

Abstract
The glycoprotein A33 (GPA33) is a colon cancer antigen. Phase I trials with 131I and 125I monoclonal antibody A33 in colon carcinoma patients showed excellent localization to colorectal cancer and some evidence of tumor response. Using DNA microarrays, we have identified the GPA33 gene as a target of PPARγ in HT29-Cl.16E colon cancer cells. Treatment of HT29-Cl.16E, Caco2, SW1116 and LS174T colon cancer cells with the PPARγ agonist GW7845 induced a 2- to 6-fold increase in GPA33 mRNA as determined by real-time PCR. This induction was also found in HT29-Cl.16E cells treated with rosiglitazone and ciglitazone and was prevented by cotreatment with the PPARγ antagonist GW9662, indicating that this regulation was PPARγ dependent. No canonical PPAR responsive element was found in the GPA33 promoter. We therefore analyzed the expression of transcription factors involved in GPA33 expression. CDXl, CDX2 and KLF5 expression was not modified by PPARγ activation. By contrast, a significant increase in KLF4 was seen, both at mRNA and protein levels. Furthermore, chromatin immunoprecipitation studies demonstrated that an increased amount of KLF4 protein was bound to the GPA33 promoter in cells treated with rosiglitazone. Finally, downregulation of KLF4 expression by siRNA reduced rosiglita-zone-induced GPA33 expression. This indicates that PPARγ activation induces KLF4 expression, which in turn increases GPA33 expression. We also demonstrate that PPARγ activation leads to increased (p21WAF1/Cip1 and keratin 19) or decreased (cyclin D1) expression of known KLF4 targets, suggesting that KLF4 is a nodal player in a network of PPARγ-regulated genes.
Keywords: GPA33, PPARγ, KLF4, regulation, colon cancer
Peroxisome proliferator-activated receptor gamma (PPARγ) is a member of the nuclear receptor superfamily of ligand-activated transcriptional factors. PPARγ is expressed throughout the gastrointestinal epithelium from duodenum to rectum and plays a regulator role in differentiated functions of intestinal epithelial cells.1–3 Furthermore, numerous studies showed that PPARγ is expressed in a variety of malignant tissues including prostate, breast and colon. The implication of PPARγ in colorectal carcinogenesis is still debated. In fact, contrasting with the observation of an increase in the number and burden of naturally occurring intestinal tumors in APCMin mice fed with a diet containing a PPARγ agonist,4,5 several models suggest that PPARγ agonists have colonic anticancer activity. In vitro, treatment of colorectal carcinoma cell lines with PPARγ ligands induces cell-cycle blockade resulting in the inhibition of cell proliferation, stimulation of cell differentiation and/or promotion of cell death.6,7In vivo, thiazolidinediones, synthetic PPARγ agonists, decrease the development of tumors derived from colon cancer cells in xenograft models,8–10 suppress colon carcinogenesis induced by azoxymethane in mice11 and are able to reduce the number of chemically induced aberrant crypt foci, which are early precursor lesions of colon cancer.12 Consistent with these findings, PPARγ heterozygous knockout mice (PPARγ+/−) have an increased susceptibility to develop tumors, including colon tumors, after administration of a carcinogen.13 These data, together with the antiproliferative activity of PPARγ ligands observed in many human colorectal cell lines, suggest that these molecules may have promise as anticancer drugs.
In an effort to identify PPARγ gene targets in colon cancer cells, we used microarray technology. The differentiated cell line HT29-Cl.16E, a clonal derivative from the HT29 cell line,14 was grown on filters and cultured for 24 hr in the presence or in the absence of a PPARγ agonist. RNA was then extracted, amplified and hybridized to pan-genomic DNA microarrays. This allowed us to identify the GPA33 gene as a potential PPARγ target. The GPA33 gene encodes a 43-kDa transmembrane glycoprotein15 of the junctional adhesion molecule family,16 with homology to the immunoglobulin superfamily.15,17,18 GPA33 consists of two extracellular immunoglobulin domains, a single transmembrane domain and a short intracellular tail containing four acylation sites.15,18 Extensive immunohistochemical analysis has shown that the antigen is present on the basolateral surfaces of pyloric stomach, small intestine and colon epithelial cells,19 and that it is homogeneously expressed by >95% of colon cancers.19,20
The GPA33 structure is consistent with a putative role as a cell adhesion molecule or a novel cell surface receptor, but no function has been assigned to date. However, the restricted pattern of expression in normal tissue makes this antigen a possible target for immunotherapy of colorectal carcinomas. Phase I and II trials with 131I and 125I humanized murine monoclonal antibody A33 in patients with colon carcinoma showed excellent localization to colorectal cancer and some evidence of tumor response.21–23
Here, we demonstrate that the GPA33 gene is regulated by PPARγ activation. This regulation is mediated by PPARγ, but is indirect, and involves Krüppel-like factor 4 (KLF4), also known as gut-enriched Krüppel-like factor (GKLF). KLF4 is a member of the KLF family of zinc-finger-containing transcription factors.24,25 It is expressed in epithelial cells of the gastrointestinal tract,26 where it plays important roles in differentiation and cell maturation.27,28 PPARγ activation regulates the expression of known KLF4 targets, suggesting that KLF4 is a nodal player in a network of PPARγ-regulated genes.
Material and methods
Human colonic cancer cell lines
Several human colonic cancer cell lines were used. The differentiated cell lines, HT29-Cl.16E and Caco2, were grown on trans-well filters (12-well Transwell Clear, 0.45 μm porosity, Corning-Costar, Cambridge, MA). The nondifferentiated cell lines, SW1116 and LS174T cells, were grown on plastic. All these cell lines were cultured in DMEM (InVitrogen, Cergy Pontoise, France) supplemented with 10% fetal bovine serum (FBS, InVitrogen) and used at postconfluency (SW1116 and LS174T) or until full differentiation (HT29-Cl.16E and Caco2).
Receptor ligands
The following synthetic ligands were used in our studies to determine the specificity and selectivity of GPA33 induction: rosiglitazone (PPARγ agonist; 10 μM), GW7845 (PPARγ agonist; 5 μM), ciglitazone (PPARγ agonist; 20 μM), GW9662 (irreversible PPARγ antagonist; 10 μM), WY14643 (PPARα agonist; 20 μM), 9-cis retinoic acid (RXR agonist; 10 μM).
