

Abstract
Thiazolidinediones (TZDs) are peroxisome proliferators–activated receptor gamma (PPARγ) activators that exhibit antihypertensive and vasculoprotective effects. Here we describe the use of Tie2Cre/flox and SM22Cre/flox mice, which respectively lacked PPARγ in the endothelium and the smooth muscle, to study vascular function of PPARγ. Rosiglitazone (RGZ) induced a similar blood pressure (BP)–lowering effect in deoxycorticosterone acetate (DOCA) salt–treated PPARγ f/f and SM22Cre/flox mice, whereas Tie2Cre/flox mice were completely resistant to this effect. The femoral arteries lacking endothelial PPARγ exhibited increased reactivity to various vasoconstrictors without a significant alteration in acetylcholine-induced relaxation. In sharp contrast, the vasculature lacking smooth muscle PPARγ had blunted sensitivity to α1-adrenergic agents but enhanced sensitivity to acetylcholine. Our results demonstrated endothelium but not smooth muscle as the site for TZD-induced BP-lowering effect and also uncovered distinct functions of endothelial and smooth muscle PPARγ in regulation of vascular tone.
Keywords: PPARγ, rosiglitazone, endothelial cells, smooth muscle cells, blood pressure
Introduction
Peroxisome proliferators–activated receptor-γ (PPARγ) is a member of the superfamily of nuclear receptor ligand–activated transcription factors that regulate gene transcription through binding to PPAR-responsive elements of the target genes. Compelling evidence from both human and animal studies has established a pivotal role of this nuclear receptor in the control of glucose and lipid metabolism (Berger et al. 2005; Evans et al. 2004). The synthetic PPARγ ligands, which are thiazolidinediones (TZDs) including rosiglitazone (RGZ) and pioglitazone, are widely prescribed and highly effective for the treatment of type 2 diabetes. Apart from the metabolic action, TZDs exert blood pressure (BP)-lowering effects independent of the insulin-sensitizing effects. In several experimental and clinical settings of type 2 diabetes, TZDs improved BP control and insulin resistance, and the decline of blood pressure further correlated with the improvement in insulin sensitivity (Raji et al. 2003). Although dissociation between the two phenomena in metabolic syndrome might be difficult, another insulin sensitizer, metformin, had no beneficial effect on BP (Komers and Vrana 1998). In line with this finding, TZDs are reported to lower BP in nondiabetic hypertensive rats (Atkins et al. 2005; Diep et al. 2002) and even in normotensive rats (Song et al. 2004). The mechanisms of these observations are poorly characterized. PPARγ is expressed in all components of the vascular system including endothelial cells (ECs) (Inoue et al. 1998; Satoh et al. 1999), vascular smooth muscle cells (VSMCs) (Staels et al. 1998), and monocytes/macrophages (Fernandez 2008). The mechanisms may involve improvement of endothelial dysfunction (Diep et al. 2002), attenuation of smooth muscle contraction (Buchanan et al. 1995), and inhibition of proliferation and inflammation (Fukunaga et al. 2001; Marx et al. 1998). Because of these beneficial effects, TZDs hold great promise for treatment of hypertension and vascular disease associated with or not associated with diabetes. Therefore, it is critically important to understand the molecular mechanism of vascular actions of PPARγ. The aim of the present study was to determine TZD-induced BP-lowering effect as well as the vessel reactivity in mice lacking PPARγ in ECs (Tie2Cre/flox) or in SMCs (SM22Cre/flox).
Materials and Methods
Animals
SM22Cre/flox and Tie2Cre/flox mice were produced by crossing PPARγf/f mice with Tie2-Cre and SM22-Cre mice, respectively. PPARγf/f mice contain two loxP sites inserted into introns 1 and 2 of the PPARγ gene flanking the critical exon 2 by homologous recombination in embryonic stem (ES) cells. The floxed mice were crossed with Tie2-Cre (Kisanuki et al. 2001) and SM22-Cre mice (Holtwick et al. 2002), respectively, to yield mice homologous for the floxed allele and heterozygous for the Cre transgene (termed Tie2Cre/flox and SM22Cre/flox). Each Cre strain has been used to target respective vascular cells (Tie2Cre for ECs and SM22Cre for SMCs) (Boucher et al. 2003; Frutkin et al. 2006; Hernando et al. 2007; Miano et al. 2004; Xin et al. 2007). Littermates that were homozygous for the floxed PPARγ gene, but without the Cre transgene, were used as control mice. Genotypes were confirmed by PCR analysis, as previously described (Nicol et al. 2005; Zhang et al. 2005). Both Tie2Cre/flox and SM22Cre/flox mice were born at the expected Mendelian ratio, and neither mutant had gross morphological abnormalities in adults. Of note, sporadic alopecia was found in young but not adult Tie2Cre/flox mice, as previously described (Wan et al. 2007). Analyses of DNA recombination and mRNA expression of the PPARγ gene in the freshly isolated ECs and VSMCs confirmed the selective deletion in ECs in Tie2Cre/flox mice and in SMCs in SM22Cre/flox mice (data not shown). All male mice at three to four months of age were maintained under a twelve-hour light/dark (LD) cycle (lights on at 6:00 a.m. and lights off at 6:00 p.m.). Animals were maintained on a twelve-hour light/dark cycle, and all procedures were in compliance with the guidelines approved by the University of Utah Institutional Animal Care and Use Committee.
