

Abstract
Here, we investigate the effects of endothelial nitric oxide synthase (eNOS) on angiogenesis, neurogenesis, neurotrophic factor expression, and neurological functional outcome after stroke. Wild-type and eNOS knock-out (eNOS-/-) mice were subjected to permanent occlusion of the right middle cerebral artery. eNOS-/- mice exhibited more severe neurological functional deficit after stroke than wild-type mice. Decreased subventricular zone (SVZ) progenitor cell proliferation and migration, measured using bromodeoxyuridine, Ki-67, nestin, and doublecortin immunostaining in the ischemic brain, and decreased angiogenesis, as demonstrated by reduced endothelial cell proliferation, vessel perimeter, and vascular density in the ischemic border, were evident in eNOS-/- mice compared with wild-type mice. eNOS-deficient mice also exhibited a reduced response to vascular endothelial growth factor (VEGF)-induced angiogenesis in a corneal assay. ELISAs showed that eNOS-/- mice have decreased brain-derived neurotrophic factor (BDNF) expression but not VEGF and basic fibroblast growth factor in the ischemic brain compared with wild-type mice. In addition, cultured SVZ neurosphere formation, proliferation, telomerase activity, and neurite outgrowth but not cell viability from eNOS-/- mice were significantly reduced compared with wild-type mice. BDNF treatment of SVZ cells derived from eNOS-/- mice restored the decreased neurosphere formation, proliferation, neurite outgrowth, and telomerase activity in cultured eNOS-/- SVZ neurospheres. SVZ explant cell migration also was significantly decreased in eNOS-/- mice compared with wild-type mice. These data indicate that eNOS is not only a downstream mediator for VEGF and angiogenesis but also regulates BDNF expression in the ischemic brain and influences progenitor cell proliferation, neuronal migration, and neurite outgrowth and affects functional recovery after stroke.
Keywords: eNOS, angiogenesis, neurogenesis, BDNF, neural progenitor cells, focal cerebral ischemia
Introduction
Nitric oxide (NO) is a reactive molecule with numerous physiological and pathophysiological roles affecting the nervous and cardiovascular systems. NO produced by endothelial nitric oxide synthase (eNOS) has a crucial role in the regulation of systemic blood pressure, vascular tone, vascular remodeling, and angiogenesis (Murohara et al., 1998; Rudic et al., 1998). Enhanced eNOS phosphorylation has a broad range of effects, including the promotion of proliferation of endothelial cells and angiogenesis (Murohara et al., 1998; Rudic et al., 1998). eNOS mutant mice are hypertensive and exhibit an increase in infarction volume at 24 h after stroke, and eNOS is neuroprotective after stroke (Huang et al., 1996). Mice deficient in eNOS show reduced long-term potentiation (Doreulee et al., 2003), and the mortality rate is increased in aged eNOS-deficient mice (Dere et al., 2002). However, the effects of eNOS on regulating neuronal progenitor cell proliferation, survival, migration, and neurite outgrowth and the effects of eNOS on functional recovery after stroke have not been investigated.
Brain-derived neurotrophic factor (BDNF) is involved in neuronal survival, migration, morphological and biochemical differentiation, and modulation of synaptic function in the CNS (Ferrer et al., 2001; Gorski et al., 2003). Recent studies show that NO regulates BDNF expression, which affects neural progenitor cell proliferation and differentiation in the mammalian brain (Canossa et al., 2002; Cheng et al., 2003). NO mediates BDNF-induced growth-associated protein 43 (GAP-43) mRNA expression (Klocker et al., 2001). BDNF exerts a neuroprotective effect via downregulation of neuronal NOS (nNOS) activity (Sharma et al., 1998). These observations highlight the potentially important role of NOS in modulating BDNF secretion and the need to investigate the effect of eNOS on regulating BDNF expression in the brain after stroke.
In the present study, we demonstrate that eNOS-deficient mice have reduced functional recovery after stroke, concomitant with reduced angiogenesis and neuronal and progenitor cell migration. We provide evidence that a reduction of BDNF in eNOS-/- mice may be responsible for the deficits in functional recovery and reduced brain plasticity.
Materials and Methods
The middle cerebral artery occlusion model and experiment groups. Adult male wild-type C57BL/6J mice (age, 2-3 months; weight, 24-28 g) were purchased from Charles River Laboratories (Wilmington, MA). Adult male C57BL/6J eNOS knock-out (eNOS-/-) mice (age 2-3 months, weight 24-28 g) were provided by Dr. Edward G. Shesely (Hypertension and Vascular Research Division, Henry Ford Hospital). Mice were anesthetized with halothane. Permanent right middle cerebral artery occlusion (MCAo) was induced by advancing a 6-0 surgical nylon suture (8.0-9.0 mm determined by body weight) with an expanded (heated) tip from the external carotid artery into the lumen of the internal carotid artery to block the origin of the MCA (Mao et al., 1999). The experimental groups consisted of group 1, wild-type mice subjected to permanent MCAo (n = 11), and group 2, eNOS-/- mice subjected to permanent MCAo (n = 14). Functional tests were performed on groups 1 and 2. To identify newly formed DNA in ischemic brain, mice received injections of bromodeoxyuridine (BrdU) (100 mg/kg in 0.007N NaOH physiological saline; Sigma, St. Louis, MO) intraperitoneally starting 1 d after MCAo and daily for 7 consecutive days. Groups 1 and 2 were killed 2 h after the last BrdU injection at 7 d after MCAo for immunostaining. Group 3 consisted of wild-type mice subjected to permanent MCAo (n = 6), and group 4 consisted of eNOS-/- mice subjected to permanent MCAo (n = 6). Blood pressure was measured in groups 3 and 4 before the animals were killed at 3 d after MCAo. The left femoral artery of each mouse was cannulated for arterial blood pressure (Protocol Systems). Mice in groups 3 and 4 were killed at 3 d after MCAo, and brain tissue extracts were obtained for ELISA.
Behavioral tests. A battery of behavioral tests [a modified neurological severity score (mNSS) and foot-fault tests] were performed before MCAo and at 1, 3, and 7 d after MCAo in all mice of groups 1 and 2.
mNSS. Neurological function is graded on a scale of 0-14 (normal score, 0; maximal deficit score, 14). mNSS is a composite of motor, sensory, reflex, and balance tests (Chen et al., 2001). In the severity scores of injury, one score point is awarded for the inability to perform the test or for the lack of a tested reflex; thus, the higher score, the more severe the injury.
Foot-fault test. For locomotor assessment, mice were tested for placement dysfunction of forelimbs with the modified foot-fault test (Hernandez and Schallert, 1988). Mice were placed on elevated hexagonal grids of different sizes (1-3 cm) with their paws on the wire while moving along the grid. With each weight-bearing step, the paw may fall or slip between the wire. This was recorded as a foot fault. The total number of steps (movement of each forelimb) was counted, and the total number of foot faults for each forelimb was recorded. The percentage of foot faults of the left paw to total steps were determined.
Histological assessment. Mice from groups 1 and 2 subjected to MCAo were killed at day 7 after MCAo. Brains were fixed by transcardial perfusion with saline, followed by perfusion and immersion in 4% paraformaldehyde, and the brains were embedded in paraffin. Seven coronal sections of tissue were processed and stained with hematoxylin and eosin for calculation of the volume of cerebral infarction (Swanson et al., 1990). The indirect lesion area, in which the intact area of the ipsilateral hemisphere was subtracted from the area of the contralateral hemisphere, was calculated (Swanson et al., 1990). Lesion volume is presented as a volume percentage of the lesion compared with the contralateral hemisphere.