Rosiglitazone and GW7845 were generously provided by GlaxoSmithKline (Research Triangle Park, NC), and the other ligands were obtained from Sigma-Aldrich (Lyon, France). All synthetic ligands were dissolved in DMSO. The final DMSO concentration in culture medium of all experiments was kept constant at 0.05%. Unless otherwise stated, cells were exposed to these ligands for 24 hr.
cDNA synthesis and real-time PCR
Total RNA was extracted from cells and colonic tissues using TRIzol® protocol (http://www.invitrogen.com/search.cfm). cDNA were synthesized as previously detailed.29
Primers were designed from the sequence of the human cDNAs using the Universal ProbeLibrary Assay Design Center (https://www.roche-applied-science.com/sis/rtpcr/upl). They were selected for binding to separate exons to avoid false positive results arising from the amplification of contaminating genomic DNA. We verified that all amplifications did not yield any product when reverse transcriptase was omitted in the cDNA synthesis reaction. The sequences of these primers are available upon request. PCR amplifications were performed using the LC480 SYBR Green I Master mix (Roche Diagnostics, Meylan, France) in a Rotorgene 3000 instrument (Corbett Research, BioLabo, Archamps, France). The reaction mixture contained 10 μl of the supplied 2× mix, 0.5 μl of each primer (final concentration 0.25 μM each), 9 μl of the template (cDNA diluted 1/40). The cycling conditions were as follows: denaturation for 5 min at 95°C; amplification for 35 cycles, with denaturation for 5 s at 95°C, annealing for 5 s and extension for 5 s at 72°C. To exclude primer–dimer artifacts, fluorescence was not measured at the end of the extension step, but a separate detection step was added (10 s) at a temperature above the melting point of primer–dimers and below the melting point of the specific PCR product. A standard curve was generated with serial dilutions of pooled cDNAs samples. The amount of transcripts was calculated from these standard curves using the RotorGene software. For each sample, the ratio between the relative amount of each specific transcript and GAPDH was then calculated to compensate for variations in quantity or quality of starting mRNA as well as for differences in reverse transcriptase efficiency. Initial DNA micro-array experiments demonstrated that PPARγ activation did not alter GAPDH expression level. Expression of β-actin and β2-microglobulin was also unaffected, and similar results were obtained when β-actin was used to normalize expression in quantitative RT-PCR experiments (not shown). Each amplification was performed in triplicate.
Western blot analysis
Total proteins were extracted from cultured cells using standard RIPA buffer. Nuclear proteins were prepared using the Qproteome cell compartment kit (QIAGEN, Courtaboeuf, France). Proteins were separated by SDS-PAGE and transferred to PVDF membranes as described.29 Membranes were probed with the A33 monoclonal antibody (gift from Joan Heath) and the H-180 anti-KLF4 antibody (Santa Cruz Biotechnology, Santa Cruz, CA). Equal protein sample loading was monitored by incubating the same membrane filter with an anti-α/β tubulin (2148, Cell Signalling, Danvers, MA) or an anti-lamin A/C antibody (sc7293, Santa Cruz Biotechnology). The probed proteins were detected by using IRD700 and IRD800 conjugated secondary antibodies and the membranes were scanned using a LICOR Odyssey infrared imager (Sciencetec, Les Ulis, France).
Chromatin immunoprecipitation assay
HT29-Cl.16E cells (2× 107) were treated with DMSO or rosiglitazone (10 μM) for 24 hr and were subjected to chromatin immunoprecipitation with the EZ ChIP Assay kit (Upstate Cell Signaling Solutions). Briefly, formaldehyde was used to crosslink proteins with DNA, and cells were lysed in SDS lysis buffer. The cell lysate was sonicated to shear the DNA. Chromatin samples were precleared with protein G agarose for 2 hr at 4°C and immunoprecipitated overnight at 4°C with normal mouse IgG or an anti-KLF4 antibody (H-180) bound to protein G agarose. Formaldehyde crosslinking was reversed by incubation at 65°C followed by proteinase K digestion and DNA purification. Quantitative PCR were then performed as described earlier, using specific primers. The region between −212 and −50 nucleotides of the GPA33 promoter (163 bp), which contains a KLF4 binding site,30 was amplified with the following primers: 5′-GGG GGA GAC TTC CTG TTT TC-3′(sense) and 5′-TCA GAG GTA GGG TTC CAA GC-3′(antisense). A 241-bp fragment of the FABP1 promoter was amplified as negative control with the following primers: 5′-AAG GCT TCC TGC TTG ACT GA-3′(sense) and 5′-GCT CCC TCT TCA CGC ATG CA-3′(antisense).
Downregulation of KLF4 expression using siRNA
RNA interference was achieved using a validated human KLF4 (NM_004235) siRNA (Qiagen) or the AllStars negative control siRNA (Qiagen). This validated negative control siRNA has no homology to any known mammalian gene and it has been shown to ensure minimal nonspecific effects on gene expression. HT29 cells were seeded to ~50% confluence in 24-well plates in triplicate. On the following day, transfections were performed with the use of INTERFERin (Ozyme, Saint-Quentinen-Yvelines, France), according to the manufacturer’s recommended protocol. Briefly, siRNA were diluted in serum-free medium. INTERFERin was added (2 μl for 100 μl of diluted siRNA). After incubation at room temperature for 10 min, the mixture was added to culture wells containing 0.5 ml of complete cell culture medium. The final concentration of siRNA was 10 nM. After 24 hr, cells were treated with DMSO (control) or rosiglitazone (10 μM). Cells were lysed and RNA was extracted 24 hr later.
Statistical analysis
Results are expressed as means ± SEM. Differences between group means are analyzed by Student’s t-test and are considered significant at p < 0.05.
Results
GPA33 is increased in colon cancer cell lines treated with GW7845
We first analyzed the regulation of the GPA33 gene in the following human colon cancer cell lines: 2 differentiated cell lines cultured on porous filters (HT29-Cl.16E and Caco2) and 2 nondifferentiated cell lines cultured on plastic (SW1116 and LS174T). These cell lines which all expressed PPARγ31 were treated with a synthetic PPARγ agonist, GW7845.32 As a control of induction, we measured the expression level of the FABP1 (fatty acid binding protein 1) gene, a bona fide PPARγ target gene. An increase in FABP1 level (1.6- to 33.4-fold) was found in the different cell lines (Fig. 1). In all these cell lines, an increased expression of GPA33 was seen upon GW7845 treatment, ranging from 1.6-fold in LS174T cells to 5.6-fold in HT29-Cl.16E cells (Fig. 1). A more detailed analysis of regulation mechanisms involved was performed using HT29-Cl.16E cells as a model.
Figure 1.
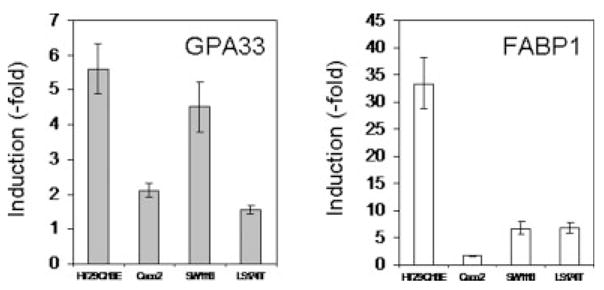
PPARγ activation induces GPA33 expression in colon cancer cells. RNA was extracted from colon cancer cell lines treated for 24 hr with DMSO or GW7845 (10 μM). GPA33 and FABP1 expression was measured by real-time PCR and normalized to GAPDH. Results presented are mean ± SD of 3 different experiments.