Animal Experimental Protocol
The radiotelemetric device was implanted into male three-or four-month-old PPARγf/f, SM22Cre/flox, and Tie2Cre/flox mice through catheterization of the carotid artery (Model#: TA11PA-C20, Data Sciences International, St. Paul, MN, USA) as previously described (Jia et al. 2006). Animals were allowed to recover from surgery for one week. Following collection of baseline mean arterial pressure (MAP) for three days, animals were implanted with subcutaneous twenty-one-day-release pellets containing 50 mg deoxycorticosterone acetate (DOCA) (Innovative Research of America, Sarasota, FL, USA). At the time of pellet implantation, mice were transferred from standard chow and tap water to gelled diets containing 1.5% NaCl and saline as drinking fluid. After five days of this treatment, mice were divided randomly to receive the gelled diets with or without RGZ incorporated (320 mg/kg diet). Daily MAP was recorded as mean values of four-hour recordings from 9:00 a.m. to 1:00 p.m.
Vascular Function
Mice were anesthetized with 2%–5% isoflurane, the chest was opened, and the heart was excised. Next, femoral arteries were isolated distal to the bifurcation of the internal iliac artery. During dissection, tissues were bathed in ice-cold, oxygenated (95% O2/5% CO2), normal physiological saline solution (pH 7.35–7.45). Once isolated and free of adherent tissue, femoral arteries were mounted on a wire-type myograph while immersed in a temperature-controlled, 8 mL tissue bath. When the tissue bath reached 37°C, tension on femoral arteries was increased 50 mg every two minutes to 200 mg. Thirty minutes later, a series of internal circumference–active tension curves was constructed to determine the vessel diameter that evoked the greatest tension development (Lmax) to 100 mM KCl. Thirty minutes later, receptor-mediated vasocontractile responses to phenylephrine (PE, 10−8–10−5 M), potassium chloride (KCl, 10–100 mM), and angiotensin II (Ang II, 0.1–1000 μM) were assessed. Next, after arteries were precontracted to 65% of maximal PE-induced contraction and tension was stable, responses to: acetylcholine (ACh, 10−8–10−4 M); sodium nitroprusside (SNP, 10−9–10−4 M); NG monomethyl-L-arginine (L-NMMA, 10−3 M) and ACh (10−8–10−4 M) in vessels incubated with L-NMMA (10−3 M) were performed. All tension data were recorded continuously by a computer through an analog-to-digital interface card (Biopac Systems Inc., Santa Barbara, CA, USA) that allowed for subsequent off-line quantitative analyses (Symons et al. 2006). Reagents for all experiments were purchased from Sigma-Aldrich unless otherwise specified.
Statistical Analysis
Values shown represent means ± standard error (SE). Statistical analysis was performed by repeated-measures analysis of variance (ANOVA) and Bonferroni posttests, with a P value of < .05 being considered statistically significant.
Results
RGZ-induced BP-lowering Effect
To evaluate the relative importance of EC and VSMC PPARγ in TZD-induced BP-lowering effect, daily MAP in DOCA salt–treated PPARγ f/f, SM22Cre/flox, and Tie2Cre/flox mice was monitored before and during a five-day RGZ treatment. The hypertension responses to DOCA salt were not different among the three mouse strains. However, the two vascular PPARγ-null mice had similar hypertensive responses to DOCA salt but displayed distinct sensitivities to RGZ-induced lowering effect as compared to their floxed controls. A five-day RGZ treatment induced a similar BP-lowering effect in DOCA salt–treated PPARγ f/f (on day 11: 139.2 ± 2.7 vs. 126.1 ± 2.9 mmHg, p < .05) and SM22Cre/flox mice (on day 11: 137.4 ± 2.1 vs. 126.2 ± 3.2 mmHg, p < .05), whereas Tie2Cre/flox mice were resistant to this effect (on day 11: 133.29 ± 3.48 vs. 131.12 ± 2.6 mmHg, p > .05) (Figure 1).