Immunohistochemistry. BrdU, telomerase reverse transcriptase (TERT) (a catalytic subunit of telomerase), von Willebrand Factor (vWF), doublecortin (DCX), nestin, and Ki-67 immunostaining was performed. Briefly, two standard paraffin blocks were obtained from the center of the lesion, corresponding to coronal coordinates for bregma -1 to 1 mm. A series of 6-μm-thick sections at various levels (50 μm interval) were cut from the two blocks. A mouse monoclonal antibody against BrdU (1:100; Roche Molecular Biochemicals, Indianapolis, IN), TERT (mouse anti-telomerase, 1:500 dilution; Calbiochem, La Jolla, CA), vWF (goat polyclonal IgG antibody, 1:200 dilution; Santa Cruz Biotechnology, Santa Cruz, CA), DCX, a protein expressed in migrating neurons (C-18, goat polyclonal IgG antibody, 1:200 dilution; Santa Cruz Biotechnology), nestin, a marker of neuroepithelial stem cells (1:100 dilution; BD Biosciences, Franklin Lake, NJ), and Ki-67, a marker of proliferating cells (1:300 dilution; Novus) were used. In addition, BrdU (Cy5) with DCX (FITC) double staining was performed (Chen et al., 2004). Control experiments consisted of staining brain coronal tissue sections as outlined above but omitting the primary antibodies, as described previously (Li et al., 1998).
Brain endothelial cell proliferation. For measurement of endothelial cell proliferation, five slides from each block of a BrdU- and TERT-immunostained coronal section were digitized using a 20× objective (BX40; Olympus Optical, Tokyo, Japan) via the microcomputer imaging device (MCID) computer imaging analysis system (Imaging Research, St. Catharines, Ontario, Canada). The number of BrdU- and TERT-immunoreactive endothelial cells (nuclei on the lumen of vessel) within a total of 10 enlarged and thin-walled vessels located in the ischemic border area were counted in each section (Chen et al., 2003). Data are presented as the percentage of the number of BrdU- or TERT-immunoreactive cells within 10 vessels per total endothelial cell number.
Vascular perimeter and density measurement. For measurement of vascular perimeters and density, five slides from each block of a vWF-immunostained coronal section, with each slide containing eight fields of view within the cortex and striatum from the ischemic border area (Fig. 1), was digitized using a 20× objective via the MCID computer imaging analysis system. The ischemic border zone is defined as the area surrounding the lesion, which morphologically differs from the surrounding normal tissue (Nedergaard et al., 1987). The total perimeter of 10 enlarged and thin-walled vessels and vascular density in the ischemic border area were measured in each referenced coronal section, as described previously (Chen et al., 2003), using the MCID computer imaging analysis system (length trace function). The total perimeter of 10 enlarged and thin-walled vessels and vascular density in the ischemic border are provided.
Figure 1.

Quantification fields of view in the ischemic border in mice.
Quantification. For semiquantitative measurements, DCX- and nestin-positive cells in the ipsilateral subventricular zone (SVZ) or ischemic border area (as shown as Fig. 1) were digitized under a 20× objective (BX40; Olympus Optical) using a 3-CCD color video camera (DXC-970MD; Sony, Tokyo, Japan) interfaced with an MCID image analysis system. The digitalized images were then contrast enhanced to clearly differentiate positivity from background, and a thresholding procedure was established to determine the proportion of immunoreactive area within each fixed field of view (Calza et al., 2001; Chen et al., 2003). The thresholds were selected using a “set color threshold” feature in the analysis software. This feature allows the user to select pixel regions that are considered positive and applies the same threshold parameter to all pixels in the same image. After establishing a threshold parameter that covers only reasonably positive regions on a control image, the same parameter was applied to all images that were obtained at equal objectives and light intensities on slides that were processed at the same time. The data are presented as a percentage of positive immunoreactivity area within the total SVZ or ischemic border area.
ELISA. To test whether eNOS regulates growth factor expression in the ischemic brain after stroke, eNOS-/- and wild-type mice were subjected to MCAo and killed at 3 d after MCAo (groups 3 and 4 animals; n = 6 per group). Brain extracts were obtained from the ischemic border identified visually (bregma -1 to 1 mm, border region encompassing the ischemic core) at 3 d after MCAo (Nedergaard et al., 1987). Tissue blocks were dissected on ice, and wet weight was rapidly measured. The brain extracts were divided into 200 μl triplicate samples. Using ELISA kits (R & D Systems, Minneapolis, MN; Calbiochem), BDNF, vascular endothelial growth factor (VEGF), and basic fibroblast growth factor (bFGF) ELISAs were performed.
Corneal assay for angiogenesis. To test whether VEGF-induced angiogenesis requires eNOS, a corneal assay (Cao et al., 1998) was performed (n = 6 per group). The experimental groups included (1) wild-type mice and eNOS-/- controls, in which 12% hydrion (1 μl) alone was inserted 2 mm away from the limbus margin of the cornea for controls, and (2) VEGF treatment in wild-type and eNOS-/- mice, in which mixed VEGF (1 μl, 160 μg) with 12% hydrion (1 μl) was inserted 2 mm away from the limbus margin of the cornea in wild-type and eNOS-/- mice. Corneal images were captured and analyzed at 14 d after surgery under a 2.5× objective for measurement of vessel length extension (in millimeters) by the vessels using a video camera interfaced with MCID image analysis system.
SVZ neurosphere culture. SVZ cells were dissociated from normal adult male C57BL/6J and eNOS-/- mice (n = 10 per group), as reported previously (Morshead et al., 1994). The cells were plated at a density of 3 × 104 cells/ml in DMEM-F-12 medium containing 20 ng/ml epidermal growth factor (EGF) (R & D Systems) and bFGF (R & D Systems). DMEM-F-12 medium contains DMEM-F-12 (R & D Systems), l-glutamine (2 mm), glucose (0.6%), putrescine (9.6 μg/ml), insulin (0.025 mg/ml), progesterone (6.3 ng/ml), apotransferrin (0.1 mg/ml), and sodium selenite (5.2 ng/ml).
Primary sphere formation assay. Mechanically dissociated SVZ cells were plated at 3 × 104 cells/ml of proliferation medium in each well of a 24-well plate (Corning, Corning, NY), with no substrate pretreatment. The primary neurosphere formation number was counted at 7 d in vitro (DIV).
Secondary sphere formation assay. Primary neurospheres were collected and digested with 0.25% trypsin-EDTA (Invitrogen, San Diego, CA) for 5 min at 37°C. They were then gently triturated with a firenarrowed Pasteur pipette, spun down at 400 rpm for 3 min, resuspended in proliferation medium, and plated at 2 × 104 cells/ml in each well of a 24-well plate (Corning).
The secondary cultured neurosphere cells were used for measurement of neurosphere cell self-renewal, proliferation (BrdU ELISA), viability, telomerase activity, and neurite outgrowth. The SVZ neurosphere culture experiment groups (n = 8 per group) included (1) wild-type SVZ neurosphere culture alone, (2) eNOS-/- mouse SVZ neurosphere culture alone, and (3) eNOS-/- SVZ neurosphere culture treated with BDNF (50 ng/ml). The neurosphere numbers were counted at 7 DIV.