GPA33 regulation by PPARγ in HT29-Cl.16E
We next examined the effect of different ligands on GPA33 expression. All PPARγ agonists tested (rosiglitazone, GW7845 and ciglitazone) increased GPA33 transcripts (Fig. 2a). In contrast, GW9662, an irreversible PPARγ antagonist,33 did not affect GPA33 expression. More importantly, it prevented the increase induced by rosiglitazone (Fig. 2a) and by the other agonists (not shown). This indicated that the PPARγ receptor was involved in GPA33 regulation. In contrast, WY14643 (PPARα ligand) and retinoic acid (RXR ligand) did not affect GPA33 expression (Fig. 2a).
Figure 2.
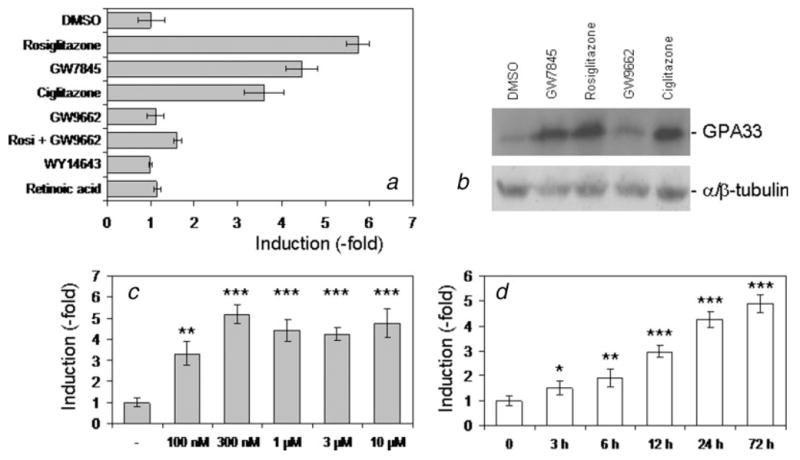
Characterization of GPA33 induction in HT29-Cl.16E. HT29-Cl.16E colon cancer cells were treated with different ligands for 24 hr. GPA33 expression was measured by real-time PCR and normalized to GAPDH (a). Proteins extracted from treated cells were analyzed by Western blotting using the A33 antibody. The α/β tubulin antibody was used as a loading control (b). HT29-Cl.16E colon cancer cells were treated with rosiglitazone at the indicated concentration for 24 hr (c), or with 10 μM for the indicated time (d). RNA was extracted and analyzed by quantitative RT-PCR. (*p < 0.05; **p < 0.01; ***p < 0.001). Results presented are mean ± SD of 3 different experiments.
This induction observed at the mRNA level was confirmed at the protein level. Total proteins were extracted from untreated or treated cells and analyzed by immunoblotting. A 4.5- to 6.2-fold increase of GPA33 was seen with all the PPARγ agonists, whereas the PPARγ antagonist had no effect (Fig. 2b).
By quantitative RT-PCR, GPA33 expression levels were shown to be increased in a concentration-dependent manner. A significant increase was observed with the lowest dose tested (100 nM), with a maximal effect reached at 300 nM (Fig. 2c). GPA33 also increased in a time-dependent manner, by 10 μM rosiglitazone (Fig. 2d). The GPA33 mRNA amount was significantly increased after a 3-h exposure to rosiglitazone, and the induction increased for up to 72 hr.
We also analyzed the mRNA levels in HT29-Cl.16E cells after rosiglitazone treatment in either presence or absence of the protein synthesis inhibitor cycloheximide (CHX). HT29-Cl.16E cells grown in the presence of CHX alone were used as a control. FABP1 induction was not blocked by CHX, in agreement with the direct effect of PPARγ on transcription of the FABP1 gene (Fig. 3). In contrast, the CHX presence abolished the rosiglitazone effect on GPA33 expression (Fig. 3). This suggested the involvement of de novo protein synthesis in the mechanism of GPA33 regulation by PPARγ.
Figure 3.
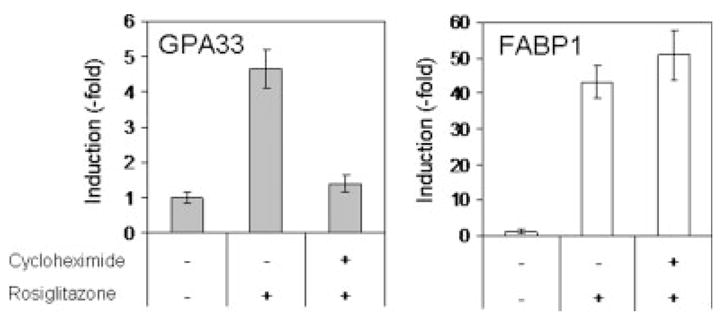
GPA33 induction requires protein synthesis. HT29-Cl.16E were treated for 24 hr with rosiglitazone (10 μM) in the absence or presence of cycloheximide (10 μg/ml). RNA was extracted and analyzed by RT-PCR. Results presented are mean ± SD of 3 different experiments.
KLF4 is induced by PPARγ in HT29-Cl.16E cells
PPARγ functions by regulating gene transcription via binding to DNA sequences known as peroxisome proliferator response elements (PPREs) located in the promoter regions of target genes. We performed an extensive analysis of the GPA33 promoter, but we did not find any PPRE in this sequence. We thus hypothesized that this regulation might be indirect, involving other factors. A comprehensive analysis of the 5′-regulatory region of the human GPA33 gene has been performed. This revealed positive cis-regulatory elements incorporating consensus Krüppel-like factor and caudal-related homeobox (CDX)-binding sites, located just upstream from the human GPA33 antigen transcription start site.34 Further analysis provided evidence that GPA33 is a target gene for the intestine-specific homeobox transcription factor, CDX1,34 and that the gut-enriched Krüppel-like factor (GKLF-KLF4) binds to the promoter region of the GPA33 gene in colonic carcinoma cells.30,34
We therefore analyzed the expression of these genes, CDX1 and KLF4, in HT29-Cl.16E cells treated with rosiglitazone. We also included 2 related genes in our series: KLF5 (IKLF, intestinal KLF) and CDX2. The quantitative RT-PCR performed clearly showed that rosiglitazone induced expression of KLF4, but the other transcription factors were not modified (Fig. 4a). Furthermore, this induction was prevented by cotreatment with the PPARγ antagonist GW9662, indicating that this induction was also PPARγ-dependent. This increase was confirmed at the protein level by immunoblotting experiments (Fig. 4b). A 2.7-fold increase was seen after 24 hr and reached 4.6-fold after 48 hr of treatment.
Figure 4.