Figure 1.
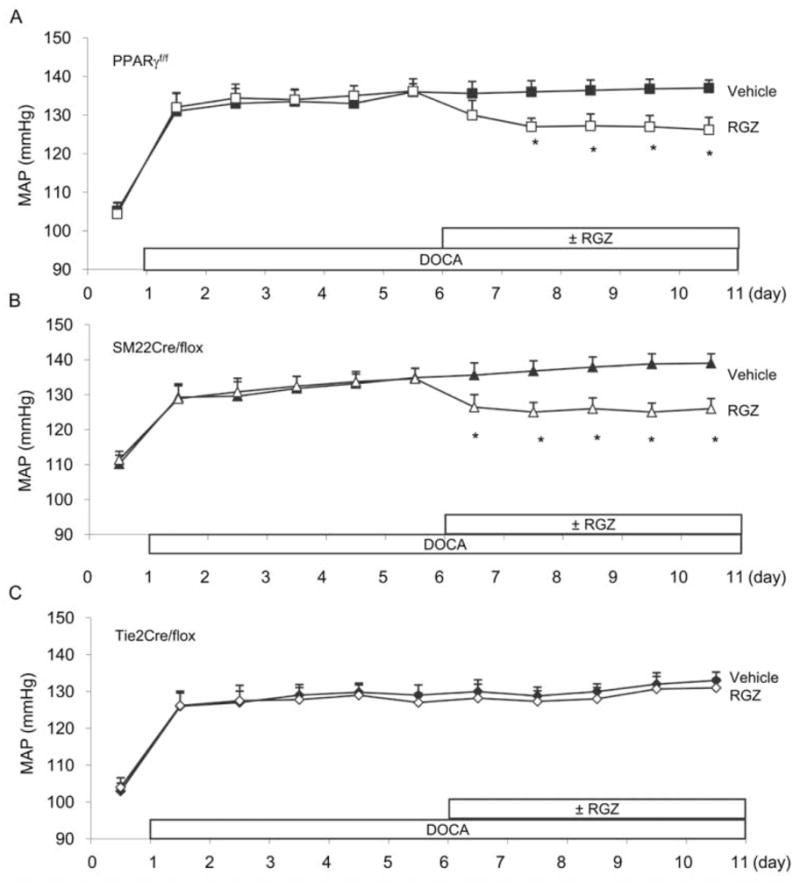
Effects of RGZ on MAP in deoxycorticosterone acetate (DOCA) salt–treated PPARγ f/f (A), SM22Cre/flox (B), and Tie2Cre/flox (C) mice. DOCA salt hypertension was induced by implantation of a 50 mg DOCA pellet in conjunction with a salt load including 1.5% NaCl in diet and 0.9% NaCl in drinking water. Five days after DOCA salt, each strain of mice was divided to receive a six-day treatment with vehicle or RGZ (320 mg/kg diet). Daily MAP was monitored by telemetry. The data from littermate floxed controls for SM22Cre/flox and Tie2Cre/flox were similar and thus were pooled. *, p < .05 versus vehicle for the corresponding period. n = 5–8 per data point.
Vessel Reactivity
Vasocontractile responses to phenylephrine (PE) but not Ang II or KCl were blunted in vessels from SM22Cre/flox versus PPARf/f mice (Figures 2A, 2B, and 2C). In contrast, femoral arteries from Tie2Cre/flox mice were more responsive to PE, Ang II, and KCl compared to vessels from PPARγf/f mice (Figures 2D, 2E, and 2F). Acetylcholine (ACh)-evoked vasorelaxation was greater in vessels from SM22Cre/flox versus PPARγf/f mice and was inhibited by NG monomethyl-L-arginine (L-NMMA) (Figures 3A–3C). Vasorelaxation responses to ACh and ACh + L-NMMA were similar between Tie2Cre/flox and PPARγf/f mice (Figures 3D–3F). Sodium nitroprusside (SNP)-induced vasorelaxation was not affected in either SM22Cre/flox or Tie2Cre/flox mice as compared with control mice (Figure 4).
Figure 2.