BrdU ELISA for SVZ neurosphere cell proliferation. To evaluate DNA synthesis, the secondary SVZ neurosphere cells were plated at 2 × 104 cells/ml in each well of a 96-well plate (Corning) cultured for 4 d (Gasparri et al., 2004). The experiment groups (n = 8 per group) included (1) wild-type SVZ neurosphere culture alone, (2) eNOS-/- mouse SVZ neurosphere culture alone, and (3) eNOS-/- SVZ neurosphere culture treated with BDNF (50 ng/ml). BrdU at 50 μm was added to the SVZ neurosphere culture medium for 12 h before fixation, and samples were processed using an ELISA for detection of BrdU incorporated into a cellular DNA kit (BrdU labeling and detection kit III; Roche Molecular Biochemicals) according to the instructions of the manufacturer. The BrdU incorporation was then quantified by colorimetric immunoassay at 450 nm by an automated microplate reader, following kit instructions. The absorbance background signal measured was subtracted from all of the values reported.
Viability of the SVZ neurosphere measurement. The secondary SVZ neurosphere cells, plated at 2.0 × 104 cells/ml in each well of a 24-well plate (Corning), were identified using fluorescence microscopy according to the procedure provided with a livedead viability-cytotoxicity kit (Molecular Probes, Eugene, OR). The experiment groups (n = 8 per group) included (1) wild-type SVZ neurosphere culture alone, (2) eNOS-/- mouse SVZ neurosphere culture alone, and (3) eNOS-/- SVZ neurosphere culture treated with BDNF (50 ng/ml). A mixture of calcein AM and ethidium homodimer was added to cover and incubate the SVZ neurosphere cells for 20 min at 37°C. Live cells (possessing intracellular esterase activity) convert calcein AM to calcein. Calcein is retained in the cell and produces green fluorescence when excited. Dead cells (with their compromised plasma membranes) allow the entrance of ethidium homodimer, which undergoes a 40-fold increase in red fluorescence after binding to nucleic acids. Therefore, the nuclei of the dead cells appear red under the fluorescence microscope. The data are presented as a percentage of dead cells to live cells.
Telomerase activity assay. To determine telomerase activity in wild-type and eNOS-/- mouse cultured SVZ neurosphere cells, the Telo TAGGG telomerase PCR ELISA kit (catalog #1854666; Roche Molecular Biochemicals) was used. A standard protocol as directed by the company was followed to conduct the assay. The experiment groups (n = 8 per group) included (1) wild-type SVZ neurosphere culture alone, (2) eNOS-/- mouse SVZ neurosphere culture alone, and (3) eNOS-/- SVZ neurosphere culture treated with BDNF (50 ng/ml) for 48 h. In brief, 2 × 105 neurosphere cells were collected and lysed. Three microliters of the supernatant were used for PCR with the following profile: 25°C, 25 min; 94°C, 5 min (94°C, 30 s; 50°C, 30 s; and 72°C, 1.5 min) for 30 cycles; 72°C, 10 min; and 4°C hold. The resulting PCR product was denatured and used for ELISA. Five microliters of the PCR product were added to the microtiter plate, and it was incubated at 37°C for 2 h while being shaken at 300 rpm. Anti-digoxigenin-peroxidase was added and then incubated at 25°C at 300 rpm for 30 min. Then, 100 μl of substrate was added and incubated at 25°C at 300 rpm for 20 min. Stop solution was added, and the plate was immediately read at 450 nm with a reference wavelength of 690 nm.
SVZ neuronal cell neurite outgrowth. To identify neurite outgrowth, the generated secondary neurospheres were passaged by mechanical dissociation and reseeded as single cells at a density of 2.0 × 104 cells/ml of differentiation media. The cells were plated directly onto lamin-coated glass coverslips in DMEM-F-12 medium containing 2% FBS and EGF, which is referred to as differentiation medium. The experiment groups (n = 8 per group) included (1) wild-type SVZ neurosphere culture alone, (2) eNOS-/- mouse SVZ neurosphere culture alone, and (3) eNOS-/- SVZ neurosphere culture treated with BDNF (50 ng/ml) for 7 d. Every 4 d, half of the medium was replaced with fresh medium. Incubation was terminated at 7 d after plating. The cells were fixed at 7 DIV for class III β-tubulin (TUJ1) immunostaining. For the axonal arbors of TUJ1-positive cells, photomicrographs were captured using a 20× objective (BX40; Olympus Optical) via the MCID computer imaging analysis system. One hundred neurons per condition were documented. The total length of neurite outgrowth was measured using the perimeter/length function on the MCID system.
SVZ tissue explant cell migration measurement. To test whether eNOS regulates neuronal cell migration in vitro (Lois et al., 1996), eNOS-/- and wild-type C57BL/6J mouse tissue explants from the SVZ were harvested and plated on Matrigel (BD Biosciences) in 24 wells with 1000 μl of Neuralbasal-A medium containing 2% B27 supplement (Invitrogen) (Leventhal et al., 1999). The average linear distance and the total area of cell migration from the explant culture edge were captured using a 10× objective (BX40; Olympus Optical) via the MCID computer imaging analysis system and measured at day 5 using the MCID software. The average of the 10 longest migration distances and the area encompassing migration were assessed in each explant culture.
Statistical analysis. Two-way ANOVA was used to test the effects on functional improvement. Data in the two groups were first analyzed by one-way ANOVA. The χ2 test was used to analysis the mortality. An unpaired Student's t test was performed to compare BrdU-, TERT-, BDNF-, VEGF-, and bFGF- and DCX-, nestin-, Ki-67-, and TUJ1-reactive cell numbers or positive cell areas and telomerase activity between the two groups. The data are presented as mean ± SD; p < 0.05 was taken as significant.
Results
Neurological functional test, lesion volume, and blood pressure
To determine whether eNOS plays a role in neurological functional impairment and recovery after stroke, eNOS-/- mice were subjected to MCAo, and a battery of functional tests were performed. Neurological functional recovery was significantly attenuated in eNOS-/- mice on mNSS and foot-fault tests at 1, 3, and 7 d after stroke compared with wild-type mice subjected to MCAo (Fig. 2) (p < 0.05). The mortality rate after MCAo was 5 of 14 in eNOS-/- mice and 2 of 11 MCAo in wild-type mice (p = 0.33). The infarction volume was not significantly different (p = 0.31) between eNOS-/- mice (20.7 ± 10.8%) and wild-type mice (16.6 ± 8.2%). Blood pressure was significantly increased in eNOS-/- mice (103.0 ± 9.0 mmHg) compared with wild-type mice (71.3 ± 3.6 mmHg; p < 0.05) at 3 d after stroke.
Figure 2.

eNOS-deficient attenuated functional recovery after stroke: wild-type (n = 11) and eNOS knock-out (n = 14) mice were subjected to permanent MCAo. Functional tests were performed before MCAo and 1, 3, and 7 d after MCAo. A, mNSS test. B, Foot-fault test. *p < 0.05 versus wild-type MCAo control.
eNOS-deficient mice exhibit decreased angiogenesis in the ischemic brain
The presence of proliferating vascular endothelial cells and enlarged and thin-walled vessels, termed “mother” vessels, is a marker of cerebral ischemic angiogenesis (Zhang et al., 2002). To test whether eNOS affects angiogenesis after stroke, endothelial cell proliferation, vascular density, and enlarged vascular perimeters in the ischemic border were measured. Figure 3A-C shows a significant (p < 0.05) decrease in the number of BrdU-immunoreactive endothelial cells in eNOS-/- mice (Fig. 3B,C) compared with wild-type mice (Fig. 3A,C) in the ipsilateral hemisphere. Figure 3D-G shows that the enlarged thin-walled vessel perimeters and vascular density are significantly decreased in the ipsilateral hemisphere of eNOS-/- mice (Fig. 3E-G) compared with wild-type mice (Fig. 3D,F,G) after stroke. TERT affects the proliferation and survival of cells (Cao et al., 2002). To determine whether eNOS-deficient cells exhibit decreased TERT expression, TERT immunostaining was performed. Figure 3H-J shows that eNOS-deficient mice exhibit significantly decreased endothelial cell TERT expression in the ischemic border (Fig. 3I,J) compared with wild-type mice (Fig. 3H,J). These data suggest that the absence of eNOS decreases angiogenesis in the ischemic brain after stroke, which may be related to a decrease of endothelial cell TERT expression.