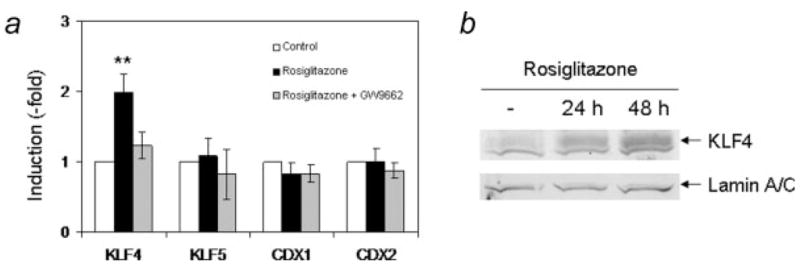
PPARγ activation specifically induces KLF4 expression. (a) HT29-Cl.16E cells were treated for 24 hr with rosiglitazone (10 μM) in the absence or presence of GW9662 (10 μM). RNA was extracted and expression of KLF4, KLF5, CDX1 and CDX2 was analyzed by RT-PCR and normalized to GAPDH (**p < 0.01). Results presented are mean ± SD of 3 different cultures. (b) Proteins extracted from untreated cells or cells treated with rosiglitazone for 24 or 48 hr were analyzed by Western blotting using an anti-KLF4 antibody. The lamin A/C antibody was used as a loading control.
An increase in KLF4 expression was also found at the RNA level in the other cell lines tested (1.5-, 2.3- and 2.5-fold increase in Caco2, LS174T and SW1116 cells, respectively; data not shown).
KLF4 is involved in PPARγ induction of GPA33 expression
Chromatin immunoprecipitation experiments were first performed to determine whether PPARγ activation led to increased binding of KLF4 to the GPA33 promoter. HT29-Cl.16E cells were treated with DMSO or rosiglitazone for 24 hr and then subjected to chromatin immunoprecipitation. Following DNA purification, quantitative PCR was performed. No differences were seen when a fragment of the FABP1 gene, which does not contain KLF4 binding sites, was amplified (Fig. 5). In contrast, Figure 5 clearly shows that upon rosiglitazone treatment, the amount of GPA33 promoter DNA immunoprecipitated with an anti-KLF4 antibody significantly increased.
Figure 5.
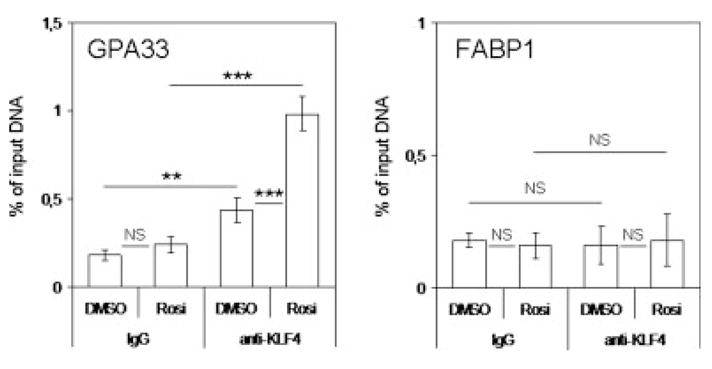
PPARγ-induced KLF4 binding to the GPA33 promoter. Chromatin immunoprecipitation analysis of KLF4 binding to the target region on the GPA33 promoter but not on the FABP1 promoter was performed as described in “Material and Methods” section. HT29-Cl.16E cells were treated for 24 hr with DMSO or rosiglitazone (10 μM) for 24 hr. Chromatin samples were subjected to immunoprecipitation using a KLF4 antibody or normal mouse IgG as a control. Quantitative PCR were then performed using specific primers. The region between −212 and −50 nucleotides of the GPA33 promoter, which contains a KLF4 binding site, was amplified. As a control, a fragment of the FABP1 gene, which does not contain KLF4 binding sites, was used. The amount of precipitated DNA was determined by real-time PCR and expressed as percentage of input DNA (NS, not significant; **p < 0.01; ***p < 0.001).
To determine whether or not GPA33 induction by PPARγ activation was due to this KLF4 increase and binding to the GPA33 promoter, we undertook a siRNA approach. KLF4 expression was significantly decreased (~2-fold) in cells transfected with a specific KLF4 siRNA as compared to cells treated with a control siRNA, both in DMSO- and in rosiglitazone-treated cells (Fig. 6). Similarly, GPA33 expression was significantly lower in cells treated with the specific KLF4 siRNA than in cells treated with the control siRNA, both in control conditions and following rosiglitazone treatment (Fig. 6). Inhibition of KLF4 expression did not affect induction of FABP1 by rosiglitazone (Fig. 6). Altogether, these data demonstrated that GPA33 induction was, at least in part, due to KLF4 increased expression.
Figure 6.

PPARγ-induced KLF4 expression is involved in GPA33 regulation. RNA interference was performed as described in “Material and Methods” section by using a validated human KLF4 siRNA or a negative control siRNA. After 24 hr, cells were treated with DMSO (control) or rosiglitazone (10 μM). RNA was extracted 24 hr later. KLF4, GPA33 and FABP1 expression was determined by real-time PCR. Data were normalized to GAPDH expression (NS, not significant; *p < 0.05; **p < 0.01).
PPARγ activation regulates additional KLF4 targets
In colon cancer cells, constitutive overexpression of KLF4 induces cell cycle arrest at the G1 phase and inhibits cell proliferation.35,36 These effects appear to be mediated through transcriptional upregulation of the negative cell-cycle-regulatory cyclin-dependent kinase inhibitor p21WAF1/Cip1 gene.37,38 On the other hand, KLF4 represses several positive cell-cycle-regulatory gene promoters, such as cyclin D1.35,39
To determine whether the PPARγ-induced KLF4 increase would also modify the expression level of other genes, we measured the expression level of known KLF4 target genes: cyclin D1, p21WAF1/Cip1 and keratin 19. Real-time PCR analysis revealed that PPARγ activation leads to an increase in p21 and Ker19 expression and a decrease in cyclin D1 expression (Fig. 7). This suggests that some of the responses affected by PPARγ are the result of changes in the activity of the KLF4 signaling network.
Figure 7.

PPARγactivation induces KLF4 target genes. RNA was extracted from colon cancer cell lines treated for 24 hr with DMSO or rosiglitazone (10 μM). Cyclin D1, p21/Cip1 and keratin 19 expression was measured by real-time PCR and normalized to GAPDH. Results presented are mean γSD of 3 different experiments (*p < 0.05).
Discussion
The biological activities of PPARγ are very broad. It is generally accepted as a master transcriptional regulator of lipid and glucose metabolism, but it has also been implicated in tumorigenesis. Indeed, PPARγ agonists, such as thiazolinediones (rosiglitazone, troglitazone and pioglitazone), prevent tumorigenesis in animal models, inhibit growth of malignant human cells and cause cell cycle arrest and apoptosis in a broad spectrum of epithelial-derived tumor cell lines.6 Several potential downstream targets of PPARγ mediating its antitumor effects have been identified in colorectal cancer cell types. The activation of PPARγ negatively regulates cell cycle progression by modulating a number of cell cycle regulators such as cyclin D1 expression in Ras-transformed intestinal epithelial cells.40 The activation of PPARγ has also been reported to inhibit tumor cell growth by upregulation of the transcriptional repressor TSC22 in colon cancer cells41 and to suppress tumor cell invasion by downregulation of matrix metalloproteinase-7.42
To provide further insights into the role of PPARγ in colorectal carcinogenesis, we sought to identify novel PPARγ-regulated genes. Using microarray technology, we identified GPA33 as a PPARγ-regulated gene in HT29-Cl.16E cells. A similar approach has been used by Gupta et al.,10 using a different cell line (MOSER S cells). They selected genes induced or repressed 2.5 times or greater after treatment with the PPARγ ligand GW7845. They identified several PPARγ-selective targets: genes linked to growth regulatory pathways (regenerating gene IA), colon epithelial cell maturation (GOB-4 and keratin 20), immune modulation (neutrophil-gelatinase-associated lipocalin) and members of the carcinoembryonic antigen family. They did not identify GPA33 as a PPARγ target. We do not know whether the GPA33 gene is expressed in this cell line or whether it is induced less than 2.5-fold.
