2005;38:382–392. doi: 10.1097/01.qai.0000152835.17747.47. [DOI] [PubMed] [Google Scholar]
- 28.Hunt PW, Harrigan PR, Huang W, Bates M, Williamson DW, McCune JM, Price RW, Spudich SS, Lampiris H, Hoh R, et al. Prevalence of CXCR4 tropism among antiretroviral-treated HIV-1-infected patients with detectable viremia. J Infect Dis. 2006;194:926–930. doi: 10.1086/507312. [DOI] [PubMed] [Google Scholar]
- 29.Johnston ER, Zijenah LS, Mutetwa S, Kantor R, Kittinunvorakoon C, Katzenstein DA. High frequency of syncytium-inducing and CXCR4-tropic viruses among human immunodeficiency virus type 1 subtype C-infected patients receiving antiretroviral treatment. J Virol. 2003;77:7682–7688. doi: 10.1128/JVI.77.13.7682-7688.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Regoes RR, Bonhoeffer S. The HIV coreceptor switch: a population dynamical perspective. Trends Microbiol. 2005;13:269–277. doi: 10.1016/j.tim.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 31.Kawakami T, Kitaura J. Mast cell survival and activation by IgE in the absence of antigen: a consideration of the biologic mechanisms and relevance. J Immunol. 2005;175:4167–4173. doi: 10.4049/jimmunol.175.7.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Presta LG, Lahr SJ, Shields RL, Porter JP, Gorman CM, Fendly BM, Jardieu PM. Humanization of an antibody directed against IgE. J Immunol. 1993;151:2623–2632. [PubMed] [Google Scholar]
- 33.Kita H, Kaneko M, Bartemes KR, Weiler DA, Schimming AW, Reed CE, Gleich GJ. Does IgE bind to and activate eosinophils from patients with allergy? J Immunol. 1999;162:6901–6911. [PubMed] [Google Scholar]
- 34.Goichberg P, Kalinkovich A, Borodovsky N, Tesio M, Petit I, Nagler A, Hardan I, Lapidot T. cAMP-induced PKCζ activation increases functional CXCR4 expression on human CD34+ hematopoietic progenitors. Blood. 2006;107:870–879. doi: 10.1182/blood-2005-03-0941. [DOI] [PubMed] [Google Scholar]
- 35.Cristillo AD, Bierer BE. Regulation of CXCR4 expression in human T lymphocytes by calcium and calcineurin. Mol Immunol. 2003;40:539–553. doi: 10.1016/s0161-5890(03)00169-x. [DOI] [PubMed] [Google Scholar]
- 36.Cristillo AD, Highbarger HC, Dewar RL, Dimitrov DS, Golding H, Bierer BE. Up-regulation of HIV coreceptor CXCR4 expression in human T lymphocytes is mediated in part by a cAMP-responsive element. FASEB J. 2002;16:354–364. doi: 10.1096/fj.01-0744com. [DOI] [PubMed] [Google Scholar]
- 37.Cole SW, Jamieson BD, Zack JA. cAMP up-regulates cell surface expression of lymphocyte CXCR4: implications for chemotaxis and HIV-1 infection. J Immunol. 1999;162:1392–1400. [PubMed] [Google Scholar]
- 38.Ganley-Leal LM, Mwinzi PN, Cetre-Sossah CB, Andove J, Hightower AW, Karanja DM, Colley DG, Secor WE. Higher percentages of circulating mast cell precursors correlate with susceptibility to reinfection with. Schistosoma mansoni Am J Trop Med Hyg. 2006;75:1053–1057. [PubMed] [Google Scholar]
- 39.Pearce EJ, Kane CM, Sun J, Taylor JJ, McKee AS, Cervi L. Th2 response polarization during infection with the helminth parasite. Schistosoma mansoni Immunol Rev. 2004;201:117–126. doi: 10.1111/j.0105-2896.2004.00187.x. [DOI] [PubMed] [Google Scholar]
- 40.Secor WE, Sundstrom JB. Below the belt: new insights into potential complications of HIV-1/schistosome coinfections. Curr Opin Infect Dis. 2007;20:519–523. doi: 10.1097/QCO.0b013e3282e9ac03. [DOI] [PubMed] [Google Scholar]