Figure 3.

eNOS-deficient mice exhibit reduced angiogenesis after stroke. Angiogenesis was indicated by BrdU-immunoreactive endothelial cells, vascular density, and perimeter in the ischemic border at 7 d after stroke. A, B, BrdU immunoreactivity in the ischemic border in wild-type (WT) control MCAo mice (A) and in eNOS-/- MCAo mice (B). D, E, vWF immunoreactivity vessels in the ischemic boundary area in wild-type mice (D) and eNOS-/- mice (E).C, F, G, Quantitative data that eNOS-deficient mice significantly (p < 0.05) decrease the number of BrdU-immunoreactive cells in vessels (C), vascular perimeter (F), and density (G) in the ipsilateral hemisphere compared with wild-type control MCAo. *p < 0.05 compared with wild-type MCAo control. H-J, TERT immunostaining in the ischemic border in wild-type mice (H) and eNOS-/- mice (I). J, Quantitative data that eNOS-deficient mice significantly (p < 0.05) decrease the number of TERT-immunoreactive cells in vessels in the ipsilateral hemisphere compared with wild-type control MCAo. K, Corneal angiogenesis assay in control wild-type mice. L, M, Corneal angiogenesis assay treated with VEGF in wild-type mice (L) and eNOS-/- mice (M), respectively. N, Quantitative data that eNOS-deficient mice significantly (p < 0.05) reduce VEGF-induced angiogenesis in the cornea compared with wild-type mice treated with VEGF. Scale bars: B, E, 100 μm; I, 50 μm.
Corneal angiogenesis assay
VEGF promotes angiogenesis. The angiogenic effect of VEGF is predominantly mediated by eNOS (Fukumura et al., 2001). To test whether eNOS-deficient mice have an impaired response to VEGF-induced angiogenesis, a corneal angiogenesis assay was performed. Figure 3K-N shows that, although VEGF induces corneal angiogenesis in wild-type mice (Fig. 3L) and eNOS-/- mice (Fig. 3M), a significant decrease in VEGF-induced angiogenesis is present in eNOS-/- mice compared with wild-type mice (Fig. 3N). These data suggest that eNOS plays an important but not a determining role in VEGF-induced angiogenesis.
eNOS-deficient mice have decreased SVZ cell proliferation and migration in the ischemic brain after stroke
BrdU, which is incorporated in the S-phase of the cell cycle, is commonly used to detect cell proliferation. Ki-67, a nuclear protein expressed in all phases of the cell cycle except the resting phase, is used as a marker of proliferation in the initial phase of adult neurogenesis (Kee et al., 2002). To test whether eNOS-deficient mice show reduced SVZ cell proliferation after stroke in the ischemic brain, Ki-67 and BrdU immunostaining were performed. Figure 4A-F shows that Ki-67- and BrdU-positive cells significantly decreased in the ipsilateral SVZ in eNOS-/- mice (Fig. 4B,C, Ki-67, E,F, BrdU) compared with wild-type mice (Fig. 4A,C, Ki-67, D,F, BrdU) at 7 d after stroke. To measure whether eNOS regulates neuronal cell migration, DCX (a migrating neuronal marker) immunostaining was performed. Figure 4G-I shows that the DCX-immunoreactive cell area significantly decreases in eNOS-/- mice (Fig. 4H,I) compared with wild-type mice (Fig. 4G,I) at 7 d after stroke. Some BrdU-positive cells (Fig. 4J) colocalized with DCX-immunoreactive cells (Fig. 4K). Nestin is a marker of immature neuroepithelial cells. Our data show that ischemia induces nestin expression in the ischemic border, and eNOS-deficient mice (Fig. 4N,O) show significantly decreased nestin expression in the ischemic border area compared with wild-type mice (Fig. 4M,O) (p < 0.05). Thus, eNOS-deficient mice exhibit decreased neuroepithelial stem cell expression. Together, these data suggest that eNOS contributes to neurogenesis and cell migration after stroke in the ischemic brain.
Figure 4.
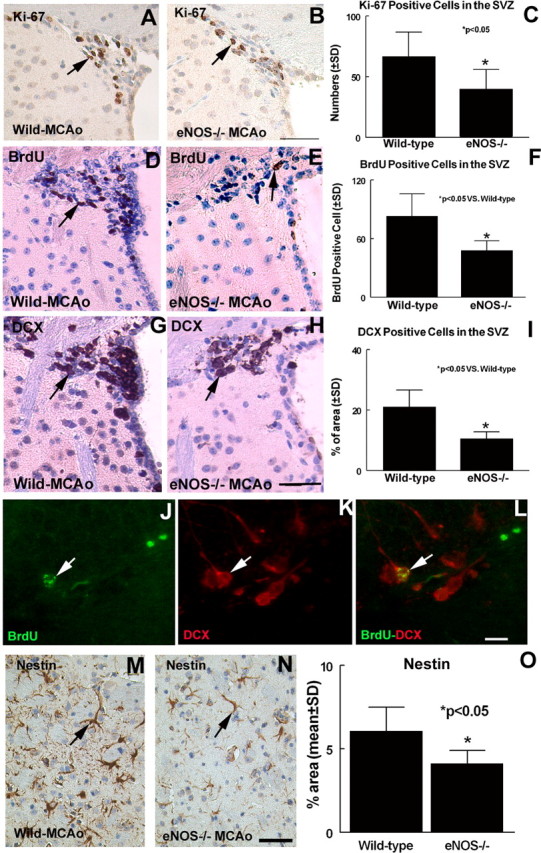
eNOS-deficient mice exhibit decreased neurogenesis after stroke. BrdU, Ki-67, DCX, and nestin are expressed in the ipsilateral SVZ at 7 d after MCAo. A, D, G, M, Ki-67 (A), BrdU (D), DCX (G), and nestin (M) expression in wild-type MCAo mice, respectively. B, E, H, N, Ki-67 (B), BrdU (E), DCX (H), and nestin (N) expression in eNOS-/- mice, respectively. C, F, I, O, Quantitative data of Ki-67-positive (C), BrdU-positive (F), DCX-positive (I), and nestin-positive (O) cell number or percentage of area in the ischemic brain. J-L, DCX (K) with BrdU (J) double immunoreactivity (red arrow, DCX-positive cell; green arrow, BrdU-positive cell). Scale bars: B, H, N, 50 μm; L, 20 μm.
eNOS-deficient mice have decreased BDNF but not VEGF and bFGF expression in the ischemic brain
To investigate the molecular mechanisms underlying reduced angiogenesis and neurogenesis in eNOS-deficient mice, growth factor (i.e., VEGF, bFGF, and BDNF) ELISAs were performed in the ischemic brain at 3 d after stroke. ELISAs reveal that eNOS-/- mice exhibit significantly decreased BDNF (1.8 ± 1.4 pg/μg tissue lysate) expression in the ischemic brain compared with wild-type mice (5.2 ± 2.1 pg/μg tissue lysate; p < 0.05) but not VEGF (eNOS-/- mice, 0.06 ± 0.03 pg/μg tissue lysate; wild-type mice, 0.03 ± 0.02 pg/μg tissue lysate; p > 0.05) and bFGF (eNOS-/- mice, 0.9 ± 0.14 pg/μg tissue lysate; wild-type mice, 1.53 ± 0.39 pg/μg tissue lysate; p > 0.05). These data suggest that decreased BDNF expression after stroke may cause the decreased neurogenesis in eNOS-/- mice.