The human and mouse GPA33 antigens are Type I transmembrane glycoproteins of the immunoglobulin superfamily.15,16 Immunohistochemical studies of normal tissues identified the large and small intestinal mucosa as the principal site of GPA33 expression.20 The expression pattern of GPA33 is characterized by robust and uniform expression by epithelial cells throughout the intestine. Tests in tumor samples demonstrated that only tumors of the gastrointestinal tract are consistently A33 antigen positive.20 The biological function of GPA33 is not yet understood. It has been suggested that it may be a novel cell surface receptor or cell adhesion molecule in the immunoglobulin superfamily.15
Our results demonstrate for the first time that, in a time- and concentration-dependent manner, GPA33 expression levels were upregulated by PPARγ ligands both at mRNA and protein levels. The induction of GPA33 expression by PPARγ agonists was observed in different human colon cancer cell lines. We also demonstrated a significant, although much lower (40% increase), induction in colon of mice receiving rosiglitazone (not shown).
This induction was PPARγ-dependent as demonstrated by the inhibitory effect of GW9662. A similar conclusion was drawn from experiments performed with mice with targeted disruption of the PPARγ gene in intestinal epithelial cells generated using a villin-Cre transgene and floxed PPARγ allele.43 We did not find an increase in GPA33 expression in animals that did not express PPARγ in their intestinal epithelial cells.
However, this regulation is indirect since the PPARγ-induced regulation of GPA33 was abolished by coincubation with cycloheximide suggesting that this regulation required de novo protein synthesis. This is in agreement with the kinetic of induction, which shows a rather slow increase of GPA33 transcripts upon rosiglitazone treatment. Among the cell lines used, the effects on GPA33 were in general more modest and different than the pattern seen for FABP1, suggesting that the regulation of GPA33 is more complex and multidimensional. In addition, we found no correlation between KLF4 and GPA33 expression levels determined by quantitative real-time PCR in cell lines and in a series of human colorectal tumors (data not shown).
The promoter region alignment revealed several conserved binding sites for transcription factors previously implicated in the regulation of gene expression in intestinal epithelial cells. A CDX-binding sequence was identified, and the binding of the intestine-specific homeobox transcription factor CDX1 was demonstrated in this site.34 In addition, potential binding sites for KLF4 and KLF5 were identified by Mao et al.30
In our model, PPARγ activation did not significantly modify CDX1, CDX2 and KLF5 expression. In contrast, it specifically increased KLF4 mRNA and protein levels. We examined the effect of cycloheximide on the expression of KLF4 mRNA in the presence of rosiglitazone. This combination resulted in an 18-fold increase in KLF4 mRNA level (data not shown). These results are in agreement with those obtained using 15-deoxy-Δ12,14 prostaglandin J2 (15d-PGJ2), a natural PPARγ ligand, and indicate that early de novo protein synthesis may not be required for induced KLF4 upregulation. Using specific inhibitors, Chen and Tseng44 demonstrated that the MEK/ERK pathway is engaged in the induction of KLF4 mRNA by 15d-PGJ2.
KLF4 does not seem to contain a potential conserved consensus of PPRE in its promoter. Chen and Tseng44 conclude that the PPARγ-induced KLF4 up-regulation is most likely to be mediated through a PPARγ-independent mechanism. They also based this conclusion on results suggesting that endogenous PPARγ in HT29 cells may not be functional or is transcriptionally inactive. We strongly disagree with these conclusions and clearly show here that PPARγ agonists always strongly induced expression of the FABP1 gene, a well-known PPARγ target. How is KLF4 upregulated upon PPARγ activation remains to be elucidated. The transcription factors, SOX9 and TCF4, recently demonstrated by Flandez et al.27 for having an effect on KLF4 expression might be good candidates.
Sodium butyrate has been shown to increase KLF4 mRNA and protein levels.27,45 We found that the treatment of HT29 cells with sodium butyrate also increased KLF4 expression. Interestingly, this treatment also induced GPA33 expression (data not shown).
KLF4 expression is altered in various models of intestinal tumorigenesis. The level of KLF4 transcript is significantly decreased in intestinal adenomas of multiple intestinal neoplasia (APCMin/+) mice and in colonic adenomas of familial adenomatous polyposis (FAP) patients.37,46 Similarly, KLF4 mRNA and protein levels are reduced in sporadic colorectal carcinomas of patients when compared with normal colonic tissues.36,47 Bearing in mind the negative effect of KLF4 on cell proliferation, it might be interesting to increase KLF4 expression in these cancer cells by activating PPARγ.
The highly tissue-specific expression of the A33 antigen presents a great opportunity for specific tumor targeting in patients with gastrointestinal cancer. Studies using nude mouse models with xenografts of the human colorectal carcinoma cell lines, SW1222, indicated that the A33 monoclonal antibody was capable of inducing tumor regression.48 In phase I and II trials, 131I- and 125I-labeled murine A33 monoclonal antibodies were shown to have antitumor effects without bowel toxicity.49,50 A genetically humanized monoclonal antibody A33 (huA33) has been generated, and phase I trials have been undertaken.21–23,51 Scott et al.21 demonstrated that huA33 localized rapidly and selectively to colorectal carcinoma in vivo, and the colon elimination half-life of huA33 was equivalent to basal colonocyte turnover.
The antiproliferative activity of PPARγ ligands observed in many human colorectal cell lines suggests that these molecules may have promise as anticancer drugs. Rosiglitazone is approved for the treatment of insulin-resistant diabetes. No significant effect could be observed when troglitazone alone was administered in patients with metastatic colorectal cancer included in a phase-II clinical trial.52 However, recent reports demonstrated a synergy between PPARγ ligands and current anticancer therapies such as fluorouracil,53 carboplatin54,55 or tamoxifen.56 Thus, PPARγ-induced GPA33 expression might increase the efficiency of anti-GPA33 monoclonal antibodies. A combination of both approaches might be effective.
In colon cancer cells, constitutive overexpression of KLF4 induces cell cycle arrest at the G1 phase and inhibits DNA synthesis and cell proliferation.35,36 These effects appear to be mediated through the regulation of genes such as p21WAF1/Cip137,38 and cyclin D1.35,39 We also demonstrated that, in addition to GPA33, PPARγ activation leads to increased p21WAF1/Cip1 and decreased cyclin D1 expression, suggesting that KLF4 is a nodal player in a network of PPARγ-regulated genes. Further work will be necessary to identify additional genes involved in this network.