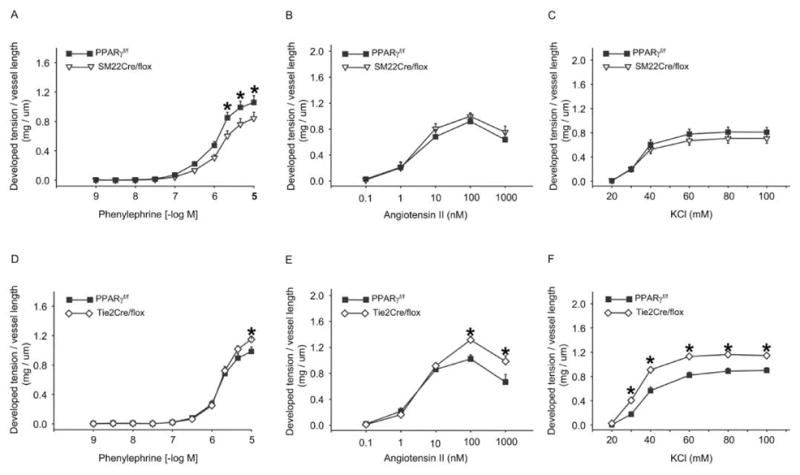
Vasoconstrictor responses in vascular PPARγ mutant mice. Phenylephrine (A, D), angiotensin II (B, E), and KCl (C, E) dose-response curves of femoral arteries from SM22Cre/flox (A, B, and C) and Tie2Cre/flox (D, E, and F) mice. *, p < .05 versus PPARγ f/f for the corresponding doses of vasoconstrictors. n = 5–9 per data point. Data are mean ± SE.
Figure 3.
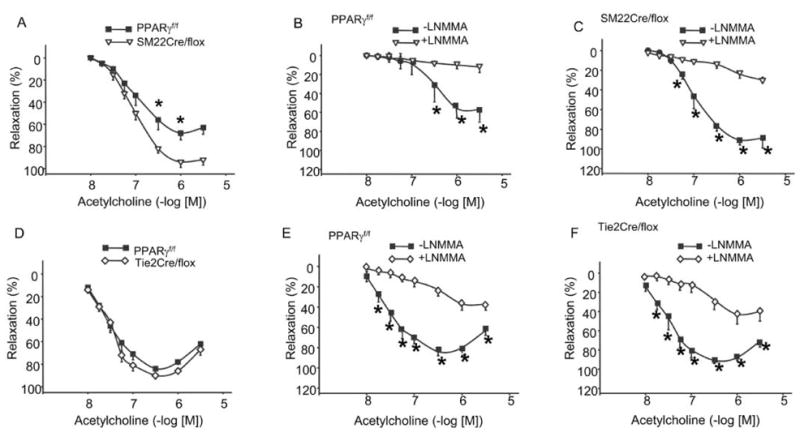
Vasodilator responses in vascular PPARγ mutant mice to acetylcholine. A and E, Acetylcholine dose-responses curves of femoral arteries in SM22Cre/flox, Tie2Cre/flox mice and their respective floxed controls. B and F, Effects of L-NMMA on acetylcholine-induced relaxation in the floxed controls for the two strains of vascular PPARγ mutant mice. C and G, Effects of L-NMMA on acetylcholine-induced relaxation in SM22Cre/flox and Tie2Cre/flox mice. D and H, Sodium nitroprusside (SNP)-induced relaxation in SM22Cre/flox, Tie2Cre/flox mice, and their respective littermate flox controls. *, p < .05 versus PPARγ f/f for the corresponding doses of vasoconstrictors. n = 5 or 6 per data point. Data are mean ± SE.
Figure 4.
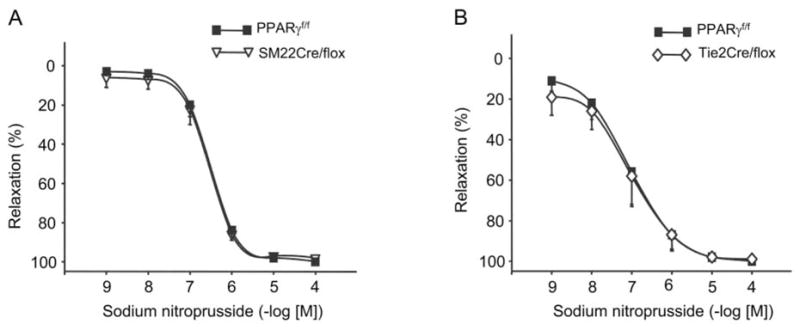
Sodium nitroprusside-induced relaxation in the femoral arteries of SM22Cre/flox (A) and Tie2Cre/flox mice (B). n = 5–6 per data point. Data are mean ± SE.