An absence of eNOS decreases SVZ neurosphere formation and progenitor cell proliferation
To further investigate whether eNOS regulates SVZ progenitor cell proliferation and survival, measurements on SVZ neurosphere cultures were performed. SVZ sphere-producing progenitor cells are self-renewing and mutipotent potential progenitor cells (Reynolds and Weiss, 1996). Putative mutipotent cells were isolated by primary neurosphere formation, and their self-renewing capacity was demonstrated by observing the number of secondary neurospheres formed (Reynolds and Weiss, 1996; Nakamura et al., 2000). Figure 5A-E shows that neurospheres obtained from eNOS-/- mice (Fig. 5B,D) exhibit a significant decrease in primary neurosphere formation compared with wild-type mice SVZ neurospheres (Fig. 5A,D)(p < 0.05). In addition, the secondary neurosphere formation number derived from eNOS-/- mice is significantly decreased compared with wild-type mice (Fig. 5E) (p < 0.05). However, the decline in neurosphere formation could result from changes in cell proliferation and/or survival of SVZ progenitor cells. To obtain further insight into the decrease of neurosphere formation in eNOS-/- mice, SVZ neurosphere proliferation and survival were measured. Using a BrdU ELISA, Figure 5F reveals that SVZ neurospheres derived from eNOS-/- mice show a decrease in BrdU-labeled cell proliferation in cultured SVZ neurospheres compared with wild-type mice (p < 0.05). Using livedead viability measurements, Figure 5G shows that SVZ neurosphere cells derived from eNOS-/- mice exhibited no significant difference in cell viability compared with wild-type mice (p > 0.05).
Figure 5.

SVZ neurosphere formation, proliferation, and survival and neurite outgrowth measurements. A-C, SVZ neurosphere formation in wild-type mouse (A), eNOS-/- mouse (B), and eNOS-/- mouse SVZ neurosphere cells treated with BDNF (C). D, E, Quantitative data of primary (D) and secondary (E) SVZ neurosphere formation. F, Quantitative data of a BrdU proliferation ELISA. G, Results of livedead viability measurement. H-J, TUJ1-positive cell neurite outgrowth in cultured SVZ neurosphere cells of wild-type mice (H), eNOS-/- mice (I), and eNOS-/- mice treated with BDNF (J). K, quantitative data of SVZ neuronal cell neurite outgrowth. Scale bar, 100 μm.
As noted above, in vivo data demonstrate that the absence of eNOS decreases BDNF secretion in the ischemic brain. To determine whether the decline in eNOS-/- neuronal progenitor cell proliferation is related to BDNF secretion, BDNF was added to SVZ neurosphere cells derived from eNOS-deficient mice. Figure 5C-G shows that BDNF significantly increases the neurosphere formation (Fig. 5C-E) and proliferation measured using a BrdU-labeling ELISA (Fig. 5F) in eNOS-/- mice SVZ neurospheres (p < 0.05). These data indicate that eNOS directly affects SVZ neurosphere formation by promoting neuronal progenitor cell proliferation but does not change cell viability. These data also suggest that the absence of eNOS decreases neural progenitor cell mutipotent potential and self-renewing capacity, and BDNF reverses eNOS-deficient reduced SVZ neurosphere formation and proliferation.
eNOS-deficient SVZ cells exhibit decreased neurite outgrowth
To test whether eNOS regulates SVZ progenitor cell neurite outgrowth, TUJ1 immunostaining was performed in cultured SVZ neurosphere cells. The total neurite outgrowth of length in 100 TUJ1-positive cells was measured. Figure 5H-K shows that eNOS-deficient cells (Fig. 5I,K) show a significant decrease in the total length of TUJ1-positive SVZ cell neurite outgrowth compared with wild-type mice (Fig. 5H,K). BDNF significantly increases the neurite outgrowth (Fig. 5J,K) in eNOS-/- mice SVZ neurospheres (p < 0.05).
eNOS-deficient SVZ neurosphere cells exhibit decreased telomerase activity
To determine telomerase activity in wild-type and eNOS-/- mouse cultured SVZ neurosphere cells, the Telo TAGGG telomerase PCR ELISA was performed. The data show that eNOS-/- mouse SVZ neurosphere cells significantly decrease telomerase activity (0.22 ± 0.01) compared with wild-type mouse SVZ neurosphere cells (0.25 ± 0.01; p < 0.05). BDNF treatment significantly increases telomerase activity (0.38 ± 0.01) compared with eNOS-/- mice. The data suggest that the absent of eNOS decreases SVZ progenitor cell telomerase activity. BDNF reverses eNOS-deficient reduced telomerase activity. The decreased telomerase activity may reduce progenitor cell proliferation.
SVZ tissue explant migration is impaired in eNOS-/- mice
To demonstrate whether eNOS regulates SVZ progenitor cell migration, SVZ explant migration was measured in both wild-type and eNOS-/- mouse SVZ explants. Figure 6 shows that SVZ tissue explants from eNOS-/- mice exhibit a significantly decreased migration distance (Fig. 6C-E)(p < 0.05) and migration area (Fig. 6F) compared with wild-type SVZ tissue explants (Fig. 6A,B,E,F)(p < 0.05). The data suggest that the absence of eNOS decreases neuronal progenitor cell migration and also decreases migrating cell numbers. These data are consistent with in vivo data.
Figure 6.

SVZ tissue explant cell migration and neurite outgrowth. A, B, Wild-type mouse SVZ explant migration at 3 DIV (A) and 5 DIV (B). C, D, eNOS-/- mouse SVZ explant migration at 3 DIV (C) and 5 DIV (D). E, F, Quantitative data of SVZ tissue explant migration distance (E) and migration area (F). Scale bar, 500 μm.
Discussion
We demonstrate for the first time that the absence of eNOS not only impairs angiogenesis but also decreases neurogenesis and attenuates neurological functional recovery after stroke. In addition, our results identify a role for eNOS in regulating neuronal progenitor cell proliferation, survival, migration, neurite out-growth, and telomerase activity in cultured SVZ progenitor cells. Our data are the first to show that eNOS-deficient mice have reduced BDNF expression in the ischemic brain after stroke, and complementary in vitro data support the hypothesis that reduced BDNF in eNOS-/- mice contributes to the impaired neurogenesis in the ischemic brain.
eNOS-deficient mice have attenuated functional recovery after stroke
eNOS has multiple effects on regulating blood flow, e.g., vasodilation of cerebral vessels supplying the ischemic penumbra and leukocyte-endothelial interactions, and is neuroprotective (Endres et al., 2003). Physical activity upregulates eNOS expression in the vasculature and protects against ischemic stroke (Endres et al., 2003). Constitutively expressed eNOS is necessary for good endothelial function and integrity (Albrecht et al., 2003), and eNOS-/- mice are hypertensive. These data are consistent with our observations that eNOS-/- mice exhibit increased blood pressure and a neurological functional deficit after stroke. There is a trend but not a statistically significant increase of infarct volumes in eNOS-/- mice compared with wild-type mice at 7 d after stroke. Huang et al. (1996) showed a significantly enlarged brain infarct volume in eNOS-/- mice at 24 h after stroke compared with wild-type mice. This discrepancy may be attributed to differences in experimental protocols used between both studies.