Collectively, our results provide potential clinical applications and will help to define the mechanisms by which PPARγ and KLF4 are involved in colorectal cancer cell biology.
Acknowledgments
Ligue Départementale Contre le Cancer; Grant numbers: 35, 44, 56; Grant sponsor: Association Pour la Recherche Contre le Cancer; Grant number: ARC-3732; Grant sponsor: Région Bretagne; Grant number: ACOMB 2627.
The A33 monoclonal antibody was kindly provided by Dr. Joan Heath (Ludwig Institute for Cancer Research, Victoria, Australia). DNA microarray experiments that allowed us to identify the GPA33 gene as a potential PPARγ-target were performed on the “plateforme Ouest-Génopole” (Dr. Marie-Dominique Anne-Galibert, Pr. Jean Mosser).
References
- 1.Lefebvre M, Paulweber B, Fajas L, Woods J, McCrary C, Colombel JF, Najib J, Fruchart JC, Datz C, Vidal H, Desreumaux P, Auwerx J. Peroxisome proliferator-activated receptor gamma is induced during differentiation of colon epithelium cells. J Endocrinol. 1999;162:331–40. doi: 10.1677/joe.0.1620331. [DOI] [PubMed] [Google Scholar]
- 2.Su W, Bush CR, Necela BM, Calcagno SR, Murray NR, Fields AP, Thompson EA. Differential expression, distribution, and function of PPAR-γ in the proximal and distal colon. Physiol Genomics. 2007;30:342–53. doi: 10.1152/physiolgenomics.00042.2007. [DOI] [PubMed] [Google Scholar]
- 3.Thompson EA. PPARgamma physiology and pathology in gastrointestinal epithelial cells. Mol Cells. 2007;24:167–76. [PubMed] [Google Scholar]
- 4.Lefebvre AM, Chen I, Desreumaux P, Najib J, Fruchart JC, Geboes K, Briggs M, Heyman R, Auwerx J. Activation of the peroxisome proliferator-activated receptor gamma promotes the development of colon tumors in C57BL/6J-APCMin/+ mice. Nat Med. 1998;4:1053–7. doi: 10.1038/2036. [DOI] [PubMed] [Google Scholar]
- 5.Saez E, Tontonoz P, Nelson MC, Alvarez JG, Ming UT, Baird SM, Thomazy VA, Evans RM. Activators of the nuclear receptor PPAR-gamma enhance colon polyp formation. Nat Med. 1998;4:1058–61. doi: 10.1038/2042. [DOI] [PubMed] [Google Scholar]
- 6.Koeffler HP. Peroxisome proliferator-activated receptor gamma and cancers. Clin Cancer Res. 2003;9:1–9. [PubMed] [Google Scholar]
- 7.Michalik L, Desvergne B, Wahli W. Peroxisome-proliferator-activated receptors and cancers: complex stories. Nat Rev Cancer. 2004;4:61–70. doi: 10.1038/nrc1254. [DOI] [PubMed] [Google Scholar]
- 8.Yoshizumi T, Ohta T, Ninomiya I, Terada I, Fushida S, Fujimura T, Nishimura G, Shimizu K, Yi S, Miwa K. Thiazolidinedione, a peroxisome proliferator-activated receptor-gamma ligand, inhibits growth and metastasis of HT-29 human colon cancer cells through differentiation-promoting effects. Int J Oncol. 2004;25:631–9. [PubMed] [Google Scholar]
- 9.Sarraf P, Mueller E, Jones D, King FJ, Deangelo DJ, Partridge JB, Holden SA, Chen LB, Singer S, Fletcher C, Spiegelman BM. Differentiation and reversal of malignant changes in colon cancer through PPARgamma. Nat Med. 1998;4:1046–52. doi: 10.1038/2030. [DOI] [PubMed] [Google Scholar]
- 10.Gupta RA, Brockman JA, Sarraf P, Willson TM, Dubois RN. Target genes of peroxisome proliferator-activated receptor gamma in colorectal cancer cells. J Biol Chem. 2001;276:29681–7. doi: 10.1074/jbc.M103779200. [DOI] [PubMed] [Google Scholar]
- 11.Osawa E, Nakajima A, Wada K, Ishimine S, Fujisawa N, Kawamori T, Matsuhashi N, Kadowaki T, Ochiai M, Sekihara H, Nakagama H. Peroxisome proliferator-activated receptor gamma ligands suppress colon carcinogenesis induced by azoxymethane in mice. Gastroenterology. 2003;124:361–7. doi: 10.1053/gast.2003.50067. [DOI] [PubMed] [Google Scholar]
- 12.Tanaka T, Kohno H, Yoshitani S, Takashima S, Okumura A, Murakami A, Hosokawa M. Ligands for peroxisome proliferator-activated receptors alpha and gamma inhibit chemically induced colitis and formation of aberrant crypt foci in rats. Cancer Res. 2001;61:2424–8. [PubMed] [Google Scholar]
- 13.Girnun GD, Smith WM, Drori S, Sarraf P, Mueller E, Eng C, Nambiar P, Rosenberg DW, Bronson RT, Edelmann W, Kucherlapati R, Gonzalez FJ, et al. APC-dependent suppression of colon carcinogenesis by PPARgamma. Proc Natl Acad Sci USA. 2002;99:13771–6. doi: 10.1073/pnas.162480299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Augeron C, Laboisse CL. Emergence of permanently differentiated cell clones in a human colonic cancer cell line in culture after treatment with sodium butyrate. Cancer Res. 1984;44:3961–9. [PubMed] [Google Scholar]
- 15.Heath JK, White SJ, Johnstone CN, Catimel B, Simpson RJ, Moritz RL, Tu G-F, Ji H, Whitehead RH, Groenen LC, Scott AM, Ritter G, et al. The human A33 antigen is a transmembrane glycoprotein and a novel member of the immunoglobulin superfamily. Proc Natl Acad Sci USA. 1997;94:469–74. doi: 10.1073/pnas.94.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnstone CN, Tebbutt NC, Abud HE, White SJ, Stenvers KL, Hall NE, Cody SH, Whitehead RH, Catimel B, Nice EC, Burgess AW, Heath JK. Characterization of mouse A33 antigen, a definitive marker for basolateral surfaces of intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2000;279:G500–G510. doi: 10.1152/ajpgi.2000.279.3.G500. [DOI] [PubMed] [Google Scholar]
- 17.Catimel B, Ritter G, Welt S, Old LJ, Cohen L, Nerrie MA, White SJ, Heath JK, Demediuk B, Domagala T, Lee FT, Scott AM, et al. Purification and characterization of a novel restricted antigen expressed by normal and transformed human colonic epithelium. J Biol Chem. 1996;271:25664–70. doi: 10.1074/jbc.271.41.25664. [DOI] [PubMed] [Google Scholar]