- 41.Collman RG, Perno CF, Crowe SM, Stevenson M, Montaner LJ. HIV and cells of macrophage/dendritic lineage and other non-T cell reservoirs: new answers yield new questions. J Leukocyte Biol. 2003;74:631–634. doi: 10.1189/jlb.0703357. [DOI] [PubMed] [Google Scholar]
- 42.Montaner LJ, Crowe SM, Aquaro S, Perno CF, Stevenson M, Collman RG. Advances in macrophage and dendritic cell biology in HIV-1 infection stress key understudied areas in infection, pathogenesis, and analysis of viral reservoirs. J Leukocyte Biol. 2006;80:961–964. doi: 10.1189/jlb.0806488. [DOI] [PubMed] [Google Scholar]
- 43.Cassol E, Alfano M, Biswas P, Poli G. Monocyte-derived macrophages and myeloid cell lines as targets of HIV-1 replication and persistence. J Leukocyte Biol. 2006;80:1018–1030. doi: 10.1189/jlb.0306150. [DOI] [PubMed] [Google Scholar]
- 44.Wu L, KewalRamani VN. Dendritic-cell interactions with HIV: infection and viral dissemination. Nat Rev Immunol. 2006;6:859–868. doi: 10.1038/nri1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gorry PR, Churchill M, Crowe SM, Cunningham AL, Gabuzda D. Pathogenesis of macrophage tropic HIV-1. Curr HIV Res. 2005;3:53–60. doi: 10.2174/1570162052772951. [DOI] [PubMed] [Google Scholar]
- 46.Galli SJ, Nakae S, Tsai M. Mast cells in the development of adaptive immune responses. Nat Immunol. 2005;6:135–142. doi: 10.1038/ni1158. [DOI] [PubMed] [Google Scholar]
- 47.Tanzola MB, Robbie-Ryan M, Gutekunst CA, Brown MA. Mast cells exert effects outside the central nervous system to influence experimental allergic encephalomyelitis disease course. J Immunol. 2003;171:4385–4391. doi: 10.4049/jimmunol.171.8.4385. [DOI] [PubMed] [Google Scholar]
- 48.Bailey JR, Sedaghat AR, Kieffer T, Brennan T, Lee PK, Wind-Rotolo M, Haggerty CM, Kamireddi AR, Liu Y, Lee J, et al. Residual human immunodeficiency virus type 1 viremia in some patients on antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4+ T cells. J Virol. 2006;80:6441–6457. doi: 10.1128/JVI.00591-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Llewellyn N, Zioni R, Zhu H, Andrus T, Xu Y, Corey L, Zhu T. Continued evolution of HIV-1 circulating in blood monocytes with antiretroviral therapy: genetic analysis of HIV-1 in monocytes and CD4+ T cells of patients with discontinued therapy. J Leukocyte Biol. 2006;80:1118–1126. doi: 10.1189/jlb.0306144. [DOI] [PubMed] [Google Scholar]
- 50.Westby M, Lewis M, Whitcomb J, Youle M, Pozniak AL, James IT, Jenkins TM, Perros M, van der Ryst E. Emergence of CXCR4-using human immunodeficiency virus type 1 (HIV-1) variants in a minority of HIV-1-infected patients following treatment with the CCR5 antagonist maraviroc is from a pretreatment CXCR4-using virus reservoir. J Virol. 2006;80:4909–4920. doi: 10.1128/JVI.80.10.4909-4920.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seminario MC, Saini SS, MacGlashan DW, Jr, Bochner BS. Intracellular expression and release of FcεRI α by human eosinophils. J Immunol. 1999;162:6893–6900. [PubMed] [Google Scholar]
- 52.Taylor RJ, Schols D, Wooley DP. Restricted entry of R5 HIV Type 1 strains into eosinophilic cells. AIDS Res Hum Retroviruses. 2004;20:1244–1253. doi: 10.1089/aid.2004.20.1244. [DOI] [PubMed] [Google Scholar]