Discussion
A component of the vasculoprotective properties afforded to TZDs is their ability to lower BP. This characteristic likely explains the paradox that fluid retention evoked by TZDs is not associated with elevated BP (Guan et al. 2005; Song et al. 2004; Zhang et al. 2005). The present study characterized the vascular site responsible for TZD-induced BP lowering using SM22Cre/flox and Tie2Cre/flox. Specifically, RGZ-induced hypotension in mice with DOCA salt hypertension was dependent on PPARγ in ECs but not SMCs. These findings are consistent with a previous study, wherein endothelial-restricted PPARγ deficiency partially blunted the BP-lowering effect induced by RGZ, as assessed by tail cuff plethymography (Nicol et al. 2005). In the latter study, BP lowering was evaluated in the context of diet-induced obesity, which made it difficult to discern whether this was a direct effect or secondary to improved insulin sensitivity. Additionally, obesity-induced hypertension is not readily produced in mice. For example, diet-induced obesity in mice has been associated with no change (Tallam et al. 2005) or a modest increase in BP (approximately 10 mmHg) (Rao et al. 2007; Williams et al. 2003), as assessed by telemetry. A further consideration is the method wherein BP was evaluated. For example, stress imposed by restraint and/or tail cuff inflation might exacerbate obesity-induced hypertension (Nicol et al. 2005), as reported in a rat model of fructose-induced hypertension (D’Angelo et al. 2005). Another major extension of the study by Nicol et al. (2005) is the parallel assessment of involvement of VSMC PPARγ. In VSMCs, TZDs are reported to attenuate the agonist-induced rise of intracellular calcium (Buchanan et al. 1995) and the activation of phosphatidylinositol 3-kinase/Akt (Atkins et al. 2005) and voltage-gated (L-type) calcium channel (Zhang et al. 1994), and to inhibit proliferative and inflammatory responses (Hsueh and Law, 2001; Law et al. 2000), all of which may contribute to TZD-induced BP lowering effect. Surprisingly, the involvement of VSMC can not be confirmed by the present study.
The distinct role of PPARγ in the two vascular cell types is further demonstrated by results we obtained from vascular function studies using isolated vessels. The vasculature from Tie2Cre/flox mice exhibited enhanced responses to receptor and nonreceptor–dependent vasoconstrictors, albeit with a well-preserved response to ACh, suggesting that EC PPARγ may exert a vasculoprotective effect, primarily via attenuating vasoconstriction rather than enhancing endothelium-dependent vasorelaxation. This finding is unexpected, given that TZDs improve endothelial dysfunction in Ang II-infused rats (Diep et al. 2002) and in fat-fed rabbits (Tao et al. 2003). In contrast to Tie2Cre/flox, SM22Cre/flox mice exhibited blunted sensitivity to PE but enhanced ACh-evoked vasorelaxation. The enhanced vasorelaxation appeared to be nitric oxide (NO)–dependent, given the responsiveness to L-NNMA. These findings are similar to those reported previously in whole-body PPARγ KO mice (Duan et al. 2007). The distinct actions of PPARγ in the two vascular cell types suggest that a “yin–yang” relationship might exist in blood vessels to maintain vascular homeostasis. For instance, deletion of PPARγ in one cell type perturbs vascular responsiveness in the other, revealing that crosstalk exists between the two vascular cells. The underling mechanism of this phenomenon is unclear and represents an attractive area for future research. However, our results disagree with a recent study by Halabi et al., who report that transgenic expression of dominant-negative mutations of PPARγ in the smooth muscle leads to reduced vasorelaxation and enhanced vasoconstriction in the aorta (Halabi et al. 2008). The finding that the different phenotypes resulted from an entire gene deletion versus mutagenesis of a single amino acid may indicate that different regions in the PPARγ protein may serve distinct functions in the control of vascular tone.
In summary, the present study employed a cell-specific approach to study the vascular function of PPARγ. EC PPARγ deficiency completely blocked TZD-induced BP-lowering effect, whereas SMC PPARγ deficiency was without an effect. Surprisingly, the vasculature lacking EC PPARγ exhibited an overall enhanced response to vasoconstrictors, whereas the vasculature lacking SMC PPARγ displayed blunted phenylephrine-induced vasoconstriction but enhanced ACh-induced vasorelaxation. These results offer a new insight into distinct functions of EC and SMC PPARγ in regulation of BP and vascular tone.