eNOS mediates angiogenesis
eNOS regulates both angiogenesis and vasculogenesis (Duda et al., 2004). Molecules able to increase NO levels induce endothelial cell proliferation and migration in vivo and in vitro (Ziche and Morbidelli, 2000; Cooke, 2003). Endothelium-derived NO plays a critical role in the regulation of vascular function and structure and is a downstream mediator of the angiogenic response to numerous vascular growth factors (Babaei et al., 1998). eNOS plays a prominent role in VEGF-induced proliferation of endothelial cells and angiogenesis (Fukumura et al., 2001). VEGF upregulation of eNOS expression has been shown to be mediated via VEGF receptor 2, although there is controversy about which of the VEGF receptors triggers the release of NO in endothelial cells (Kroll and Waltenberger, 1999). NOS inhibitors block VEGF-induced endothelial cell migration and proliferation (Ziche et al., 1997). In the present study, we demonstrated that cerebral ischemic tissue in eNOS-/- mice has decreased endothelial cell proliferation, vascular density, and perimeter after stroke in the ischemic border but did not exhibit reduced VEGF expression in the ischemic brain. In addition, eNOS-/- mice showed an angiogenic response, albeit a reduced response to VEGF-induced angiogenesis in the corneal assay. These results are consistent with previous studies that have demonstrated that eNOS-derived NO acts as a downstream signal for VEGF-induced angiogenesis (Ziche et al., 1997). Our data suggest that reduced angiogenesis in eNOS-/- mice is not attributable to a decrease in VEGF.
eNOS regulates neurogenesis
The adult SVZ contains neural stem and progenitor cells that generate neurons and glia throughout adulthood (Gage, 2000). Neuronal regeneration is present after stroke (Zhang et al., 2001b). The regeneration of new neurons in the adult is primarily restricted to the SVZ and the subgranular zone of the hippocampus (Gage, 2000). eNOS is expressed not only in endothelial cells but also in the neurons of brain (Shin et al., 2004). NO regulates neuronal progenitor cell proliferation and differentiation (Zhang et al., 2001a; Reif et al., 2004) and is a putative participant in synaptic plasticity, and exogenous NO elicits brain plasticity (Zhang et al., 2001a). Therefore, it is reasonable to consider that eNOS deficiency may reduce neurogenesis in the ischemic brain. Nestin is an intermediate filament protein expressed in neural stem cells, as well as in lineage-constricted progenitors, immature neurons, and some radial glia neuroepithelial cells (Fukuda et al., 2003). We found that eNOS-/- mice had reduced nestin expression in the ischemic border. eNOS-/- mice also exhibited decreased SVZ progenitor cell proliferating activity, as shown by BrdU labeling and Ki-67 immunostaining. Postmitotic neurons derived from neural progenitor cells subsequently migrate away from the SVZ. Newly generated neurons migrate from the SVZ to the ischemic striatum after stroke (Zhang et al., 2001b). Our data show that eNOS-/- mice exhibit a decrease of doublecortin-positive cell expression (neuronal migration) in the SVZ after stroke. Thus, eNOS contributes to neuronal precursor cell proliferation and migration after stroke.
eNOS regulates telomerase activity
Telomerase is a reverse transcriptase that maintains chromosome ends (telomeres) during successive cell divisions in mitotic cells (Fu et al., 2002). Telomerase plays a major role in the control of replicative capacity, a critical property for successful angiogenesis and maintenance of endothelial integrity. Stable expression of TERT results in endothelial cells with a younger phenotype and with greater amounts of eNOS and NO activity. Exogenous NO increases telomerase activity and delays endothelial cell senescence in culture (Vasa et al., 2000). TERT expression is increased in embryonic hippocampal neurons by BDNF (Fu et al., 2002). Downregulation of TERT in the adult brain contributes to increased neuronal vulnerability in various age-related neurodegenerative disorders (Fu et al., 2000). The telomerase activity in stem and progenitor cells correlates with their proliferative capacity, and telomerase is active in neuronal precursor cells of the adult SVZ (Caporaso et al., 2003). Our data show that eNOS-/- mice exhibit decreased endothelial cell and SVZ progenitor cell proliferation, as well as decreased endothelial and SVZ progenitor cell telomerase activity. BDNF increases SVZ neurosphere cell telomerase activity and proliferation. Inhibition of telomerase activity promotes decreases in cell proliferation (Haendeler et al., 2003), which may be related to the attenuated angiogenesis and neurogenesis after stroke in eNOS-/- mice.
Molecular mechanism underlying eNOS regulation neurogenesis
BDNF is a mitogenic factor, promotes the migration of granule cells, and enhances SVZ neuronal progenitor cell proliferation (Borghesani et al., 2002). Our data show that eNOS-/- mice exhibit decreased BDNF expression in the ischemic brain. Therefore, an absence of eNOS is associated with a downregulation of BDNF expression, which possibly reduces neurogenesis after stroke. However, the mechanism by which eNOS regulates BDNF secretion is not fully understood. Previous studies demonstrated that adult neurogenesis in the subgranular zone occurs within an angiogenic niche (Palmer et al., 2000; Louissaint et al., 2002). The newly activated and expanded vasculature substantially increases BDNF production and release, whose induction is both spatially and temporally associated with recruitment of new neurons (Leventhal et al., 1999) and also stimulates and attracts neuronal cell migration (Borghesani et al., 2002; Zhang et al., 2003). Therefore, decreased neurogenesis present in eNOS-/- mice may be caused by impaired angiogenesis and endothelial cell proliferation, which subsequently decreases BDNF expression after stroke in eNOS-/- mice. In addition, nNOS regulates BDNF secretion, and BDNF effects on GAP-43 mRNA expression are mediated by an NO-dependent mechanism (Klocker et al., 2001). Another possibility is that eNOS or NO directly regulates BDNF secretion and expression. However, in the in vivo study, we were unable to tease out the affect of the influence of the angiogenic niche on neurogenesis in the ischemic brain from a direct coupling of eNOS and BDNF. To elucidate the effects of eNOS on neurogenesis alone, SVZ neurosphere cultures were used. Putative multipotent progenitor cells can be isolated using the neurosphere formation assay, and successive neurosphere proliferation and differentiation assays have been used to show that SVZ progenitor cells self-renew, their multipotency giving rise to both neurons and glia (Haughey et al., 2002). Our data demonstrate that eNOS-deficient SVZ cells have decreased SVZ progenitor cell proliferation and migration. However, addition of BDNF in the culture rescues the reduced SVZ neurosphere formation, proliferation, and neurite outgrowth in eNOS-/- cells. eNOS appears to have a direct effect on regulating SVZ neuronal progenitor cell proliferation and migration. There are two possible sources for eNOS in SVZ neurospheres: (1) neural stem and progenitor cells are capable of producing endothelial and mural cells (Oishi et al., 2004) and thus progenitor endothelial cells produced by the neurosphere express eNOS; and (2) eNOS is expressed not only in endothelial cells but also in neurons (Dinerman et al., 1994).
Our data show that adult eNOS-deficient mice have decreased BDNF secretion in the ischemic brain. However, Canossa et al. (2002) reported in studies with cultured embryonic hippocampal neurons that l-N-ω-nitro-l-arginine methyl ester, a nonspecific NOS inhibitor, increased BDNF secretion, and an NO donor decreased BDNF secretion. In contrast to our in vivo studies in adult cerebral tissue, their studies were performed in vitro with embryonic cells. Thus, the apparent discrepancy may be resolved by the different experimental conditions of the contrasting studies.