- 18.Ritter G, Cohen LS, Nice EC, Catimel B, Burgess AW, Moritz RL, Ji H, Heath JK, White SJ, Welt S, Old LJ, Simpson RJ. Characterization of posttranslational modifications of human A33 antigen, a novel palmitoylated surface glycoprotein of human gastrointestinal epithelium. Biochem Biophys Res Commun. 1997;236:682–6. doi: 10.1006/bbrc.1997.6966. [DOI] [PubMed] [Google Scholar]
- 19.Garin-Chesa PSJ, Welt S, Real FX, Rettig WJ, Old LJ. Organ-specific expression of the colon cancer antigen A33, a cell surface target for antibody-based therapy. Int J Oncol. 1996;9:465–71. doi: 10.3892/ijo.9.3.465. [DOI] [PubMed] [Google Scholar]
- 20.Sakamoto J, Kojima H, Kato J, Hamashima H, Suzuki H. Organ-specific expression of the intestinal epithelium-related antigen A33, a cell surface target for antibody-based imaging and treatment in gastrointestinal cancer. Cancer Chemother Pharmacol. 2000;46(Suppl):S27–S32. doi: 10.1007/pl00014045. [DOI] [PubMed] [Google Scholar]
- 21.Scott AM, Lee F-T, Jones R, Hopkins W, Macgregor D, Cebon JS, Hannah A, Chong G, Paul U, Papenfuss A, Rigopoulos A, Sturrock S, et al. A phase I trial of humanized monoclonal antibody A33 in patients with colorectal carcinoma: biodistribution, pharmacokinetics, and quantitative tumor uptake. Clin Cancer Res. 2005;11:4810–17. doi: 10.1158/1078-0432.CCR-04-2329. [DOI] [PubMed] [Google Scholar]
- 22.Welt S, Ritter G, Williams C, Cohen LS, John M, Jungbluth A, Richards EA, Old LJ, Kemeny NE. Phase I study of anticolon cancer humanized antibody A33. Clin Cancer Res. 2003;9:1338–46. [PubMed] [Google Scholar]
- 23.Chong G, Lee FT, Hopkins W, Tebbutt N, Cebon JS, Mountain AJ, Chappell B, Papenfuss A, Schleyer P, Paul U, Murphy R, Wirth V, et al. Phase I trial of 131I-huA33 in patients with advanced colorectal carcinoma. Clin Cancer Res. 2005;11:4818–26. doi: 10.1158/1078-0432.CCR-04-2330. [DOI] [PubMed] [Google Scholar]
- 24.Dang DT, Pevsner J, Yang VW. The biology of the mammalian Kruppel-like family of transcription factors. Int J Biochem Cell Biol. 2000;32:1103–21. doi: 10.1016/s1357-2725(00)00059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bieker JJ. Kruppel-like factors: three fingers in many pies. J Biol Chem. 2001;276:34355–8. doi: 10.1074/jbc.R100043200. [DOI] [PubMed] [Google Scholar]
- 26.Shields JM, Christy RJ, Yang VW. Identification and characterization of a gene encoding a gut-enriched Kruppel-like factor expressed during growth arrest. J Biol Chem. 1996;271:20009–17. doi: 10.1074/jbc.271.33.20009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flandez M, Guilmeau S, Blache P, Augenlicht LH. KLF4 regulation in intestinal epithelial cell maturation. Exp Cell Res. 2008;314:3712–23. doi: 10.1016/j.yexcr.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Evans PM, Liu C. Roles of Krupel-like factor 4 in normal homeostasis, cancer and stem cells. Acta Biochim Biophys Sin (Shanghai) 2008;40:554–64. doi: 10.1111/j.1745-7270.2008.00439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blanchot-Jossic F, Jarry A, Masson D, Bach-Ngohou K, Paineau J, Denis MG, Laboisse CL, Mosnier JF. Up-regulated expression of ADAM17 in human colon carcinoma: co-expression with EGFR in neoplastic and endothelial cells. J Pathol. 2005;207:156–63. doi: 10.1002/path.1814. [DOI] [PubMed] [Google Scholar]
- 30.Mao Z, Shan S, Zhu Y, Yi X, Zhang H, Shang Y, Tong T. Transcriptional regulation of A33 antigen expression by gut-enriched Kruppel-like factor. Oncogene. 2003;22:4434–43. doi: 10.1038/sj.onc.1206508. [DOI] [PubMed] [Google Scholar]
- 31.Bouancheau D, Buecher B, Jarry A, Simon B, Masson D, Cassagnau E, Hamelin R, Laboisse CL, Bezieau S, Denis MG. The PPAR γK422Q mutation does not contribute to troglitazone inefficiency in colon cancer treatment. Cancer Lett. 2005;224:111–16. doi: 10.1016/j.canlet.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 32.Suh N, Wang Y, Williams CR, Risingsong R, Gilmer T, Willson TM, Sporn MB. A new ligand for the peroxisome proliferator-activated receptor-gamma (PPAR-gamma), GW7845, inhibits rat mammary carcinogenesis. Cancer Res. 1999;59:5671–3. [PubMed] [Google Scholar]
- 33.Huang JT, Welch JS, Ricote M, Binder CJ, Willson TM, Kelly C, Witztum JL, Funk CD, Conrad D, Glass CK. Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxy-genase. Nature. 1999;400:378–82. doi: 10.1038/22572. [DOI] [PubMed] [Google Scholar]
- 34.Johnstone CN, White SJ, Tebbutt NC, Clay FJ, Ernst M, Biggs WH, Viars CS, Czekay S, Arden KC, Heath JK. Analysis of the regulation of the A33 antigen gene reveals intestine-specific mechanisms of gene expression. J Biol Chem. 2002;277:34531–9. doi: 10.1074/jbc.M204865200. [DOI] [PubMed] [Google Scholar]
- 35.Chen ZY, Shie J-L, Tseng C-C. Up-regulation of gut-enriched Kruppel-like factor by interferon-γ in human colon carcinoma cells. FEBS Lett. 2000;477:67–72. doi: 10.1016/s0014-5793(00)01764-6. [DOI] [PubMed] [Google Scholar]
- 36.Shie J-L, Chen ZY, O’brien MJ, Pestell RG, Lee M-E, Tseng C-C. Role of gut-enriched Kruppel-like factor in colonic cell growth and differentiation. Am J Physiol Gastrointest Liver Physiol. 2000;279:G806–G814. doi: 10.1152/ajpgi.2000.279.4.G806. [DOI] [PubMed] [Google Scholar]
- 37.Zhang W, Geiman DE, Shields JM, Dang DT, Mahatan CS, Kaestner KH, Biggs JR, Kraft AS, Yang VW. The gut-enriched Kruppel-like factor (Kruppel-like factor 4) mediates the transactivating effect of p53 on the p21WAF1/Cip1 promoter. J Biol Chem. 2000;275:18391–8. doi: 10.1074/jbc.C000062200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen X, Whitney EM, Gao SY, Yang VW. Transcriptional profiling of Kruppel-like factor 4 reveals a function in cell cycle regulation and epithelial differentiation. J Mol Biol. 2003;326:665–77. doi: 10.1016/S0022-2836(02)01449-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shie JL, Chen ZY, Fu M, Pestell RG, Tseng CC. Gut-enriched Kruppel-like factor represses cyclin D1 promoter activity through Sp1 motif. Nucleic Acids Res. 2000;28:2969–76. doi: 10.1093/nar/28.15.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kitamura S, Miyazaki Y, Hiraoka S, Nagasawa Y, Toyota M, Takakura R, Kiyohara T, Shinomura Y, Matsuzawa Y. PPARgamma agonists inhibit cell growth and suppress the expression of cyclin D1 and EGF-like growth factors in ras-transformed rat intestinal epithelial cells. Int J Cancer. 2001;94:335–42. doi: 10.1002/ijc.1470. [DOI] [PubMed] [Google Scholar]