Acknowledgments
This work was supported by Merit Review from the Department of Veterans Affairs as well as RO-1 HL079453 from National Institute of Health (to T. Yang).
Abbreviations
- ACh
acetylcholine
- Ang II
angiotensin II
- BP
blood pressure
- Cre
Cre recombinase
- DOCA
deoxycorticosterone acetate
- ECs
endothelial cells
- L-NMMA
NG monomethyl-L-arginine
- MAP
mean arterial pressure
- PE
phenylephrine
- PPARγ
peroxisome proliferators–activated receptor gamma
- RGZ
rosiglitazone
- SM22Cre/flox
mice homozygous for the PPARγ floxed allele and heterozygous the SM22-Cre transgene (smooth muscle PPARγ deficient)
- SNP
sodium nitroprusside
- Tie2Cre/flox
mice homozygous for the PPARγ floxed allele and heterozygous the Tie2-Cre transgene (endothelial PPARγ deficient)
- TZD
thiazolidinedione
- VSMCs
vascular smooth muscle cells
References
- Atkins KB, Northcott CA, Watts SW, Brosius FC. Effects of PPAR-gamma ligands on vascular smooth muscle marker expression in hypertensive and normal arteries. Am J Physiol Heart Circ Physiol. 2005;288:H235–43. doi: 10.1152/ajpheart.00643.2004. [DOI] [PubMed] [Google Scholar]
- Berger JP, Akiyama TE, Meinke PT. PPARs: therapeutic targets for metabolic disease. Trends Pharmacol Sci. 2005;26:244–51. doi: 10.1016/j.tips.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Boucher P, Gotthardt M, Li WP, Anderson RG, Herz J. LRP: role in vascular wall integrity and protection from atherosclerosis. Science. 2003;300:329–32. doi: 10.1126/science.1082095. [DOI] [PubMed] [Google Scholar]
- Buchanan TA, Meehan WP, Jeng YY, Yang D, Chan TM, Nadler JL, Scott S, Rude RK, Hsueh WA. Blood pressure lowering by pioglitazone. Evidence for a direct vascular effect. J Clin Invest. 1995;96:354–60. doi: 10.1172/JCI118041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angelo G, Elmarakby AA, Pollock DM, Stepp DW. Fructose feeding increases insulin resistance but not blood pressure in Sprague-Dawley rats. Hypertension. 2005;46:806–11. doi: 10.1161/01.HYP.0000182697.39687.34. [DOI] [PubMed] [Google Scholar]
- Diep QN, El Mabrouk M, Cohn JS, Endemann D, Amiri F, Virdis A, Neves MF, Schiffrin EL. Structure, endothelial function, cell growth, and inflammation in blood vessels of angiotensin II-infused rats: role of peroxisome proliferator-activated receptor-gamma. Circulation. 2002;105:2296–302. doi: 10.1161/01.cir.0000016049.86468.23. [DOI] [PubMed] [Google Scholar]
- Duan SZ, Ivashchenko CY, Whitesall SE, D’Alecy LG, Duquaine DC, Brosius FC, 3rd, Gonzalez FJ, Vinson C, Pierre MA, Milstone DS, Mortensen RM. Hypotension, lipodystrophy, and insulin resistance in generalized PPARgamma-deficient mice rescued from embryonic lethality. J Clin Invest. 2007;117:812–22. doi: 10.1172/JCI28859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med. 2004;10:355–61. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- Fernandez AZ. Peroxisome proliferator-activated receptors in the modulation of the immune/inflammatory response in atherosclerosis. PPAR Res. 2008;2008:285842. doi: 10.1155/2008/285842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frutkin AD, Shi H, Otsuka G, Leveen P, Karlsson S, Dichek DA. A critical developmental role for tgfbr2 in myogenic cell lineages is revealed in mice expressing SM22-Cre, not SMMHC-Cre. J Mol Cell Cardiol. 2006;41:724–31. doi: 10.1016/j.yjmcc.2006.06.067. [DOI] [PubMed] [Google Scholar]
- Fukunaga Y, Itoh H, Doi K, Tanaka T, Yamashita J, Chun TH, Inoue M, Masatsugu K, Sawada N, Saito T, et al. Thiazolidinediones, peroxisome proliferator-activated receptor gamma agonists, regulate endothelial cell growth and secretion of vasoactive peptides. Atherosclerosis. 2001;158:113–19. doi: 10.1016/s0021-9150(01)00430-0. [DOI] [PubMed] [Google Scholar]