Summary
In this study, an analysis of the angiogenesis and neurogenesis in eNOS-/- mice after stroke supports the hypothesis that eNOS is important not only for the regulation of angiogenesis but also for aspects of brain plasticity, such as the promotion of neuronal progenitor cell proliferation, migration, neurite outgrowth, and telomerase activity. eNOS is thus an important target gene for new therapeutic strategies in brain ischemia.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grants PO1 NS23393 and RO1 NS047682. We thank Dr. Edward G. Shesely (Hypertension and Vascular Research Division, Henry Ford Hospital, Detroit, MI) for providing eNOS-/- mice.
Correspondence should be addressed to Dr. Michael Chopp, Neurology Research, Henry Ford Hospital, Education and Research Building, Room 3056, 2799 West Grand Boulevard, Detroit, MI 48202. E-mail:chopp@neuro.hfh.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/252366-10$15.00/0
References
- Albrecht EW, Stegeman CA, Heeringa P, Henning RH, van Goor H (2003) Protective role of endothelial nitric oxide synthase. J Pathol 199: 8-17. [DOI] [PubMed] [Google Scholar]
- Babaei S, Teichert-Kuliszewska K, Monge JC, Mohamed F, Bendeck MP, Stewart DJ (1998) Role of nitric oxide in the angiogenic response in vitro to basic fibroblast growth factor. Circ Res 82: 1007-1015. [DOI] [PubMed] [Google Scholar]
- Borghesani PR, Peyrin JM, Klein R, Rubin J, Carter AR, Schwartz PM, Luster A, Corfas G, Segal RA (2002) BDNF stimulates migration of cerebellar granule cells. Development 129: 1435-1442. [DOI] [PubMed] [Google Scholar]
- Calza L, Giardino L, Giuliani A, Aloe L, Levi-Montalcini R (2001) Nerve growth factor control of neuronal expression of angiogenetic and vasoactive factors. Proc Natl Acad Sci USA 98: 4160-4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canossa M, Giordano E, Cappello S, Guarnieri C, Ferri S (2002) Nitric oxide down-regulates brain-derived neurotrophic factor secretion in cultured hippocampal neurons. Proc Natl Acad Sci USA 99: 3282-3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Linden P, Farnebo J, Cao R, Eriksson A, Kumar V, Qi JH, Claesson-Welsh L, Alitalo K (1998) Vascular endothelial growth factor C induces angiogenesis in vivo. Proc Natl Acad Sci USA 95: 14389-14394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Li H, Deb S, Liu JP (2002) TERT regulates cell survival independent of telomerase enzymatic activity. Oncogene 21: 3130-3138. [DOI] [PubMed] [Google Scholar]
- Caporaso GL, Lim DA, Alvarez-Buylla A, Chao MV (2003) Telomerase activity in the subventricular zone of adult mice. Mol Cell Neurosci 23: 693-702. [DOI] [PubMed] [Google Scholar]
- Chen J, Li Y, Wang L, Zhang Z, Lu D, Lu M, Chopp M (2001) Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke 32: 1005-1011. [DOI] [PubMed] [Google Scholar]
- Chen J, Zhang ZG, Li Y, Wang Y, Wang L, Jiang H, Zhang C, Lu M, Katakowski M, Feldkamp CS, Chopp M (2003) Statins induce angiogenesis, neurogenesis, and synaptogenesis after stroke. Ann Neurol 53: 743-751. [DOI] [PubMed] [Google Scholar]
- Chen J, Li Y, Zhang R, Katakowski M, Gautam SC, Xu Y, Lu M, Zhang Z, Chopp M (2004) Combination therapy of stroke in rats with a nitric oxide donor and human bone marrow stromal cells enhances angiogenesis and neurogenesis. Brain Res 1005: 21-28. [DOI] [PubMed] [Google Scholar]
- Cheng A, Wang S, Cai J, Rao MS, Mattson MP (2003) Nitric oxide acts in a positive feedback loop with BDNF to regulate neural progenitor cell proliferation and differentiation in the mammalian brain. Dev Biol 258: 319-333. [DOI] [PubMed] [Google Scholar]
- Cooke JP (2003) NO and angiogenesis. Atheroscler Suppl 4: 53-60. [DOI] [PubMed] [Google Scholar]
- Dere E, De Souza Silva MA, Topic B, Fiorillo C, Li JS, Sadile AG, Frisch C, Huston JP (2002) Aged endothelial nitric oxide synthase knockout mice exhibit higher mortality concomitant with impaired open-field habituation and alterations in forebrain neurotransmitter levels. Genes Brain Behav 1: 204-213. [DOI] [PubMed] [Google Scholar]
- Dinerman JL, Dawson TM, Schell MJ, Snowman A, Snyder SH (1994) Endothelial nitric oxide synthase localized to hippocampal pyramidal cells: implications for synaptic plasticity. Proc Natl Acad Sci USA 91: 4214-4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doreulee N, Sergeeva OA, Yanovsky Y, Chepkova AN, Selbach O, Godecke A, Schrader J, Haas HL (2003) Cortico-striatal synaptic plasticity in endothelial nitric oxide synthase deficient mice. Brain Res 964: 159-163. [DOI] [PubMed] [Google Scholar]
- Duda DG, Fukumura D, Jain RK (2004) Role of eNOS in neovascularization: NO for endothelial progenitor cells. Trends Mol Med 10: 143-145. [DOI] [PubMed] [Google Scholar]
- Endres M, Gertz K, Lindauer U, Katchanov J, Schultze J, Schrock H, Nickenig G, Kuschinsky W, Dirnagl U, Laufs U (2003) Mechanisms of stroke protection by physical activity. Ann Neurol 54: 582-590. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Krupinski J, Goutan E, Marti E, Ambrosio S, Arenas E (2001) Brain-derived neurotrophic factor reduces cortical cell death by ischemia after middle cerebral artery occlusion in the rat. Acta Neuropathol (Berl) 101: 229-238. [DOI] [PubMed] [Google Scholar]
- Fu W, Killen M, Culmsee C, Dhar S, Pandita TK, Mattson MP (2000) The catalytic subunit of telomerase is expressed in developing brain neurons and serves a cell survival-promoting function. J Mol Neurosci 14: 3-15. [DOI] [PubMed] [Google Scholar]
- Fu W, Lu C, Mattson MP (2002) Telomerase mediates the cell survival-promoting actions of brain-derived neurotrophic factor and secreted amyloid precursor protein in developing hippocampal neurons. J Neurosci 22: 10710-10719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda S, Kato F, Tozuka Y, Yamaguchi M, Miyamoto Y, Hisatsune T (2003) Two distinct subpopulations of nestin-positive cells in adult mouse dentate gyrus. J Neurosci 23: 9357-9366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumura D, Gohongi T, Kadambi A, Izumi Y, Ang J, Yun CO, Buerk DG, Huang PL, Jain RK (2001) Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc Natl Acad Sci USA 98: 2604-2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage FH (2000) Mammalian neural stem cells. Science 287: 1433-1438. [DOI] [PubMed] [Google Scholar]
- Gasparri F, Mariani M, Sola F, Galvani A (2004) Quantification of the proliferation index of human dermal fibroblast cultures with the ArrayScan high-content screening reader. J Biomol Screen 9: 232-243. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Zeiler SR, Tamowski S, Jones KR (2003) Brain-derived neurotrophic factor is required for the maintenance of cortical dendrites. J Neurosci 23: 6856-6865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haendeler J, Hoffmann J, Rahman S, Zeiher AM, Dimmeler S (2003) Regulation of telomerase activity and anti-apoptotic function by protein-protein interaction and phosphorylation. FEBS Lett 536: 180-186. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Liu D, Nath A, Borchard AC, Mattson MP (2002) Disruption of neurogenesis in the subventricular zone of adult mice, and in human cortical neuronal precursor cells in culture, by amyloid beta-peptide: implications for the pathogenesis of Alzheimer's disease. Neuromol Med 1: 125-135. [DOI] [PubMed] [Google Scholar]