- 41.Gupta RA, Sarraf P, Brockman JA, Shappell SB, Raftery LA, Willson TM, Dubois RN. Peroxisome proliferator-activated receptor gamma and transforming growth factor-beta pathways inhibit intestinal epithelial cell growth by regulating levels of TSC-22. J Biol Chem. 2003;278:7431–8. doi: 10.1074/jbc.M208076200. [DOI] [PubMed] [Google Scholar]
- 42.Sunami E, Tsuno NH, Kitayama J, Saito S, Osada T, Yamaguchi H, Tomozawa S, Tsuruo T, Shibata Y, Nagawa H. Decreased synthesis of matrix metalloproteinase-7 and adhesion to the extracellular matrix proteins of human colon cancer cells treated with troglitazone. Surg Today. 2002;32:343–50. doi: 10.1007/s005950200049. [DOI] [PubMed] [Google Scholar]
- 43.Adachi M, Kurotani R, Morimura K, Shah Y, Sanford M, Madison BB, Gumucio DL, Marin HE, Peters JM, Young HA, Gonzalez FJ. Peroxisome proliferator activated receptor γ in colonic epithelial cells protects against experimental inflammatory bowel disease. Gut. 2006;55:1104–13. doi: 10.1136/gut.2005.081745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen ZY, Tseng C-C. 15-Deoxy-{Delta}12,14 prostaglandin J2 up-regulates Kruppel-like factor 4 expression independently of peroxisome proliferator-activated receptor γ by activating the mitogen-activated protein kinase kinase/extracellular signal-regulated kinase signal transduction pathway in HT-29 colon cancer cells. Mol Pharmacol. 2005;68:1203–13. doi: 10.1124/mol.105.014944. [DOI] [PubMed] [Google Scholar]
- 45.Chen ZY, Rex S, Tseng C-C. Kruppel-like factor 4 is transactivated by butyrate in colon cancer cells. J Nutr. 2004;134:792–8. doi: 10.1093/jn/134.4.792. [DOI] [PubMed] [Google Scholar]
- 46.Dang DT, Bachman KE, Mahatan CS, Dang LH, Giardiello FM, Yang VW. Decreased expression of the gut-enriched Kruppel-like factor gene in intestinal adenomas of multiple intestinal neoplasia mice and in colonic adenomas of familial adenomatous polyposis patients. FEBS Lett. 2000;476:203–7. doi: 10.1016/s0014-5793(00)01727-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ghaleb AM, Yang VW. The pathobiology of Kruppel-like factors in colorectal cancer. Curr Colorectal Cancer Rep. 2008;4:59–64. doi: 10.1007/s11888-008-0011-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Antoniw P, Farnsworth AP, Turner A, Haines AM, Mountain A, Mackintosh J, Shochat D, Humm J, Welt S, Old LJ, Yarranton GT, King DJ. Radioimmunotherapy of colorectal carcinoma xenografts in nude mice with yttrium-90 A33 IgG and Tri-Fab (TFM) Br J Cancer. 1996;74:513–24. doi: 10.1038/bjc.1996.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Welt S, Divgi CR, Kemeny N, Finn RD, Scott AM, Graham M, Germain JS, Richards EC, Larson SM, Oettgen HF. Phase I/II study of iodine 131-labeled monoclonal antibody A33 in patients with advanced colon cancer. J Clin Oncol. 1994;12:1561–71. doi: 10.1200/JCO.1994.12.8.1561. [DOI] [PubMed] [Google Scholar]
- 50.Welt S, Scott AM, Divgi CR, Kemeny NE, Finn RD, Daghighian F, Germain JS, Richards EC, Larson SM, Old LJ. Phase I/II study of iodine 125-labeled monoclonal antibody A33 in patients with advanced colon cancer. J Clin Oncol. 1996;14:1787–97. doi: 10.1200/JCO.1996.14.6.1787. [DOI] [PubMed] [Google Scholar]
- 51.Welt S, Ritter G, Williams C, Cohen LS, Jungbluth A, Richards EA, Old LJ, Kemeny NE. Preliminary report of a phase I study of combination chemotherapy and humanized A33 antibody immunotherapy in patients with advanced colorectal cancer. Clin Cancer Res. 2003;9:1347–53. [PubMed] [Google Scholar]
- 52.Kulke MH, Demetri GD, Sharpless NE, Ryan DP, Shivdasani R, Clark JS, Spiegelman BM, Kim H, Mayer RJ, Fuchs CS. A phase II study of troglitazone, an activator of the PPARgamma receptor, in patients with chemotherapy-resistant metastatic colorectal cancer. Cancer J. 2002;8:395–9. doi: 10.1097/00130404-200209000-00010. [DOI] [PubMed] [Google Scholar]
- 53.Zhang YQ, Tang XQ, Sun L, Dong L, Qin Y, Liu HQ, Xia H, Cao JG. Rosiglitazone enhances fluorouracil-induced apoptosis of HT-29 cells by activating peroxisome proliferator-activated receptor gamma. World J Gastroenterol. 2007;13:1534–40. doi: 10.3748/wjg.v13.i10.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Girnun GD, Chen L, Silvaggi J, Drapkin R, Chirieac LR, Padera RF, Upadhyay R, Vafai SB, Weissleder R, Mahmood U, Naseri E, Buckley S, et al. Regression of drug-resistant lung cancer by the combination of rosiglitazone and carboplatin. Clin Cancer Res. 2008;14:6478–86. doi: 10.1158/1078-0432.CCR-08-1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Girnun GD, Naseri E, Vafai SB, Qu L, Szwaya JD, Bronson R, Alberta JA, Spiegelman BM. Synergy between PPARgamma ligands and platinum-based drugs in cancer. Cancer Cell. 2007;11:395–406. doi: 10.1016/j.ccr.2007.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu HN, Noh EM, Lee YR, Roh SG, Song EK, Han MK, Lee YC, Shim IK, Lee SJ, Jung SH, Kim JS, Youn HJ. Troglitazone enhances tamoxifen-induced growth inhibitory activity of MCF-7 cells. Biochem Biophys Res Commun. 2008;377:242–7. doi: 10.1016/j.bbrc.2008.09.111. [DOI] [PubMed] [Google Scholar]