- Guan Y, Hao C, Cha DR, Rao R, Lu W, Kohan DE, Magnuson MA, Redha R, Zhang Y, Breyer MD. Thiazolidinediones expand body fluid volume through PPARgamma stimulation of ENaC-mediated renal salt absorption. Nat Med. 2005;11:861–66. doi: 10.1038/nm1278. [DOI] [PubMed] [Google Scholar]
- Halabi CM, Beyer AM, de Lange WJ, Keen HL, Baumbach GL, Faraci FM, Sigmund CD. Interference with PPAR gamma function in smooth muscle causes vascular dysfunction and hypertension. Cell Metab. 2008;7:215–26. doi: 10.1016/j.cmet.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernando E, Charytonowicz E, Dudas ME, Menendez S, Matushansky I, Mills J, Socci ND, Behrendt N, Ma L, Maki RG, Pandolfi PP, Cordon-Cardo C. The AKT-mTOR pathway plays a critical role in the development of leiomyosarcomas. Nat Med. 2007;13:748–53. doi: 10.1038/nm1560. [DOI] [PubMed] [Google Scholar]
- Holtwick R, Gotthardt M, Skryabin B, Steinmetz M, Potthast R, Zetsche B, Hammer RE, Herz J, Kuhn M. Smooth muscle-selective deletion of guanylyl cyclase-A prevents the acute but not chronic effects of ANP on blood pressure. Proc Natl Acad Sci U S A. 2002;99:7142–47. doi: 10.1073/pnas.102650499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsueh WA, Law RE. PPARgamma and atherosclerosis: effects on cell growth and movement. Arterioscler Thromb Vasc Biol. 2001;21:1891–95. doi: 10.1161/hq1201.100261. [DOI] [PubMed] [Google Scholar]
- Inoue I, Shino K, Noji S, Awata T, Katayama S. Expression of peroxisome proliferator-activated receptor alpha (PPAR alpha) in primary cultures of human vascular endothelial cells. Biochem Biophys Res Commun. 1998;246:370–74. doi: 10.1006/bbrc.1998.8622. [DOI] [PubMed] [Google Scholar]
- Jia Z, Zhang A, Zhang H, Dong Z, Yang T. Deletion of microsomal prostaglandin E synthase-1 increases sensitivity to salt loading and angiotensin II infusion. Circ Res. 2006;99:1243–51. doi: 10.1161/01.RES.0000251306.40546.08. [DOI] [PubMed] [Google Scholar]
- Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–42. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- Komers R, Vrana A. Thiazolidinediones—tools for the research of metabolic syndrome X. Physiol Res. 1998;47:215–25. [PubMed] [Google Scholar]
- Law RE, Goetze S, Xi XP, Jackson S, Kawano Y, Demer L, Fishbein MC, Meehan WP, Hsueh WA. Expression and function of PPARgamma in rat and human vascular smooth muscle cells. Circulation. 2000;101:1311–18. doi: 10.1161/01.cir.101.11.1311. [DOI] [PubMed] [Google Scholar]
- Marx N, Schonbeck U, Lazar MA, Libby P, Plutzky J. Peroxisome proliferator-activated receptor gamma activators inhibit gene expression and migration in human vascular smooth muscle cells. Circ Res. 1998;83:1097–103. doi: 10.1161/01.res.83.11.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miano JM, Ramanan N, Georger MA, de Mesy Bentley KL, Emerson RL, Balza RO, Jr, Xiao Q, Weiler H, Ginty DD, Misra RP. Restricted inactivation of serum response factor to the cardiovascular system. Proc Natl Acad Sci U S A. 2004;101:17132–37. doi: 10.1073/pnas.0406041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol CJ, Adachi M, Akiyama TE, Gonzalez FJ. PPARgamma in endothelial cells influences high fat diet-induced hypertension. Am J Hypertens. 2005;18:549–56. doi: 10.1016/j.amjhyper.2004.10.032. [DOI] [PubMed] [Google Scholar]
- Raji A, Seely EW, Bekins SA, Williams GH, Simonson DC. Rosiglitazone improves insulin sensitivity and lowers blood pressure in hypertensive patients. Diabetes Care. 2003;26:172–78. doi: 10.2337/diacare.26.1.172. [DOI] [PubMed] [Google Scholar]