- Hernandez TD, Schallert T (1988) Seizures and recovery from experimental brain damage. Exp Neurol 102: 318-324. [DOI] [PubMed] [Google Scholar]
- Huang Z, Huang PL, Ma J, Meng W, Ayata C, Fishman MC, Moskowitz MA (1996) Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-l-arginine. J Cereb Blood Flow Metab 16: 981-987. [DOI] [PubMed] [Google Scholar]
- Kee N, Sivalingam S, Boonstra R, Wojtowicz JM (2002) The utility of Ki-67 and BrdU as proliferative markers of adult neurogenesis. J Neurosci Methods 115: 97-105. [DOI] [PubMed] [Google Scholar]
- Klocker N, Jung M, Stuermer CA, Bahr M (2001) BDNF increases the number of axotomized rat retinal ganglion cells expressing GAP-43, L1, and TAG-1 mRNA—a supportive role for nitric oxide? Neurobiol Dis 8: 103-113. [DOI] [PubMed] [Google Scholar]
- Kroll J, Waltenberger J (1999) A novel function of VEGF receptor-2 (KDR): rapid release of nitric oxide in response to VEGF-A stimulation in endothelial cells. Biochem Biophys Res Commun 265: 636-639. [DOI] [PubMed] [Google Scholar]
- Leventhal C, Rafii S, Rafii D, Shahar A, Goldman SA (1999) Endothelial trophic support of neuronal production and recruitment from the adult mammalian subependyma. Mol Cell Neurosci 13: 450-464. [DOI] [PubMed] [Google Scholar]
- Li Y, Jiang N, Powers C, Chopp M (1998) Neuronal damage and plasticity identified by microtubule-associated protein 2, growth-associated protein 43, and cyclin D1 immunoreactivity after focal cerebral ischemia in rats. Stroke 29: 1972-1981. [DOI] [PubMed] [Google Scholar]
- Lois C, Garcia-Verdugo JM, Alvarez-Buylla A (1996) Chain migration of neuronal precursors. Science 271: 978-981. [DOI] [PubMed] [Google Scholar]
- Louissaint Jr A, Rao S, Leventhal C, Goldman SA (2002) Coordinated interaction of neurogenesis and angiogenesis in the adult songbird brain. Neuron 34: 945-960. [DOI] [PubMed] [Google Scholar]
- Mao Y, Yang GY, Zhou LF, Stern JD, Betz AL (1999) Focal cerebral ischemia in the mouse: description of a model and effects of permanent and temporary occlusion. Brain Res Mol Brain Res 63: 366-370. [DOI] [PubMed] [Google Scholar]
- Morshead CM, Reynolds BA, Craig CG, McBurney MW, Staines WA, Morassutti D, Weiss S, van der Kooy D (1994) Neural stem cells in the adult mammalian forebrain: a relatively quiescent subpopulation of subependymal cells. Neuron 13: 1071-1082. [DOI] [PubMed] [Google Scholar]
- Murohara T, Asahara T, Silver M, Bauters C, Masuda H, Kalka C, Kearney M, Chen D, Symes JF, Fishman MC, Huang PL, Isner JM (1998) Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J Clin Invest 101: 2567-2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Sakakibara S, Miyata T, Ogawa M, Shimazaki T, Weiss S, Kageyama R, Okano H (2000) The bHLH gene Hes1 as a repressor of the neuronal commitment of CNS stem cells. J Neurosci 20: 283-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M, Gjedde A, Diemer NH (1987) Hyperglycaemia protects against neuronal injury around experimental brain infarcts. Neurol Res 9: 241-244. [DOI] [PubMed] [Google Scholar]
- Oishi K, Kobayashi A, Fujii K, Kanehira D, Ito Y, Uchida MK (2004) Angiogenesis in vitro: vascular tube formation from the differentiation of neural stem cells. J Pharmacol Sci 96: 208-218. [DOI] [PubMed] [Google Scholar]
- Palmer TD, Willhoite AR, Gage FH (2000) Vascular niche for adult hippocampal neurogenesis. J Comp Neurol 425: 479-494. [DOI] [PubMed] [Google Scholar]
- Reif A, Schmitt A, Fritzen S, Chourbaji S, Bartsch C, Urani A, Wycislo M, Mossner R, Sommer C, Gass P, Lesch KP (2004) Differential effect of endothelial nitric oxide synthase (NOS-III) on the regulation of adult neurogenesis and behaviour. Eur J Neurosci 20: 885-895. [DOI] [PubMed] [Google Scholar]
- Reynolds BA, Weiss S (1996) Clonal and population analyses demonstrate that an EGF-responsive mammalian embryonic CNS precursor is a stem cell. Dev Biol 175: 1-13. [DOI] [PubMed] [Google Scholar]
- Rudic RD, Shesely EG, Maeda N, Smithies O, Segal SS, Sessa WC (1998) Direct evidence for the importance of endothelium-derived nitric oxide in vascular remodeling. J Clin Invest 101: 731-736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma HS, Nyberg F, Westman J, Alm P, Gordh T, Lindholm D (1998) Brain derived neurotrophic factor and insulin like growth factor-1 attenuate upregulation of nitric oxide synthase and cell injury following trauma to the spinal cord: an immunohistochemical study in the rat. Amino Acids 14: 121-129. [DOI] [PubMed] [Google Scholar]
- Shin T, Weinstock D, Castro MD, Hamir AN, Wampler T, Walter M, Kim HY, Acland H (2004) Immunohistochemical localization of endothelial and inducible nitric oxide synthase within neurons of cattle with rabies. J Vet Med Sci 66: 539-541. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR (1990) A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab 10: 290-293. [DOI] [PubMed] [Google Scholar]
- Vasa M, Breitschopf K, Zeiher AM, Dimmeler S (2000) Nitric oxide activates telomerase and delays endothelial cell senescence. Circ Res 87: 540-542. [DOI] [PubMed] [Google Scholar]
- Zhang H, Vutskits L, Pepper MS, Kiss JZ (2003) VEGF is a chemoattractant for FGF-2-stimulated neural progenitors. J Cell Biol 163: 1375-1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Zhang L, Zhang Z, Wang Y, Lu M, Lapointe M, Chopp M (2001a) A nitric oxide donor induces neurogenesis and reduces functional deficits after stroke in rats. Ann Neurol 50: 602-611. [DOI] [PubMed] [Google Scholar]
- Zhang RL, Zhang ZG, Zhang L, Chopp M (2001b) Proliferation and differentiation of progenitor cells in the cortex and the subventricular zone in the adult rat after focal cerebral ischemia. Neuroscience 105: 33-41. [DOI] [PubMed] [Google Scholar]
- Zhang ZG, Zhang L, Jiang Q, Chopp M (2002) Bone marrow-derived endothelial progenitor cells participate in cerebral neovascularization after focal cerebral ischemia in the adult mouse. Circ Res 90: 284-288. [DOI] [PubMed] [Google Scholar]
- Ziche M, Morbidelli L (2000) Nitric oxide and angiogenesis. J Neurooncol 50: 139-148. [DOI] [PubMed] [Google Scholar]
- Ziche M, Morbidelli L, Choudhuri R, Zhang HT, Donnini S, Granger HJ, Bicknell R (1997) Nitric oxide synthase lies downstream from vascular endothelial growth factor-induced but not basic fibroblast growth factor-induced angiogenesis. J Clin Invest 99: 2625-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]