- Rao R, Hao CM, Redha R, Wasserman DH, McGuinness OP, Breyer MD. Glycogen synthase kinase 3 inhibition improves insulin-stimulated glucose metabolism but not hypertension in high-fat-fed C57BL/6J mice. Diabetologia. 2007;50:452–60. doi: 10.1007/s00125-006-0552-5. [DOI] [PubMed] [Google Scholar]
- Satoh H, Tsukamoto K, Hashimoto Y, Hashimoto N, Togo M, Hara M, Maekawa H, Isoo N, Kimura S, Watanabe T. Thiazolidinediones suppress endothelin-1 secretion from bovine vascular endothelial cells: a new possible role of PPARgamma on vascular endothelial function. Biochem Biophys Res Commun. 1999;254:757–63. doi: 10.1006/bbrc.1998.0126. [DOI] [PubMed] [Google Scholar]
- Song J, Knepper MA, Hu X, Verbalis JG, Ecelbarger CA. Rosiglitazone Activates Renal Sodium- and Water-Reabsorptive Pathways and Lowers Blood Pressure in Normal Rats. J Pharmacol Exp Ther. 2004;308:426–33. doi: 10.1124/jpet.103.058008. [DOI] [PubMed] [Google Scholar]
- Staels B, Koenig W, Habib A, Merval R, Lebret M, Torra IP, Delerive P, Fadel A, Chinetti G, Fruchart JC, Najib J, Maclouf J, Tedqui A. Activation of human aortic smooth-muscle cells is inhibited by PPARalpha but not by PPARgamma activators. Nature. 1998;393:790–93. doi: 10.1038/31701. [DOI] [PubMed] [Google Scholar]
- Symons JD, Rutledge JC, Simonsen U, Pattathu RA. Vascular dysfunction produced by hyperhomocysteinemia is more severe in the presence of low folate. Am J Physiol Heart Circ Physiol. 2006;290:H181–91. doi: 10.1152/ajpheart.00765.2005. [DOI] [PubMed] [Google Scholar]
- Tallam LS, Stec DE, Willis MA, da Silva AA, Hall JE. Melanocortin-4 receptor-deficient mice are not hypertensive or salt-sensitive despite obesity, hyperinsulinemia, and hyperleptinemia. Hypertension. 2005;46:326–32. doi: 10.1161/01.HYP.0000175474.99326.bf. [DOI] [PubMed] [Google Scholar]
- Tao L, Liu HR, Gao E, Teng ZP, Lopez BL, Christopher TA, Ma XL, Batinic-Haberle I, Willette RN, Ohlstein EH, Yue TL. Antioxidative, antinitrative, and vasculoprotective effects of a peroxisome proliferator-activated receptor-gamma agonist in hypercholesterolemia. Circulation. 2003;108:2805–11. doi: 10.1161/01.CIR.0000097003.49585.5E. [DOI] [PubMed] [Google Scholar]
- Wan Y, Saghatelian A, Chong LW, Zhang CL, Cravatt BF, Evans RM. Maternal PPAR gamma protects nursing neonates by suppressing the production of inflammatory milk. Genes Dev. 2007;21:1895–908. doi: 10.1101/gad.1567207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams TD, Chambers JB, Roberts LM, Henderson RP, Overton JM. Diet-induced obesity and cardiovascular regulation in C57BL/6J mice. Clin Exp Pharmacol Physiol. 2003;30:769–78. doi: 10.1046/j.1440-1681.2003.t01-1-03808.x. [DOI] [PubMed] [Google Scholar]
- Xin M, Small EM, van Rooij E, Qi X, Richardson JA, Srivastava D, Nakagawa O, Olson EN. Essential roles of the bHLH transcription factor Hrt2 in repression of atrial gene expression and maintenance of postnatal cardiac function. Proc Natl Acad Sci U S A. 2007;104:7975–80. doi: 10.1073/pnas.0702447104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Sowers JR, Ram JL, Standley PR, Peuler JD. Effects of pioglitazone on calcium channels in vascular smooth muscle. Hypertension. 1994;24:170–75. doi: 10.1161/01.hyp.24.2.170. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zhang A, Kohan DE, Nelson RD, Gonzalez FJ, Yang T. Collecting duct-specific deletion of peroxisome proliferator-activated receptor gamma blocks thiazolidinedione-induced fluid retention. Proc Natl Acad Sci U S A. 2005;102:9406–11. doi: 10.1073/pnas.0501744102. [DOI] [PMC free article] [PubMed] [Google Scholar]